Structural and Metabolic Remodeling of Skeletal Muscle in Heart Failure with Reduced Ejection Fraction: A Review: Beyond the Failing Heart
Mamata Chaudhari, Jamila Makhloufi, Benjamin Doelling, Raveena Kataria, Aruni Bhatnagar, Dinesh Kalra, Shahid Pervez Baba

TL;DR
Heart failure with reduced ejection fraction affects skeletal muscle, leading to fatigue and exercise intolerance due to metabolic and structural changes.
Contribution
This review highlights skeletal muscle metabolic remodeling as a central factor in heart failure-related exercise intolerance.
Findings
Skeletal muscle in heart failure shows mitochondrial dysfunction and impaired ATP production.
Oxidative and carbonyl stress worsen muscle dysfunction in heart failure patients.
Exercise training is effective in restoring skeletal muscle health in heart failure.
Abstract
Heart failure (HF) with reduced ejection fraction is a systemic disorder that extends beyond cardiac dysfunction and involves peripheral organs, particularly skeletal muscle. Exercise intolerance and fatigue are the hallmark manifestations of HF that strongly predict morbidity and mortality. Accumulating evidence suggests that intrinsic skeletal muscle abnormalities are key contributors to exercise intolerance in HF. In HF, skeletal muscle undergoes metabolic remodeling characterized by shifts in fiber type composition, mitochondrial dysfunction, and increased oxidative stress. Mitochondrial dysfunction, characterized by decreased mitochondrial density, impaired biogenesis, and reduced respiratory capacity, further compromises skeletal muscle performance. These alterations impair adenosine triphosphate (ATP) generation via oxidative phosphorylation, forcing reliance on less efficient…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
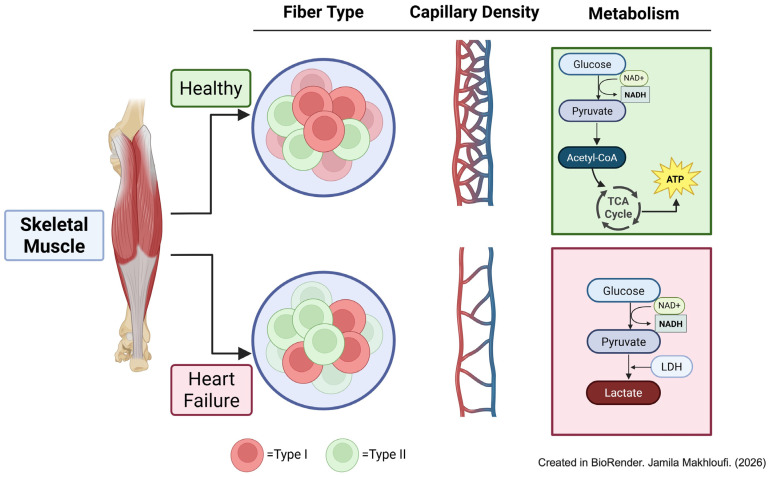 Figure 1
Figure 1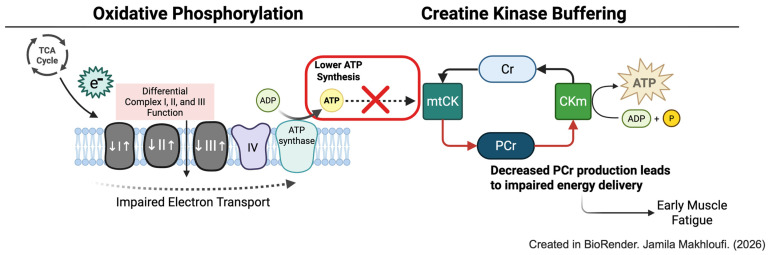 Figure 2
Figure 2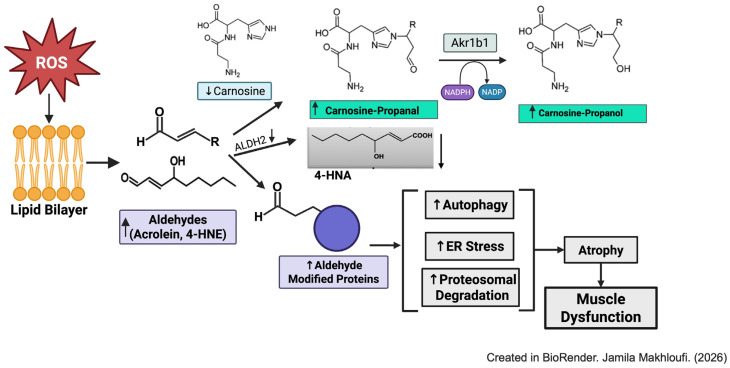 Figure 3
Figure 3- —National Institutes of Health
- —University of Louisville, School of Medicine
- —NIH
- —AHA predoctoral fellowship
- —NIH F31 fellowship
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiovascular and exercise physiology · Cardiovascular Function and Risk Factors · Muscle Physiology and Disorders
1. Introduction
Heart failure (HF) with reduced ejection fraction is a multifaceted clinical syndrome characterized by the heart’s inability to generate adequate cardiac output to meet the body’s metabolic demands. Among its systemic manifestations, exercise intolerance is a cardinal hallmark that profoundly diminishes quality of life and serves as a predictor of adverse outcomes. Traditionally, exercise intolerance in HF patients was primarily viewed through the lens of central hemodynamic impairment. However, cardiotonic agents intended to improve symptoms and exercise intolerance in these patients failed to improve exercise capacity despite improving hemodynamics [1,2]. This observation helped shift the focus toward peripheral factors.
Accumulating evidence demonstrates that skeletal muscle dysfunction and altered metabolism play a central role to the symptomatology of exercise intolerance in HF. Wilson et al. first reported this concept by showing that, during dobutamine infusion, HF patients exhibited increased cardiac output and leg blood flow during exercise, yet displayed no improvements in maximal exercise duration, peak systemic VO_2_, or lactate kinetics, indicating that intrinsic skeletal muscle defects substantially contribute to exertional fatigue in HF [3].
The purpose of this review is to summarize the structural, metabolic, and molecular alterations in skeletal muscle in HF with reduced ejection fraction. Furthermore, we will discuss emerging therapeutic interventions aimed at restoring skeletal muscle function and improving exercise tolerance in this patient population. Although, approximately half of all HF patients have preserved ejection fraction (HFpEF) and exhibit impaired exercise performance. Addressing the complex pathophysiological heterogeneity and the contribution of individual components to exercise intolerance in this patient population is beyond the scope of this review. Therefore, this review focuses on the contribution of skeletal muscle pathology to exercise intolerance in HF with reduced fraction.
2. Alterations in Skeletal Muscle Structure and Fiber Type During Heart Failure
Myocytes are the basic cellular units of skeletal muscle and contain a highly organized array of contractile proteins within sarcomeres, the fundamental functional units of striated muscle. Each sarcomere spans a few micrometers from Z-line to Z-line and consists of thick myosin and thin actin arranged in a repeating pattern that determines the contractile properties. The thin filament contains the regulatory proteins tropomyosin (Tm) and troponin (Tn), which mediate Ca^2+^-dependent regulation of contraction. When Ca^2+^ binds to Tn, Tm repositions, exposing myosin binding sites on actin and enabling cross-bridge cycling. Titin, a large elastic protein extending from the Z-line to the M-line, maintains sarcomere integrity and contributes to passive stiffness. In the relaxed state, myosin heads adopt a low-energy “super relaxed” conformation stabilized by Mg-ATP and low cytosolic Ca^2+^. Activation initiates asynchronous cross-bridging interactions between myosin heads and actin filaments, driven by ATP hydrolysis and regulated by Ca^2+^ binding [4].
Alterations in contractile protein composition and kinetics contribute significantly to the reduced skeletal muscle strength in HF. Reduced force production in HF is partly due to loss of myosin heavy chain (MHC) content and diminished myofibrillar density. In rat models of coronary artery ligation, maximal specific force, MHC abundance, and Ca^2+^ sensitivity of contraction is markedly reduced in diaphragm fibers [5]. Similarly, in HF patients, single myofibril studies reveal decreased MHC content, fewer cross-bridges, and lower maximal Ca^2+^-activated tension [6,7]. Beyond quantitative deficits, intrinsic alteration in myosin kinetics exacerbates functional decline. In vitro motility assays using purified myosin isolated from HF skeletal muscle show slowed cross-bridge cycling characterized by prolonged actomyosin attachment times, reduced Ca^2+^ sensitivity, and altered viscoelastic properties, indicating intrinsic molecular defects in contractile performance [8,9,10].
Skeletal muscle fibers are categorized into slow-twitch (Type I) and fast-twitch (Type II) based on their MHC isoforms. Fast-twitch fibers are further subdivided into IIa, IIx, and IIb. Type I and IIa fibers rely primarily on oxidative metabolism, whereas IIx and IIb rely on glycolytic metabolism [4]. Both human and animal studies demonstrate consistent remodeling of MHC composition in HF, profoundly affecting muscle function. In the rat coronary ligation model, expression of fast MHC isoforms (MHC IIx and IIb) increases in gastrocnemius and plantaris muscles, correlating with HF severity [11]. In dogs with chronic HF, type I fibers decrease while glycolytic type II fibers increase [12]. Mancini et al. reported increased type IIb fibers, selective atrophy of type II fiber, and appearance of type IIc fibers [13]. Histological studies reveal intramyocellular lipid accumulation endomysial fibrosis within type I fibers and type II fiber hypotrophy [14]. Lipkin et al. further described atrophy of both type I and II fibers, increased interstitial cellularity, lipid deposition, and acid phosphatase staining in HF biopsies [15]. This shift in the fiber type correlates with HF severity, higher NYHA functional class associated with increased MHCII expression and decreased MHCI abundance [16,17,18]. Collectively, these studies indicate maladaptive remodeling marked by loss of oxidative fibers, expansion of glycolytic fibers, and impaired contractile protein composition (Figure 1).
3. Skeletal Muscle Metabolism
3.1. Insulin Resistance and Exercise Intolerance
Insulin resistance is a common metabolic abnormality in HF and is strongly associated with reduced exercise capacity and adverse clinical outcomes [19,20]. Impaired insulin sensitivity correlates with skeletal muscle weakness and increased mortality risk [19]. In HF, skeletal muscle glucose uptake is reduced by ~20% compared with healthy individuals, and this impairment is consistently observed in both ischemic heart disease and idiopathic dilated cardiomyopathy. These observations suggest that systemic insulin resistance may arise secondary to HF rather than being directly related to ventricular dysfunction.
Although reduced skeletal muscle blood flow and abnormal sympathetic nervous system are present in HF, insulin resistance is primarily attributed to intrinsic metabolic and biochemical defects in the skeletal muscle. Normally, insulin signaling in skeletal muscle involves a cascade of phosphorylation events, leading to the activation of insulin receptor substrates (IRS), AKT phosphorylation, and the translocation of GLUT4 to the plasma membrane, facilitating glucose uptake. In HF, insulin signaling defects do not occur at the IRS-1/PI-3kinase/AKT phosphorylation levels. Instead, there is a reduction in GLUT4 transport protein in skeletal muscle [21]. Physiological hyperinsulinemia increases tyrosine phosphorylation of IRS and IRS-associated PI3-K; however, skeletal muscle glucose uptake in skeletal muscle remains unchanged [22]. Similarly, in mouse models of myocardial infarction (MI), protein levels and phosphorylation status of IRS-β, IRS-1, and PI-3K remain unchanged under both baseline and insulin-stimulated conditions, but serine phosphorylation of AKT and GLUT4 translocation are decreased in insulin-stimulated skeletal muscle [23]. Increased reactive oxygen species generated via NADPH oxidase have been reported to impair insulin signaling at the Akt level in HF skeletal muscle [23]. Recent findings also show that skeletal muscle-specific microRNA-133b, which targets the 3′-untranslated region (UTR) of KLF15- a transcription factor regulating GLUT4, is increased in the skeletal muscle of HF mice. Lowering miR-133b levels in skeletal myocytes improves glucose uptake and restores GLUT4 abundance in skeletal muscle [24].
Insulin resistance is additionally associated with reduced coronary flow reserve (CFR), a marker of coronary micro vessel formation [25], and improving insulin sensitivity via exercise training improves CFR [26]. However, it is not yet known whether improvements in CFR through exercise training directly translate into downstream enhancements in skeletal muscle metabolic function. Nonetheless, insulin resistance is intractably linked with skeletal muscle dysfunction and exercise intolerance.
3.2. Shift in Glucose Metabolism
In healthy individuals, skeletal muscle uses both glucose and fatty acids to produce ATP through oxidative phosphorylation; approximately 50% of absorbed muscle glucose is oxidized, 35% is stored, and 15% is released as lactate and pyruvate [27]. In HF skeletal muscle, however, metabolism shifts away from an efficient oxidative metabolism towards a less efficient glycolytic metabolism, unrelated to blood flow [28,29]. Phosphorus-31 (31-P) nuclear magnetic resonance studies demonstrate that, although baseline skeletal muscle pH is normal, HF patients experience a more rapid decline in pH during exercise and delayed metabolic recovery compared with normal subjects [30,31]. Lactate levels and lactate dehydrogenase activity are elevated, whereas oxidative enzyme activity, such as citrate synthetase, is decreased [32]. High-throughput RNA-Seq analysis of skeletal muscle from HF patients further confirms downregulation of lactate dehydrogenase C expression [33]. Collectively, these findings indicate a metabolic shift characterized by increased glycolytic and reduced oxidative enzyme activity in HF skeletal muscle.
4. Mitochondrial Defects in Skeletal Muscle
4.1. Ultrastructural Alteration in Mitochondria
Mitochondrial dysfunction occurs in skeletal muscle during the early, progressive, and late phases of heart failure. Severe heart failure patients exhibit reduced mitochondrial volume density, cristae surface density, and smaller mitochondria, indicating decreased oxidative capacity [34]. Mitochondria from the HF skeletal muscle present a hydropic degeneration, membrane disruption, and are smaller compared with the normal subjects [35]. Decreased mitochondrial volume correlates with peak exercise (VO_2_) and VO_2_ at anaerobic threshold [34]. Similarly, murine models of HF show reduced mitochondrial cristae density and increased mitochondrial damage, parallel with a decrease in exercise capacity and mitochondrial respiration in the skeletal muscle fibers [36].
4.2. Mitochondrial DNA and Biogenesis
Mitochondria contain their own DNA (mtDNA), encoding 13 proteins essential for mitochondrial respiration, packaged with nuclear encoded proteins, including ATPase family AAA domain-containing protein 3 (ATAD3), mitochondrial transcription factor A (TFAM), and POLG (DNA polymerase gamma, catalytic subunit), forming an mtDNA–protein complex. Among these, TFAM is critical for maintaining mitochondrial function. Although several studies in human and mouse models of heart failure report no changes in the mtDNA in the skeletal muscle; TFAM expression is significantly reduced in the angiotensin-induced heart failure models [37] and in rats subjected to coronary ligation [38]. Interestingly, while TFAM expression remains unaltered in skeletal muscle of human heart failure patients compared to healthy subjects, resistance exercise training increases TFAM expression, correlating with improvements in the muscle strength [39].
Deletion of Tfam in the skeletal muscle causes myopathy, accumulation of abnormal mitochondria, and reduced muscle forces [40], while TFAM overexpression protects against atrophy changes during disuse [41]. Recent evidence shows that sodium-glucose cotransporters (SGLT2) inhibitors promote mitochondrial biogenesis by upregulating TFAM expression [42]. Moreover, SGLT2-inhibitor therapy in heart failure patients improves peak VO_2_ consumption and oxygen uptake efficiency [43,44].
4.3. Mitochondrial Dynamics
Mitochondria undergo continuous remodeling through fusion and fission. Mitochondrial fusion allows two mitochondria to merge at the outer and inner membrane interfaces, a process mediated by three GTPases: mitofusin1 (MFN1), mitofusin2 (MFN2) on the outer membrane, and optic atrophy protein (OPA1) on the inner membrane. MFN1 and 2 belong to the dynamin-related proteins (DRP) superfamily, while OPA1 is the mammalian ortholog of Mgm1 [45,46,47]. Outer mitochondrial fusion is facilitated by MFN1 and MFN2, while inner membrane fusion is mediated by OPA1 and specific inner mitochondrial components, such as cardiolipin [47]. Mitochondrial fission, on the other hand, divides a mitochondrion into two smaller mitochondria, which is crucial for removing damaged organelles and prepares the mitochondria to be removed via mitophagy. Fission is primarily coordinated by dynamin-related protein, a large dynamin-like GTPase. Other proteins involved in the fission process include dynamin 2, human mitochondrial dynamics proteins 49 and 51, mitochondrial fission factor 1 protein (MFP1), and mitochondrial fission factor (MFF) [47,48,49,50]. In addition to the fusion and fission, mitochondrial quality control is maintained by mitochondria-specific autophagy termed mitophagy. Here, PTEN-induced putative kinase (PINK1), activates Parkin, an E3 ubiquitin ligase, which ubiquitinates outer mitochondrial membrane protein of damaged mitochondria. This ubiquitination promotes the recruitment of downstream autophagy adaptor proteins, such as P62 and Beclin 1, which interact with LC3 and facilitate the selective degradation of impaired mitochondria via autophagy [51,52,53,54,55].
In the gastrocnemius muscle of mice subjected to coronary artery ligation, the balance between mitochondrial fission and fusion is disrupted, with a lower ratio of OPA19 (fusion) and DRP1 (fission) protein levels. This imbalance is driven by reduced OPA1 protein content, suggesting increased mitochondrial fragmentation [56]. Interestingly, sex-specific divergences are observed in human heart failure patients. While transcript levels of MFN2 and DRP1 remain unchanged in the skeletal muscle of male HF patients, OPA1 expression is uniquely lower in female HF patients [57,58]. However, expression of F1S1, a regulator of mitochondrial fission, is unaffected, indicating a shift in equilibrium toward mitochondrial fragmentation and network disruption. OPA1 expression correlates with whole-body oxygen consumption (VO_2_ peak), suggesting that impaired mitochondrial fusion not only reduces mitochondrial capacity but also contributes to skeletal muscle dysfunction and worsens exercise tolerance.
Very few studies have directly examined the role of PINK1-Parkin pathway in regulating the skeletal muscle mitophagy, and therefore its specific contribution remains incompletely defined. Deletion of PINK1 in skeletal muscle reduces LC3-II flux in mitochondria-enriched skeletal muscle following endurance exercise, suggesting that Parkin may be required for exercise-induced autophagy; however, these studies did not directly assess mitophagy [59]. Similarly, studies using mice expressing the fluorescent reporter gene pMitotimer and subjected to acute treadmill showed that PINK1 did not accumulate in the mitochondria-enriched fractions, indicating that PINK1 is dispensable for exercise-induced mitophagy [60]. Despite these advances, information regarding the molecular drivers of mitochondrial fragmentation and mechanisms regulating mitochondrial quality in skeletal muscle during HF is unknown. Furthermore, no study has been able to clearly demonstrate that PINK1-Parkin-mediated mitochondrial ubiquitylation occurs in skeletal muscle either in normal or HF conditions. Additionally, the sex differences in mitochondrial dynamics of HF patients warrant further investigation to develop a personalized therapeutic strategy.
4.4. Defects in Oxidative Phosphorylation (OXPHOS)
OXPHOS is the primary mechanism of ATP generation. It is comprised of five inner mitochondrial–protein complexes: complex I, complex II, dimeric complex III, complex IV, and complex V. And it is comprised of two electron carriers: membrane-embedded hydrophobic ubiquinone and soluble cytochrome c. Together, these form an electron transport chain (ETC). Complexes I, III, and IV, function as proton pumps. Complex II reduces ubiquinone and serves as electron transporter for complex III, which reduces cytochrome c that, in turn, shuttles to complex IV and donates its electron for the final reduction of oxygen. The generated proton is used by the ATP synthase, which phosphorylates ADP to ATP.
Previous work assessing mitochondrial respiratory rates in skeletal muscle biopsies of HF patients and correlating these with whole-body exercise capacity using cardiopulmonary exercise test (CPET) has shown that mitochondrial respiration (complex I, II, and IV) as well as electron transport chain (ETC) capacity correlates strongly with VO_2 peak_, lactate threshold (VO_2LT_), and peak circulatory power CirP_peak_. Importantly, the mitochondrial ETC capacity contributes around 50% of the variability in VO_2LT_ peak, a key prognostic marker of exercise limitation in HF [61].
Numerous studies have reported OXPHOS dysregulation in skeletal muscle during the development of HF. In rats subjected to pressure overload-induced HF, a biphasic response of mitochondrial function was observed during the transition from compensated hypertrophy to heart failure [62]. During the first 6 weeks of compensated hypertrophy, state 3 respiration in the gastrocnemius and soleus increases, followed by a decline between 10 and 20 weeks of TAC. Notably, the ADP/O ratios remained unchanged during the transition from hypertrophy to heart failure. Similarly, activities of complex I and II activities are elevated in the gastrocnemius and soleus muscles during the compensated stage of cardiac remodeling but decrease substantially with the onset of HF (Figure 2). These changes in the activities of these complexes have been attributed to either changes in the mRNA and protein expression or impaired assembly of respiratory complex subunits to super complex. In human HF patients, decreases in complex activities are accompanied by a decrease in mRNA and protein expressions of complex I, complex II, and complex III [63,64]. Studies in pacing-induced HF in dogs further reveal that impaired oxidative phosphorylation is not only restricted to defects localized in the ETC but also in the components of the phosphorylation system, such as reduced ANT2 and increased ANT1 expression [65].
5. Phosphocreatine Depletion
The ATP generated by oxidative phosphorylation is transferred to mitochondrial creatine kinase (mi-CK), present on the outer surface of the inner mitochondrial membrane. mi-CK transfers the phosphate moiety of ATP to creatine, producing phosphocreatine (PCr) [66]. PCr plays a critical role as a rapid high-energy phosphate reservoir in skeletal muscle through CK, enabling immediate ATP production during periods of high-energy demand [67]. It is also crucial for cellular energy homeostasis during muscle contraction. PCr serves as an energy carrier for the connecting sites of energy production with energy utilization via subcellular compartmentalization. Several studies in both human subjects and animal models of HF have reported significantly reduced storage of PCr, accompanied by faster PCr depletion and slower recovery in the skeletal muscle under exercise conditions [68,69,70]. Muscle tissue from heart failure mice and humans showed mi-CK activity had a dramatic decrease, of approximately 60–80% compared with the controls (Figure 2) [71,72]. Nagai et al. further reported there is a significant correlation between the PCr breakdown and peakV(O)2 in both the arms and legs, as well as a close relationship between the ventilatory anaerobic threshold and the PCr breakdown [28]. These findings suggest that impaired PCr metabolism is closely linked to exercise intolerance in HF patients.
6. Oxidative Stress and Lipid Peroxidation in the Skeletal Muscle
Oxidative stress arises from an imbalance between oxidant production and antioxidant defenses, leading to excessive generation of reactive oxygen species (ROS), including superoxide anion (O_2_^−^), hydroxyl radical (•OH), and hydrogen peroxide (H_2_O_2_). ROS are generated through several processes, such as the mitochondrial respiratory chain, NAD(P)H oxidase, and xanthine oxidases (XO). In addition, impaired antioxidant capacity, resulting from decreased expression of antioxidant enzymes, further exacerbates oxidative stress.
Extensive clinical and experimental evidence shows that HF is associated with increased oxidative stress in skeletal muscle. In patients with HF, inducible NOS (iNOS) expression is elevated in skeletal muscle and inversely correlates with maximal oxygen uptake and exercise tolerance [73,74]. Moreover, xanthine oxidase (XO), which converts xanthine to uric acid while generating superoxide and hydrogen peroxide, is activated in the slow-twitch muscle fibers (type I and IIa) during the acute phase of myocardial infarction in mice [36]. This activation contributes to mitochondrial dysfunction, reduced exercise capacity, and apoptosis. Notably, pharmacological inhibition of XO with febuxostat prevents exercise intolerance without affecting left ventricular function or remodeling [36]. Studies in animal models of HF further show that overexpression of superoxide dismutase in the skeletal muscle or antioxidant supplementation improves muscle function [75,76,77]. However, clinical trials of antioxidants therapies have largely failed to demonstrate benefit in HF patients [77].
Excessive ROS generation induces the formation of highly reactive lipid peroxidation products, such as acrolein, 4-hydroxnonenal (4-HNE), and malondialdehyde, produced through the oxidation of polyunsaturated fatty acids (PUFA) in cellular membranes. These aldehydes possess two reactive carbonyl groups that form covalent adducts with amino acid side chains and nucleophilic sites of DNA, forming aldehyde-modified proteins and DNA adducts [78,79]. Lipid-derived aldehydes activate catabolic pathways including the ubiquitin proteosome pathway (UPS), autophagy, and inflammation, leading to muscle degradation [78,80]. They also impair membrane integrity, disrupt mitochondrial function and ATP production, promote apoptosis, and contribute to sarcopenia [81]. Detoxification of these lipid peroxidation products occurs primarily via enzymatic reduction by aldehyde dehydrogenase (ALDH2) and aldose reductase (Akr1b1) [82]. Additionally, aldehydes are scavenged through conjugation with the nucleophile glutathione [83] and endogenous histidyl dipeptides such as carnosine (β-alanine-histidine) and anserine (β-alanine-N^π^-histidine), as shown in Figure 3 [84,85]. To determine whether these toxic aldehydes accumulate in the skeletal muscle during HF, we recently showed that mice subjected to transaortic constriction for 12 weeks exhibit significantly elevated levels of acrolein and 4-HNE protein adducts in the skeletal muscle, accompanied by reduced skeletal muscle weight, strength, and decreased ALDH2 expression. Importantly, both carnosine levels and the expression of enzyme carnosine synthase, the enzyme which synthesizes carnosine, were reduced in the skeletal muscles of HF mice [80]. These findings suggest that skeletal muscle carnosine may serve as a diagnostic marker of skeletal muscle dysfunction during HF.
7. Therapeutic Interventions Targeting Skeletal Muscle in Heart Failure
Exercise training is an evidence-based intervention that reverses skeletal muscle dysfunction in HF. Previous studies in patients with HF have shown that exercise training reduces inflammatory cytokine levels in skeletal muscle and improves muscle function [86,87,88]. However, due to limited efficacy and durability, exercise cannot be a standalone intervention in these patients.
Emerging pharmacological therapies aim to address the metabolic and molecular derangements observed in HF skeletal muscle. Mitochondria-targeted antioxidants, ACE inhibitors, calcium sensitizers, and metabolic modulators have been tested and continue to be explored to restore energy homeostasis and improve skeletal performance in HF patients. A recent report in heart failure mice showed that targeting mitochondrial redox signaling by modulating the SIRT3–SOD2 axis in the skeletal muscle improves exercise tolerance. Treatment with Honokiol, a SIRT3 activator, resulted in SOD2 deacetylation and activation reduced mitochondrial ROS, restored oxidative capacity, and significantly improved exercise performance. Furthermore, skeletal muscle-targeted SIRT3 overexpression via AAV9 improved exercise capacity, confirming that the modulation of mitochondrial quality in muscle alone is sufficient to enhance functional performance in heart failure [89]. Mitochondrial ROS are a primary contributor of oxidative stress in skeletal muscle during HF. Elamipretide, a mitochondria targeted tetrapeptide (also known as MTP-131, SS-31), crosses the plasma membrane, localizes to the inner mitochondrial membrane, binds with cardiolipin, and enhances the flux of electrons through cytochrome c. In addition, elamipretide reduces mitochondrial ROS production [90]. Treatment with elamipretide has been shown to improve mitochondrial respiratory function and restore ATP synthesis and the ATP/ADP ratio in the skeletal muscle of dogs with experimentally induced HF [91]. While elamipretide is safe and tolerated in HF patients [92,93], its impact on exercise performance has not been directly tested.
ACE inhibitors are an established intervention for chronic heart failure (CHF) that also improve skeletal muscle dysfunction in both human and animal model of HF. In HF patients, ACE inhibition increases the proportion of slow oxidative MHCI fibers and decreases fast glycolytic MHCII, changes that correlate with improved exercise tolerance [94]. Additionally, several studies have shown that testosterone supplementation improves exercise capacity, muscle strength, and glucose metabolism in HF. In a pilot study of CHF patients, intramuscular testosterone therapy improved the distance covered in a shuttle walking test [95]. Similarly, a placebo-controlled trial demonstrated that 12 months of testosterone replacement therapy in men with moderately severe CHF improved the distance covered on shuttle walking and forearm strength [96]. Short-term administration of testosterone in HF patients also improved the baseline peak oxygen consumption, ventilatory efficiency, and isometric quadricep strength [97]. Although the mechanisms by which testosterone improves muscle strength in these patients remain unclear, studies in healthy mice show that administration of testosterone increases both the number and size of type I slow oxidative fibers [98], suggesting that testosterone may improve the oxidative capacity in HF skeletal muscle.
Studies using SGLT2 inhibitors in both murine models and human HF indicate that improvements in exercise performance may result from beneficial effects on skeletal muscle pathology. In mice subjected to myocardial infarction and subsequently treated with SGLT2 inhibitor empagliflozin, improved exercise capacity was linked to increased fatty acid oxidation and oxidative phosphorylation in the skeletal muscle [99]. Similarly, HF patients treated with SGLT2 inhibitor dapagliflozin improved exercise capacity, correlated with increased tryptophan metabolism and enrichment of anti-atrophic transcriptomic profile in skeletal muscle [100].
Levosimendan, a calcium sensitizer used clinically for HF, improves forelimb grip strength and distance and time running time in mice with HF. Treatment increased the cross-sectional area of skeletal muscle fibers, mitochondrial content, and mitochondrial membrane potential, and the improvement in exercise performance was independent of changes in cardiac function [101]. Similarly, a recent study in patients with HF showed that intravenous injection of iron in iron-deficient patients improves skeletal muscle function by enhancing energy metabolism and reducing reliance on glycolytic ATP production [102].
Molecular interventions targeting muscle regeneration pathways and histidyl dipeptide metabolism also hold promise. Carnosine, an endogenous histidyl dipeptide with aldehyde-scavenging properties, is depleted in HF skeletal muscle [80]. Supplementation strategies using β-alanine or carnosine precursors may restore muscle detoxification capacity, reduce aldehyde-induced protein damage, and improve exercise performance.
8. Conclusions
Skeletal muscle dysfunction is a critical contributor to exercise intolerance in HF. Disruptions in fiber type composition, mitochondrial structure and function, oxidative phosphorylation, and insulin signaling, compounded by oxidative stress and aldehyde accumulation, create a self-perpetuating cycle of metabolic impairment, fatigue, and atrophy. Emerging pharmacological, molecular, and nutritional strategies targeting mitochondrial health, oxidative stress, and histidyl dipeptide metabolism represent promising approaches to enhance skeletal muscle performance and exercise tolerance in HF patients. Future research should be focused on elucidating molecular mechanisms driving skeletal muscle dysfunction, including sex-specific differences in mitochondrial dynamics and regulation; evaluating therapeutic strategies that restore metabolic flexibility and improve mitochondrial quality; and investigating combinatorial approaches integrating exercise, pharmacological agents, and molecular interventions to maximize functional recovery in HF skeletal muscle. Addressing these gaps is essential to developing therapies that preserve muscle function, improve exercise tolerance, and ultimately enhance quality of life and outcomes for HF patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Maskin C.S. Forman R. Sonnenblick E.H. Frishman W.H. Le Jemtel T.H. Failure of dobutamine to increase exercise capacity despite hemodynamic improvement in severe chronic heart failure Am. J. Cardiol.19835117718210.1016/S 0002-9149(83)80032-06849256 · doi ↗ · pubmed ↗
- 2Wilson J.R. Martin J.L. Ferraro N. Impaired skeletal muscle nutritive flow during exercise in patients with congestive heart failure: Role of cardiac pump dysfunction as determined by the effect of dobutamine Am. J. Cardiol.1984531308131510.1016/0002-9149(84)90085-76711433 · doi ↗ · pubmed ↗
- 3Wiener D.H. Fink L.I. Maris J. Jones R.A. Chance B. Wilson J.R. Abnormal skeletal muscle bioenergetics during exercise in patients with heart failure: Role of reduced muscle blood flow Circulation 1986731127113610.1161/01.CIR.73.6.11273698247 · doi ↗ · pubmed ↗
- 4Richard L.L. Skeletal Muscle Structure, Function, and Plasticity 3rd ed.Walters Kluwer Philadelphia, PA, USA 2010
- 5van Hees H.W. van der Heijden H.F. Ottenheijm C.A. Heunks L.M. Pigmans C.J. Verheugt F.W. Brouwer R.M. Dekhuijzen P.N. Diaphragm single-fiber weakness and loss of myosin in congestive heart failure rats Am. J. Physiol. Heart Circ. Physiol.2007293 H 819H 82810.1152/ajpheart.00085.200717449557 · doi ↗ · pubmed ↗
- 6Miller M.S. Vanburen P. Lewinter M.M. Lecker S.H. Selby D.E. Palmer B.M. Maughan D.W. Ades P.A. Toth M.J. Mechanisms underlying skeletal muscle weakness in human heart failure: Alterations in single fiber myosin protein content and function Circ. Heart Fail.2009270070610.1161/CIRCHEARTFAILURE.109.87643319919996 PMC 2782533 · doi ↗ · pubmed ↗
- 7Toth M.J. Matthews D.E. Ades P.A. Tischler M.D. Van Buren P. Previs M. Le Winter M.M. Skeletal muscle myofibrillar protein metabolism in heart failure: Relationship to immune activation and functional capacity Am. J. Physiol. Endocrinol. Metab.2005288 E 685E 69210.1152/ajpendo.00444.200415562248 · doi ↗ · pubmed ↗
- 8Coirault C. Guellich A. Barbry T. Samuel J.L. Riou B. Lecarpentier Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure Am. J. Physiol. Heart Circ. Physiol.2007292 H 1009 H 101710.1152/ajpheart.00438.200617040975 · doi ↗ · pubmed ↗