Inflammatory and Nested Testicular Sex Cord Tumors: Clinical and Molecular Characterization
Panagiotis J. Vlachostergios, Foteini Karasavvidou, Konstantinos Evmorfopoulos, Ioannis Zachos, Vassilios Tzortzis

TL;DR
This paper describes a new type of testicular tumor called IN-TSCT, characterized by specific genetic changes and distinct clinical features.
Contribution
The paper identifies IN-TSCT as a distinct subtype of testicular tumor defined by EWSR1::ATF1 gene fusions and unique clinicopathologic features.
Findings
IN-TSCT is associated with EWSR1::ATF1 gene fusions and lacks germ cell tumor markers.
The tumor exhibits aggressive behavior in some cases, including metastasis.
Molecular studies show fusion-driven oncogenesis with low tumor mutational burden.
Abstract
Inflammatory and nested testicular sex cord tumor (IN-TSCT) is a recently characterized malignant neoplasm within the spectrum of testicular sex cord–stromal tumors. Previously misclassified as Sertoli cell tumor, not otherwise specified, or as seminoma, this entity has emerged as a distinct clinicopathologic and molecular subtype defined by recurrent EWSR1::ATF1 gene fusions and a potentially aggressive clinical course. Patients most commonly present with unilateral painless testicular enlargement, and radiologic findings are typically nonspecific. Histologically, tumors demonstrate solid and nested growth patterns, epithelioid cytology with eosinophilic to clear cytoplasm, prominent hyalinized stroma, and a conspicuous inflammatory infiltrate. Immunophenotypically, tumors express sex cord–stromal markers, including steroidogenic factor-1 (SF-1) and inhibin, and frequently co-express…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
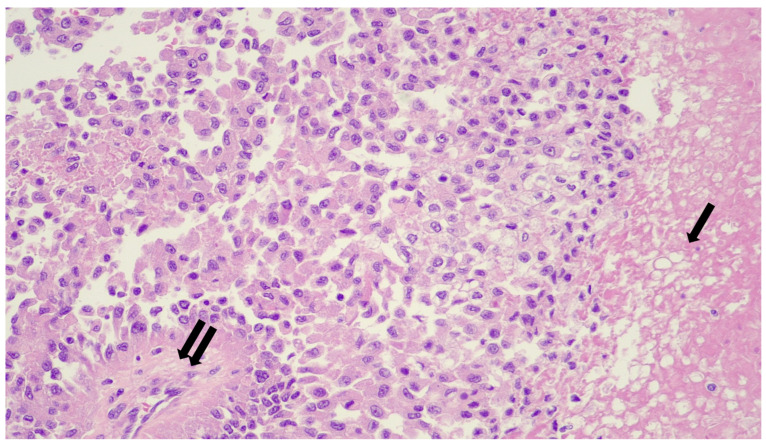 Figure 1
Figure 1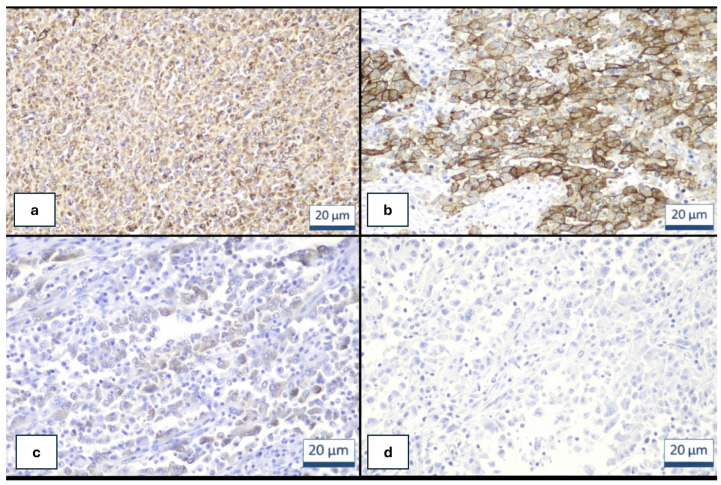 Figure 2
Figure 2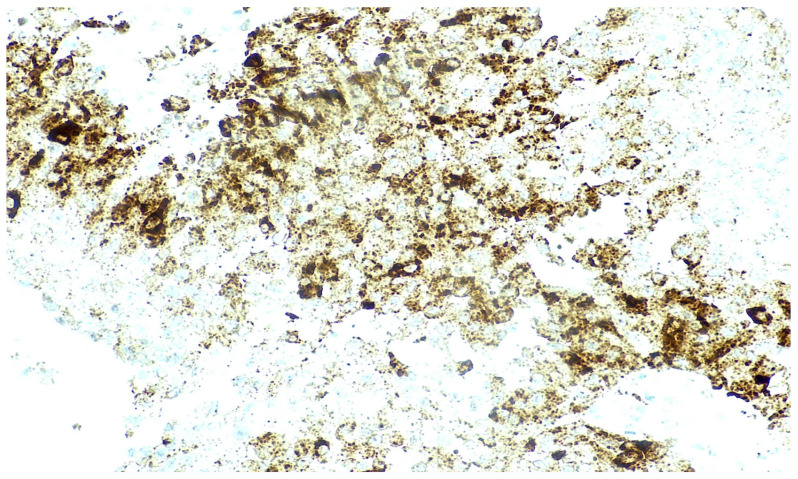 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTesticular diseases and treatments · Ovarian cancer diagnosis and treatment · Chromatin Remodeling and Cancer
1. Introduction
Testicular sex cord–stromal tumors (SCSTs) comprise a rare and heterogeneous group of neoplasms derived from the supporting cells of the testis, including Sertoli, Leydig, and granulosa cell lineages [1,2,3]. Historically, their classification has relied largely on morphologic features; however, advances in molecular pathology have revealed genetically distinct subsets within morphologically overlapping categories. Recent genomic studies have identified recurrent alterations such as CTNNB1 mutations in Sertoli cell tumors and PRKAR1A alterations in large cell calcifying Sertoli cell tumors, underscoring the importance of an integrated morphologic and molecular classification [1,2,3].
Inflammatory and nested testicular sex cord tumor (IN-TSCT) is a recently described malignant subtype of testicular sex cord–stromal tumor (TSCT) characterized by distinctive histopathologic and molecular features. These tumors typically form solid nests and sheets of epithelioid cells with granular eosinophilic to clear or vacuolated cytoplasm and are commonly accompanied by a prominent inflammatory infiltrate composed of lymphocytes, plasma cells, eosinophils, and neutrophils. The stroma is often collagenous and may show hyalinization. Tumor necrosis and local invasion are frequently observed, whereas mitotic activity is generally low [1].
Immunophenotypically, these tumors are characteristically positive for α-inhibin, steroidogenic factor-1 (SF-1), epithelial membrane antigen, and often CD30, but lack nuclear β-catenin expression. Molecularly, they are defined by recurrent EWSR1::ATF1 gene fusions or ATF1 amplification, distinguishing them from classic Sertoli cell tumors and other TSCTs. Clinically, inflammatory and nested TSCTs appear to exhibit aggressive behavior in a subset of reported cases, with metastases involving retroperitoneal and nonretroperitoneal lymph nodes, bone, the contralateral testis, and lung [1].
This entity is not yet formally included in the current World Health Organization (WHO) classification, but recent expert analyses and molecular studies support its recognition as a distinct diagnostic category because of its unique morphologic, immunophenotypic, and clinical characteristics.
In this review, we integrate clinical, histopathologic, and genomic data from the 15 cases reported to date worldwide together with a recent case from our institution (UTh), for which comprehensive next-generation sequencing (NGS), germline testing, and immunohistochemical (IHC) profiling were performed. Recognition of this entity is clinically important because it is frequently misdiagnosed as seminoma or lymphoma, which may lead to inappropriate or suboptimal management.
2. Clinical Presentation and Natural History
Patients with IN-TSCT span a broad adult age range, with reported cases occurring from the second to the eighth decades of life and a median age in the mid-30s [1]. The most common clinical presentation is unilateral painless testicular swelling or a palpable mass. Constitutional symptoms are typically absent. Serum germ cell tumor markers are usually within normal limits, reflecting the non-germ cell origin of the tumor [1].
Radiologic evaluation generally demonstrates a solid intratesticular mass without specific distinguishing imaging features. Because imaging lacks sufficient discriminatory value, histopathologic examination remains the cornerstone of diagnosis. In several early reports, a significant proportion of tumors were initially misdiagnosed as seminoma, highlighting the clinical impact of morphologic overlap and diagnostic mimicry [1,2,3,4].
These tumors may demonstrate local invasion and the potential for metastatic spread. Metastases can be present at diagnosis or may develop early during the disease course, and several reported patients have developed metastatic disease during follow-up. The retroperitoneal lymph nodes represent the most common site of metastasis, followed by extra-retroperitoneal nodal stations, lung, bone, and occasionally the contralateral testis [1,4,5,6]. Compared with other testicular sex cord–stromal tumors (TSCTs), inflammatory and nested tumors appear to exhibit more aggressive clinical behavior in a subset of reported cases, potentially associated with less favorable outcomes [1,6].
When IN-TSCT is confined to the testis, orchiectomy with or without adjuvant platinum-based chemotherapy has been reported as an effective therapeutic approach [1,7,8]. However, disease recurrence and metastatic progression have been associated with poor outcomes in some reported cases [1,7,9,10]. Nevertheless, because the total number of reported cases remains very small, currently available data are insufficient to accurately define the true metastatic risk and prognostic spectrum of this entity. Although published cases suggest that IN-TSCT may exhibit aggressive clinical behavior, including metastatic spread in a subset of patients, the overall natural history of the disease remains incompletely characterized.
3. Pathology
Gross examination typically reveals a solid, well-circumscribed intratesticular mass. Cut surfaces are variably tan-white to yellow and often show firm fibrotic or hyalinized areas. Foci of necrosis may be identified, whereas hemorrhage is uncommon but has been reported. No gross features are pathognomonic, and a definitive diagnosis requires histopathologic and molecular evaluation [2,3].
3.1. Histopathologic Features
Microscopically, IN-TSCT exhibits a combination of solid, nested, trabecular, and sheet-like architectural patterns. Tumor nests may be lobulated and are frequently separated by dense collagenous or hyalinized stroma (Figure 1). Focal discohesion within tumor nests can create pseudoglandular or pseudotubular spaces. An intratubular growth component has been described in a subset of cases, further supporting sex cord–stromal differentiation [1,7].
Tumor cells are predominantly epithelioid and display abundant eosinophilic to clear or vacuolated cytoplasm. High-power examination often demonstrates delicate cytoplasmic clearing that imparts a characteristic “spider-web” appearance. Intracytoplasmic hyaline inclusions are frequently observed but are not present in all cases. Nuclear atypia is generally mild to moderate, with smaller nuclei and less conspicuous nucleoli compared with those seen in seminoma. Mitotic activity is typically low, despite the clinically malignant behavior of the tumor [1,7].
The stroma represents a defining morphologic feature, often demonstrating dense hyalinization and stromal sclerosis. A prominent inflammatory infiltrate is consistently present and typically includes lymphocytes, plasma cells, eosinophils, and neutrophils. Occasional granulomas or aggregates of foamy histiocytes have also been described. Tumor necrosis is common and may be extensive in advanced cases [1,7].
3.2. Immunophenotype
Formalin-fixed, paraffin-embedded (FFPE) tissue sections from reported cases of IN-TSCT (n = 16) were analyzed using immunohistochemistry (IHC) according to standardized protocols, including antigen retrieval, blocking, and incubation with primary antibodies [1,8,9,10]. The antibody panel included SF-1, vimentin, inhibin, EMA, CD30, OCT3/4, SALL4, WT1, PD-L1, HER2, and ALK, with results interpreted by experienced pathologists.
Immunohistochemical studies support sex cord–stromal differentiation while also highlighting diagnostically useful aberrant expression patterns. Vimentin immunohistochemistry serves as a sensitive marker of mesenchymal and sex cord–stromal differentiation, and its expression supports the diagnosis of testicular sex cord–stromal tumors, including inflammatory and nested variants. Vimentin is consistently expressed in normal Sertoli cells, Leydig cells, and most sex cord–stromal tumors, reflecting their mesenchymal origin and differentiation profile [11,12,13].
In the context of inflammatory and nested testicular sex cord tumors, vimentin positivity may assist in distinguishing these neoplasms from epithelial tumors and germ cell tumors, which typically demonstrate absent or only focal vimentin expression [13,14,15]. However, vimentin is not specific for sex cord–stromal tumors, as it is also expressed in a broad range of mesenchymal neoplasms and certain carcinomas. Therefore, the diagnostic value of vimentin lies in its use as part of a broader immunohistochemical panel—including α-inhibin, steroidogenic factor-1 (SF-1), and CD30—to confirm sex cord–stromal lineage and to distinguish inflammatory and nested testicular sex cord tumors from histologic mimics [1,11,12]. Vimentin alone cannot reliably subclassify sex cord–stromal tumors or differentiate inflammatory and nested variants from other subtypes, but its positivity provides supportive evidence in the diagnostic workup (Figure 2a).
CD30 expression is present in the majority of reported cases and may show diffuse staining (Figure 2b). This finding represents an important diagnostic pitfall, as it may lead to misclassification as anaplastic large cell lymphoma. Tumor cells typically demonstrate diffuse nuclear expression of steroidogenic factor-1 (SF-1) and frequent positivity for WT1 and α-inhibin (Figure 2c). Diffuse epithelial membrane antigen (EMA) expression is also characteristic, an unusual feature among sex cord–stromal tumors that may create diagnostic confusion with carcinoma [1,8,9,10].
Cytokeratin expression is variable, whereas nuclear accumulation of β-catenin is absent, helping distinguish IN-TSCT from CTNNB1-mutant Sertoli cell tumors. Germ cell tumor markers—including OCT3/4, placental alkaline phosphatase (PLAP), and SALL4—are consistently negative (Figure 2d). This immunophenotypic profile is critical for differentiating IN-TSCT from seminoma and other germ cell tumors.
Anaplastic lymphoma kinase (ALK) expression in IN-TSCT represents an emerging and incompletely characterized immunophenotypic feature. Strong diffuse cytoplasmic ALK expression by immunohistochemistry has been reported in 1 of 16 cases, in the absence of detectable ALK gene rearrangement [10] (Figure 3). This pattern suggests protein overexpression rather than canonical ALK fusion-driven oncogenesis.
The therapeutic relevance of ALK expression in IN-TSCT remains uncertain. In tumors such as ALK-rearranged non-small cell lung carcinoma and anaplastic large cell lymphoma (ALCL), ALK inhibitors have demonstrated substantial clinical efficacy. However, in the absence of confirmed ALK genomic activation, there is currently no evidence supporting the use of targeted ALK inhibition in IN-TSCT.
Likewise, although strong diffuse cytoplasmic ALK expression has been reported in isolated IN-TSCT cases, genomic analyses have not identified ALK rearrangements or activating mutations. Therefore, ALK immunoreactivity alone should not be interpreted as evidence of a therapeutically targetable oncogenic driver, and ALK inhibitors currently have no established role in the management of IN-TSCT without confirmed genomic alterations.
4. Molecular Genetics
The recognition of IN-TSCT as a molecularly defined entity highlights the critical importance of tumor next-generation sequencing (NGS) in diagnostically challenging testicular neoplasms. While histomorphology and immunohistochemistry remain the foundation of diagnosis, IN-TSCT is fundamentally a fusion-driven tumor characterized by recurrent EWSR1::ATF1 gene rearrangements. Consequently, molecular confirmation is not merely adjunctive but often decisive for establishing the diagnosis. Comprehensive genomic profiling may also identify potentially actionable co-alterations in rare cases.
Among the reported cases in the literature, tumor NGS has been performed using several validated targeted sequencing platforms, including a 282-gene panel (OncoPanel) in three cases, a 523-gene panel (HopeSeq) in one case, and a 1021-gene panel with 38 fusion targets (PrimeDx) in the most recent case from our institution. The defining molecular hallmark of IN-TSCT is rearrangement of EWSR1, most commonly resulting in an EWSR1::ATF1 fusion. This in-frame fusion juxtaposes the transcriptional activation domain of EWSR1 with the DNA-binding domain of ATF1, resulting in aberrant transcriptional activation of downstream target genes. Fusion detection has been achieved using fluorescence in situ hybridization (FISH) and next-generation sequencing-based platforms.
Genomic profiling generally reveals a relatively quiet mutational landscape, characterized by low tumor mutational burden and microsatellite stability. Recurrent secondary mutations appear to be uncommon. Isolated reports have described pathogenic TP53 alterations and germline CHEK2 variants, although their biological and clinical significance remains uncertain (Table 1). Rare fusion-negative tumors demonstrating ATF1 amplification have also been described, suggesting alternative mechanisms that activate the same transcriptional pathway. Of note, SF-1 negativity has been reported in 1 of the 16 cases described to date, which is notable because most previously reported IN-TSCTs demonstrate SF-1 positivity. This observation highlights phenotypic variability and further reinforces the importance of molecular confirmation for accurate diagnosis.
Collectively, molecular findings indicate that IN-TSCT is a fusion-driven tumor with a relatively genomically quiet profile, although occasional alterations affecting homologous recombination DNA repair pathways may provide potential opportunities for therapeutic targeting.
5. Differential Diagnosis
The principal histologic differential diagnosis is seminoma, given the shared nested architecture and associated lymphoid infiltrates. However, seminoma typically exhibits fibrous septa, a relatively uniform lymphocytic inflammatory infiltrate, prominent nucleoli, and strong expression of germ cell tumor markers. In contrast, IN-TSCT demonstrates a mixed inflammatory infiltrate, hyalinized stroma, and positivity for sex cord–stromal markers.
Sertoli cell tumor, not otherwise specified, also represents an important differential diagnostic consideration. Unlike IN-TSCT, many Sertoli cell tumors harbor CTNNB1 mutations with nuclear β-catenin accumulation and lack EWSR1 rearrangements.
CD30 positivity in IN-TSCT may raise the consideration of lymphoma, particularly anaplastic large cell lymphoma (ALCL). However, demonstration of steroidogenic factor-1 (SF-1) and inhibin expression, together with the absence of leukocyte markers, readily resolves this diagnostic distinction. Additional considerations include metastatic carcinoma and paratesticular mesothelioma, although clinicopathologic correlation and molecular testing are typically decisive in establishing the correct diagnosis [1,2,3,7].
6. Comparative Genomics and Cross-Gonadal Parallels in Sex Cord–Stromal Tumors
Sex cord–stromal tumors of the testis and ovary share a common embryologic origin, arising from gonadal supporting cell lineages. Consequently, tumors arising in these organs often exhibit overlapping morphologic features, immunophenotypic profiles (Table 2), and, increasingly, shared molecular alterations.
Comparative genomic analysis has become an essential tool for refining tumor classification, clarifying lineage relationships, and identifying therapeutically relevant pathways [16]. Among ovarian SCSTs, adult granulosa cell tumor is the best-characterized entity at the molecular level. More than 95% of cases harbor the pathognomonic FOXL2 c.402C>G (p.C134W) mutation, which drives tumorigenesis through transcriptional dysregulation and altered TGF-β signaling pathways. This mutation is highly specific and is widely considered a defining diagnostic marker [17,18]. Juvenile granulosa cell tumors, in contrast, lack FOXL2 mutations and instead demonstrate alterations involving AKT1, GNAS, or DICER1, highlighting age-dependent molecular divergence within granulosa cell tumors [19,20].
Sertoli–Leydig cell tumors of the ovary provide another informative molecular parallel. A substantial subset harbors DICER1 mutations, frequently of germline origin, linking these neoplasms to the DICER1 tumor predisposition syndrome [21]. These tumors demonstrate aberrant microRNA processing and dysregulated cellular differentiation, reinforcing the importance of post-transcriptional regulatory mechanisms in sex cord–stromal tumorigenesis [22].
Testicular SCSTs share some overlapping molecular pathways but also exhibit organ-specific oncogenic drivers. For example, CTNNB1 mutations characterize a subset of Sertoli cell tumors, leading to nuclear accumulation of β-catenin and activation of the Wnt signaling pathway [23,24]. Large cell calcifying Sertoli cell tumors frequently harbor PRKAR1A alterations, particularly in patients with Carney complex [25].
Within this comparative framework, inflammatory and nested testicular sex cord tumor (IN-TSCT) appears molecularly distinct. Rather than mutation-driven oncogenesis typical of many ovarian SCSTs, IN-TSCT is defined by recurrent EWSR1::ATF1 gene fusions [1]. This places IN-TSCT within the broader family of CREB-fusion-driven neoplasms, rather than within the classical gonadal stromal tumor pathways. The transcriptional consequences of EWSR1::ATF1 fusions likely converge on cAMP-responsive and cellular stress-response signaling pathways, representing a mechanistic axis not previously central to gonadal SCST biology. Key molecular drivers across testicular and ovarian SCSTs are summarized in Table 3.
This divergence suggests that IN-TSCT may represent a fusion-defined lineage within the sex cord–stromal tumor (SCST) spectrum, analogous to other fusion-driven neoplasms such as sarcomas, and underscores the importance of molecular testing in diagnostically challenging cases.
Comparative analysis of gonadal sex cord–stromal tumors highlights both shared lineage biology and significant molecular divergence. While many ovarian SCSTs are driven by recurrent point mutations affecting transcriptional or microRNA regulatory networks, inflammatory and nested testicular sex cord tumor is characterized by a fusion-driven oncogenic mechanism centered on the EWSR1::ATF1 gene fusion. This distinction not only supports its classification as a unique molecular entity but also broadens the genomic landscape of gonadal stromal neoplasia.
Future studies integrating transcriptomic profiling and epigenetic mapping across gonadal tumor types may clarify whether CREB-family fusion-driven tumors represent a previously unrecognized developmental branch within sex cord–stromal tumorigenesis.
7. Management Strategies
The cornerstone of management is primary surgical excision via radical orchiectomy [4,5,6,26]. Given the potentially aggressive clinical behavior and risk of metastatic spread, early retroperitoneal lymph node dissection (RPLND) may be considered in patients with high-risk features or clinical evidence of nodal involvement [4,5,6,26]. The role of RPLND remains controversial; however, early intervention in patients with documented nodal metastases may improve clinical outcomes [4,5]. Although published reports suggest that IN-TSCT may demonstrate aggressive behavior, including metastatic spread in a subset of patients, the overall number of reported cases remains very small, and the true metastatic risk and prognostic spectrum are not yet well defined.
Systemic therapy outcomes are also poorly characterized. Malignant SCSTs, including IN-TSCT, appear relatively resistant to conventional germ cell tumor chemotherapy regimens. Platinum-based chemotherapy has been used in reported cases, with variable and often limited clinical benefit. CD30 expression raises the theoretical possibility of targeted therapy with brentuximab vedotin, although current evidence supporting this approach remains anecdotal. Overall, chemotherapy appears to have limited efficacy in metastatic TSCTs, including this subtype [4,5,6,26].
Emerging evidence suggests that immune checkpoint inhibitors (ICIs) may offer potential clinical benefit in selected chemotherapy-refractory metastatic cases, with partial responses reported and generally manageable toxicity profiles [27]. However, available data remain extremely limited and require further validation. In the Acosta case series [1], one patient (case no. 11) with IN-TSCT received combined immune checkpoint blockade with ipilimumab and nivolumab after failing multiple lines of chemotherapy—including carboplatin plus paclitaxel and etoposide plus cisplatin—but no clinical response was observed [1].
In the largest available series of metastatic TSCTs, two patients with chemotherapy-refractory disease were treated with ICIs. One patient with Sertoli cell tumor and retroperitoneal lymph node metastasis received sintilimab (200 mg intravenously every 3 weeks) combined with anlotinib (12 mg orally once daily on days 1–14 of a 21-day cycle) for four cycles. Another patient with Leydig cell tumor and metastases to the right lung and left adrenal gland received sintilimab monotherapy (200 mg intravenously every 3 weeks) after failure of first-line chemotherapy. Both patients achieved partial responses, with progression-free survival extending to the last follow-up at 14 and 22 months, respectively. Adverse events were mild (grade 1–2), and no severe immune-related toxicities were reported. These findings suggest that ICIs may provide clinical benefit in selected patients with chemotherapy-resistant disease, particularly given the limited efficacy of platinum-based chemotherapy in metastatic TSCTs [27].
Given the rarity of IN-TSCT, the absence of standardized treatment guidelines, and the tumor’s potential for metastatic progression, management should ideally occur within specialized multidisciplinary teams, including urologic oncologists, medical oncologists, genitourinary pathologists, and molecular tumor boards, to guide therapeutic decision-making and facilitate referral to clinical trials when available. Although responses to ICIs have been rarely reported and appear limited, the manageable safety profile observed in available cases suggests that immunotherapy may be considered in selected patients with metastatic, chemotherapy-refractory disease, with careful monitoring for immune-related adverse events [1,2,27]. Nevertheless, the role of ICIs remains investigational, and their use should be considered on a case-by-case basis in the absence of established effective systemic therapies. Furthermore, predictive biomarkers, expected response rates, and treatment durability remain undefined, particularly given the generally low tumor mutational burden observed in these tumors.
Alterations in CHEK2 may provide a theoretical rationale for targeted therapeutic strategies that exploit defects in DNA damage response pathways in IN-TSCT. CHEK2 functions as a central mediator of the ATM–CHEK2–p53 signaling cascade activated by DNA double-strand breaks and plays a key role in cell-cycle arrest and homologous recombination repair. Loss-of-function alterations in CHEK2 may impair homologous recombination proficiency and produce a phenotype analogous to “BRCAness.” In this context, inhibition of poly(ADP-ribose) polymerase (PARP) may induce synthetic lethality by further compromising DNA repair capacity. Although clinical evidence supporting PARP inhibition in IN-TSCT is currently lacking, this mechanistic rationale supports further investigation of PARP inhibitors in tumors harboring CHEK2 or other homologous recombination repair alterations. However, discussion of PARP inhibition, ALK-directed therapy, or other targeted approaches is currently based primarily on molecular rationale or extrapolation from other tumor types rather than established clinical efficacy in IN-TSCT.
Collectively, because IN-TSCT is an exceptionally rare tumor, therapeutic evidence is largely limited to individual case reports and small case series. Consequently, discussion of systemic treatment strategies must clearly distinguish between approaches directly reported in IN-TSCT and those extrapolated from broader experience with malignant SCSTs or from the biological implications of specific genomic alterations.
8. Outcomes and Prognosis
Outcomes for patients with IN-TSCT appear unfavorable in the limited number of reported cases, with relatively high rates of metastasis and recurrence described in the available literature [1,4,5,6]. Survival in advanced disease is often limited, and most patients with metastatic disease have demonstrated poor responses to conventional chemotherapy [4,5,6,9]. Early retroperitoneal lymph node dissection (RPLND) in patients with nodal metastases may improve disease-free survival, although this benefit appears to be largely restricted to patients with nodal-only disease [4,5]. Recent reports describing partial responses to ICIs in chemotherapy-resistant metastatic TSCTs are encouraging; however, their long-term efficacy and safety remain uncertain.
9. Knowledge Gaps
There are currently no standardized management guidelines for IN-TSCT, and long-term outcome data remain scarce. Additional research is needed to clarify optimal management strategies, identify prognostic factors, and define the potential role of emerging therapies such as immunotherapy and PARP inhibitors in this rare tumor type [1,2,3,27].
10. Conclusions
IN-TSCT represents a distinct, molecularly defined malignant testicular sex cord–stromal tumor characterized by recurrent EWSR1::ATF1 fusion, a unique inflammatory and nested morphology, and potentially aggressive clinical behavior. However, the limited number of reported cases precludes precise estimation of the true metastatic risk and prognostic spectrum of this disease. Accurate recognition requires integration of histologic, immunophenotypic, and molecular findings. Expanded case accrual and multicenter collaborative studies will be essential to refine prognostic stratification and establish evidence-based management strategies.
Although the available literature indicates malignant potential and clinically significant metastatic behavior in several reported cases, these observations should be interpreted with caution given the rarity of the tumor, the small number of reported patients, and the possibility of publication bias toward more aggressive presentations. The rarity of IN-TSCT and the diagnostic and therapeutic challenges associated with this entity underscore the need for continued molecular characterization and investigation of targeted therapeutic approaches.
The presence of CHEK2 alterations in some cases suggests additional layers of genomic instability that may influence therapeutic strategies. These alterations raise the possibility that tumors harboring homologous recombination repair defects could be susceptible to PARP inhibition, although this hypothesis remains speculative and requires validation in future studies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Acosta A.M. Bridge J.A. Dal Cin P.S. Sholl L.M. Cornejo K.M. Fletcher C.D.M. Ulbright T.M. Inflammatory and nested testicular sex cord tumor: A novel neoplasm with aggressive clinical behavior and frequent EWSR 1::ATF 1 gene fusions Am. J. Surg. Pathol.20234750451710.1097/PAS.000000000000202236791251 · doi ↗ · pubmed ↗
- 2Lobo J. Acosta A.M. Advances in non-germ cell tumours of the testis: Focus on new molecular developments in sex cord-stromal tumours Histopathology 20268823025110.1111/his.7000641384702 PMC 12700056 · doi ↗ · pubmed ↗
- 3Acosta A.M. Idrees M.T. Berney D.M. Colecchia M. Contemporary updates on sex cord-stromal tumors of the testis Adv. Anat. Pathol.20243112613510.1097/PAP.000000000000042338053410 · doi ↗ · pubmed ↗
- 4Silberstein J.L. Bazzi W.M. Vertosick E. Carver B.S. Bosl G.J. Feldman D.R. Bajorin D.F. Motzer R.J. Al-Ahmadie H. Reuter V.E. Clinical outcomes of local and metastatic testicular sex cord-stromal tumors J. Urol.201419241541910.1016/j.juro.2014.01.10424518791 PMC 6701173 · doi ↗ · pubmed ↗
- 5Nicolai N. Necchi A. Raggi D. Biasoni D. Catanzaro M. Piva L. Stagni S. Maffezzini M. Torelli T. FaréE. Clinical outcome in testicular sex cord stromal tumors: Testis sparing vs. radical orchiectomy and management of advanced disease Urology 20158540240610.1016/j.urology.2014.10.02125623702 · doi ↗ · pubmed ↗
- 6Banerji J.S. Odem-Davis K. Wolff E.M. Nichols C.R. Porter C.R. Patterns of care and survival outcomes for malignant sex cord stromal testicular cancer: Results from the national cancer data base J. Urol.20161961117112210.1016/j.juro.2016.03.14327036305 · doi ↗ · pubmed ↗
- 7Siegmund S.E. Sholl L.M. Tsai H.K. Yang Y. Vasudevaraja V. Tran I. Snuderl M. Fletcher C.D.M. Cornejo K.M. Idrees M.T. Clinicopathologic and molecular spectrum of testicular sex cord-stromal tumors not amenable to specific histopathologic subclassification Mod. Pathol.2022351944195410.1038/s 41379-022-01155-y 36180576 · doi ↗ · pubmed ↗
- 8Wang Y. Xi Y. Wu M. Clinicopathological characteristics and molecular genetic analysis of inflammatory and nested testicular sex cord tumor: A case report and literature review Discov. Oncol.202516190210.1007/s 12672-025-03704-841091249 PMC 12528643 · doi ↗ · pubmed ↗