Harnessing Gut Endocrine Cell Plasticity to Restore Insulin Production
Chaïma Ayachi, Tiziana Napolitano, Serena Silvano, Sophie Giorgetti-Peraldi, Ahmed Mansouri, Raphaël Rapetti-Mauss, Hugo Fofo, Valentin Lepage, Laura Etasse, Caroline Treins, Loan Tran, Patrick Collombat

TL;DR
This paper explores converting gut cells into insulin-producing cells as a potential treatment for type 1 diabetes.
Contribution
The study shows gut endocrine cells can be reprogrammed into functional insulin-producing cells using Pax4.
Findings
Ectopic Pax4 expression converts gut L-cells into insulin+ cells in vivo.
Converted cells express β-cell markers and secrete glucose-responsive insulin.
Organoid studies confirm functional glucose sensing and insulin secretion.
Abstract
Type 1 diabetes (T1D) results from autoimmune-mediated destruction of pancreatic β-cells, leading to insulin deficiency and chronic hyperglycemia. β-cell replacement represents a promising therapeutic strategy, yet the identification of a sustainable and immune-compatible cell source remains a major challenge. Here, we explore the potential of the gastrointestinal (GI) epithelium as an alternative source of β-cells through in vivo cellular reprogramming. Given the large size and highly regenerative nature of the GI tract, partial reprogramming could provide a renewable source of insulin-producing (insulin+) cells. We demonstrate that ectopic expression of Pax4 is sufficient to convert gut endocrine L-cells into insulin+ cells in vivo. Phenotypic analyses reveal that these gut-derived cells express key β-cell markers, components of the glucose-sensing machinery, and properly process…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
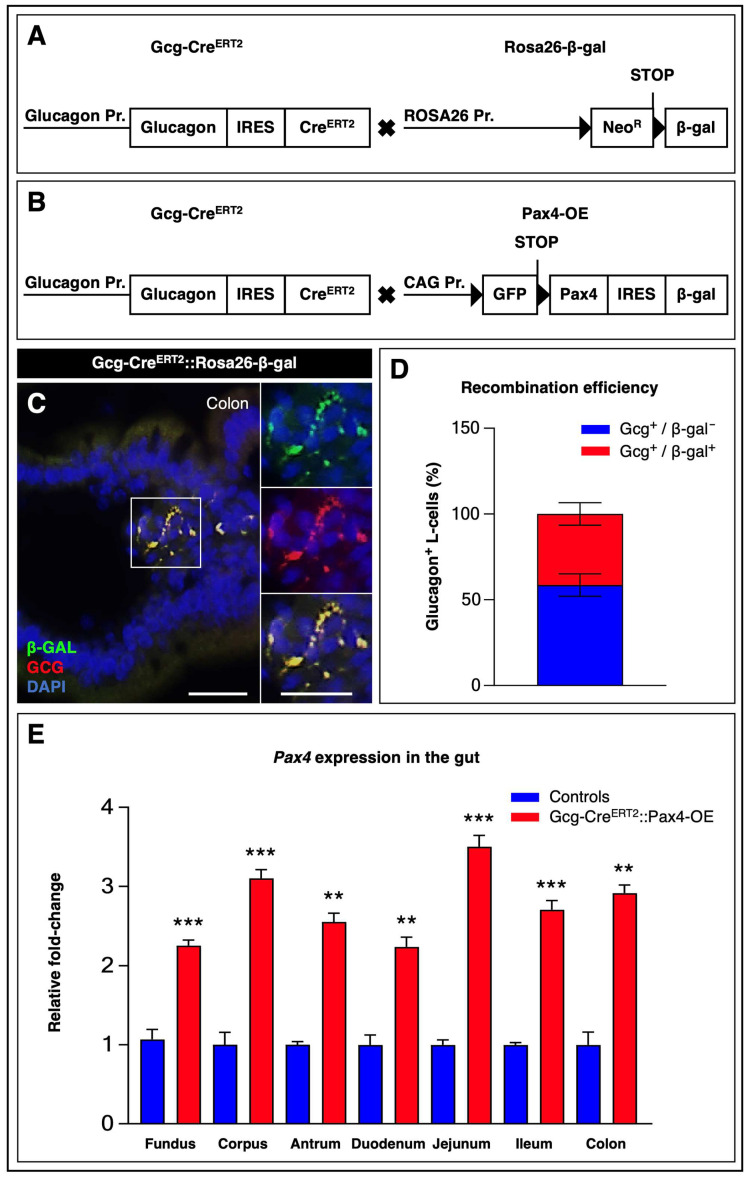 Figure 1
Figure 1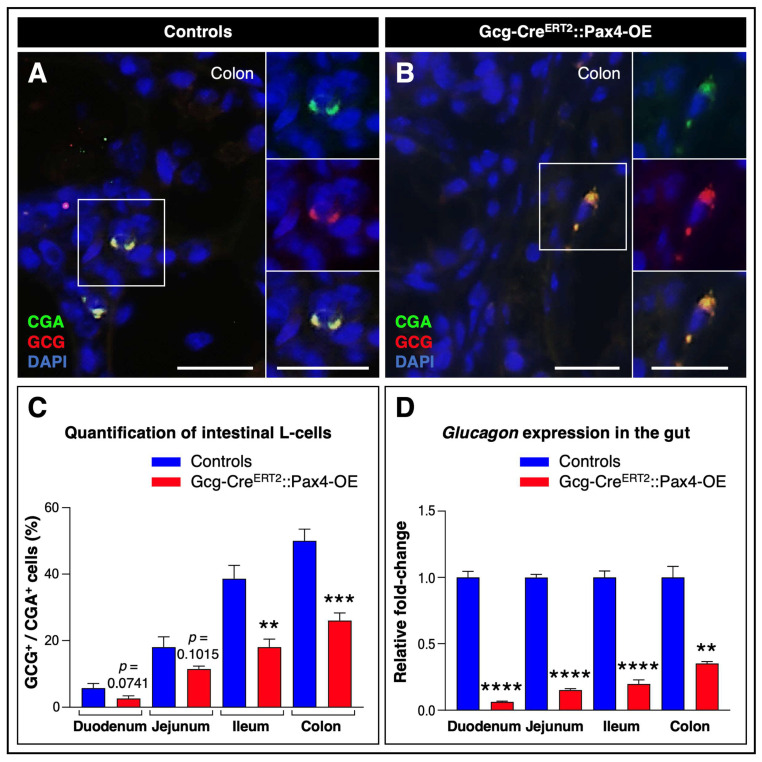 Figure 2
Figure 2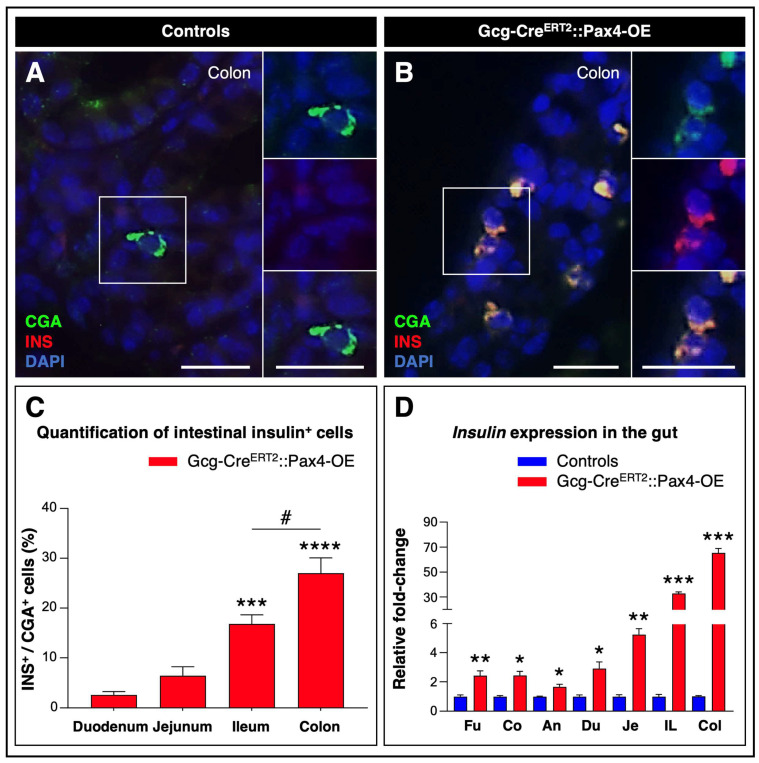 Figure 3
Figure 3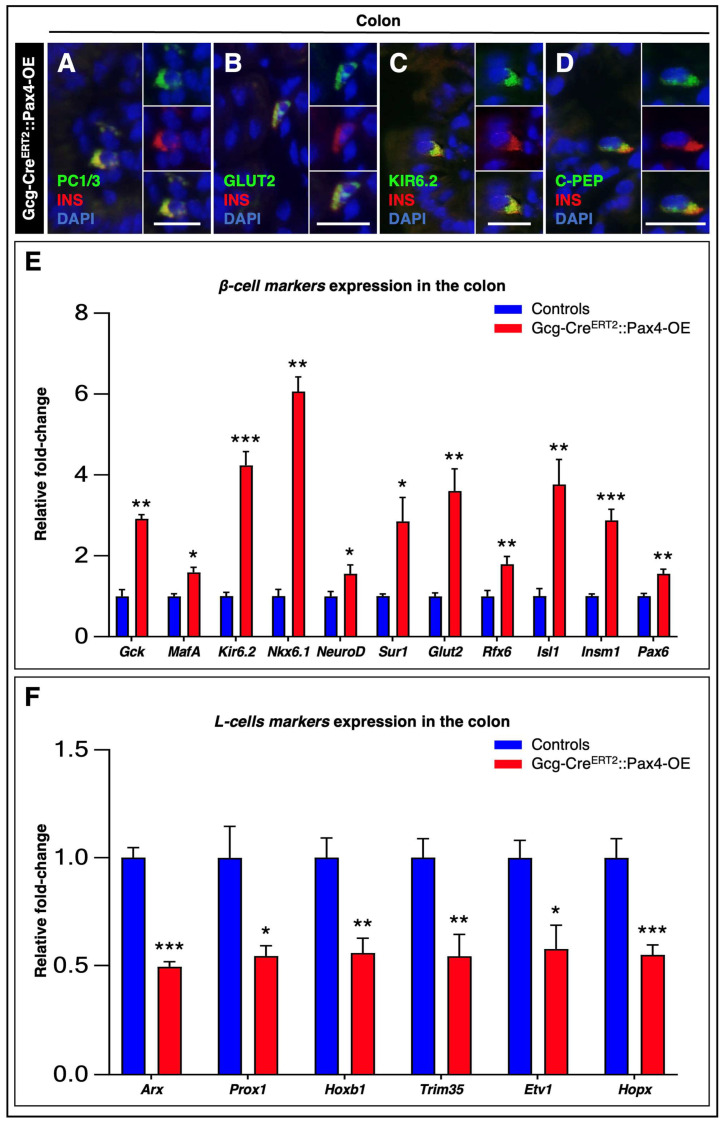 Figure 4
Figure 4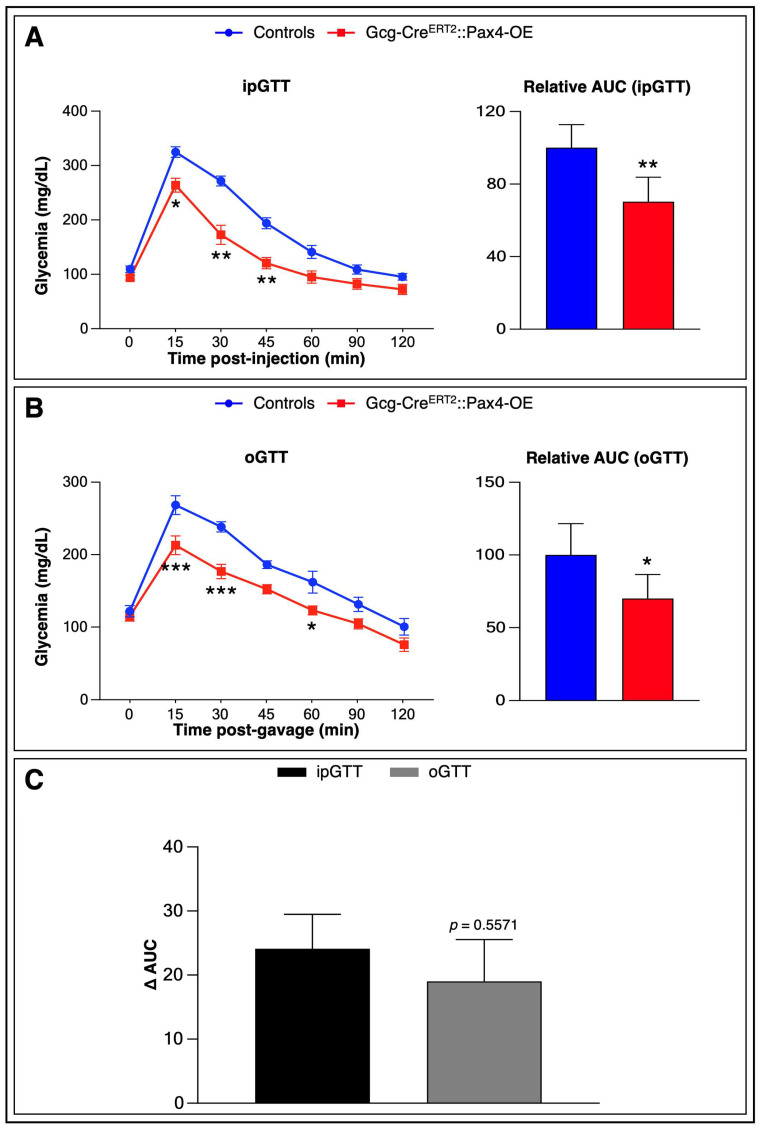 Figure 5
Figure 5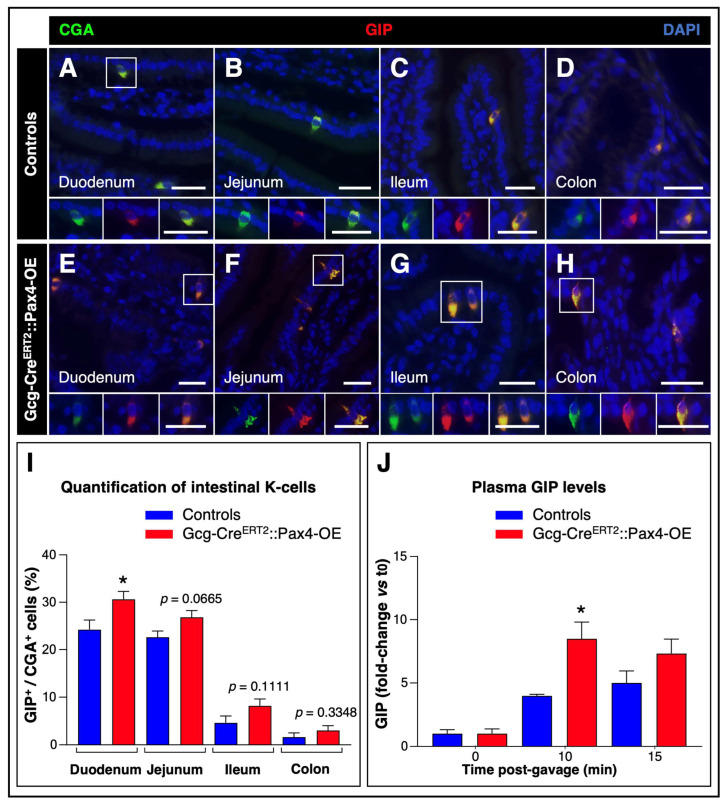 Figure 6
Figure 6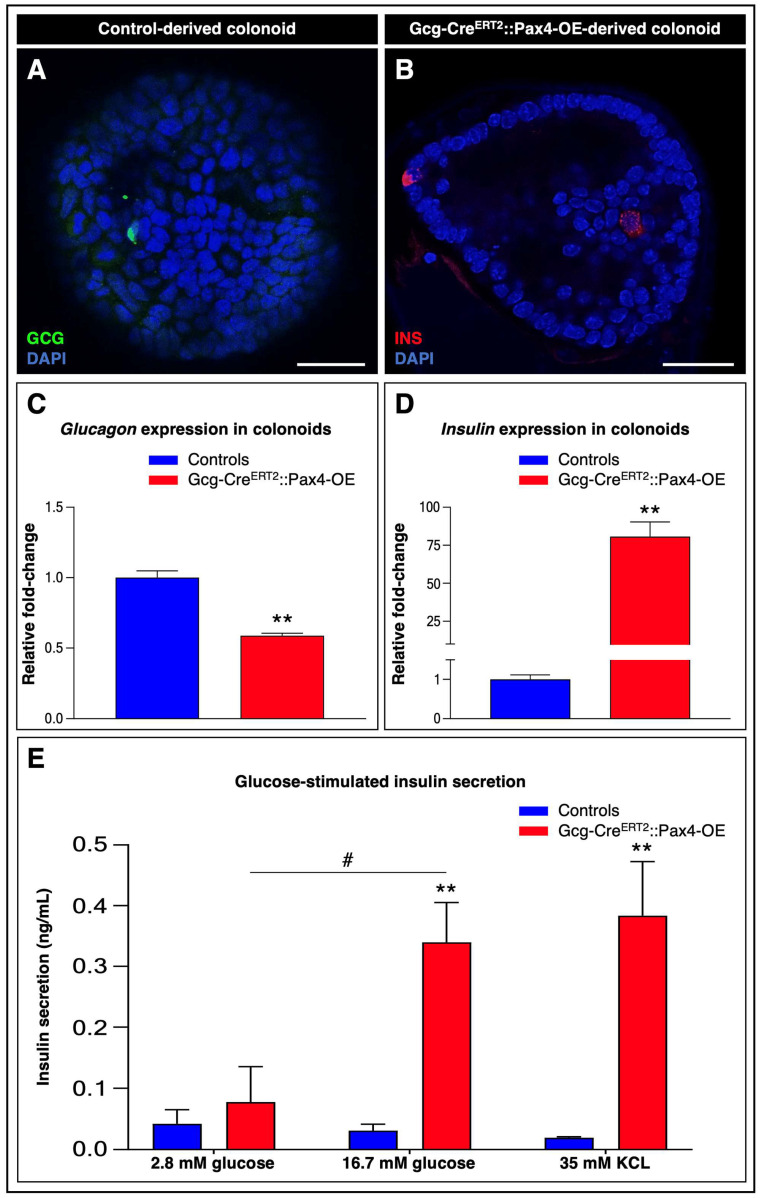 Figure 7
Figure 7- —Juvenile Diabetes Research Foundation
- —Agence Nationale pour la Recherche
- —LABEX SIGNALIFE
- —IDEX UCA Jedi
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPancreatic function and diabetes · Diabetes and associated disorders · Diabetes Management and Research
1. Introduction
The autoimmune-mediated loss of β-cell mass in type 1 diabetes (T1D) has prompted intense efforts to identify alternative sources of insulin-producing (insulin^+^) cells capable of restoring endogenous insulin secretion and normoglycemia. Among these approaches, β-cell replacement has emerged as a promising strategy, either through differentiation of embryonic stem cells or direct reprogramming of non-β cells [1,2,3,4]. However, the autoimmune nature of T1D represents a major challenge for β-cell replacement strategies, as newly generated cells may remain susceptible to immune-mediated destruction.
Reprogramming endogenous endocrine cells into glucose-responsive β-cells represents an attractive avenue for developing autologous regenerative therapies in situ. However, newly generated β-cells may remain vulnerable to autoimmune recurrence. In this context, identifying a renewable, accessible, and potentially immune-compatible source of insulin^+^ cells is of particular interest.
The gastrointestinal (GI) epithelium constitutes a highly regenerative tissue containing abundant stem and endocrine cell populations [3,5]. Given its extensive surface area and rapid epithelial turnover, reprogramming even a limited fraction of gut epithelial cells could theoretically provide a sustainable source of β-like cells without compromising gut function [6]. Previous studies have established the feasibility of generating gut-derived β-like cells through Foxo1 inhibition or overexpression of pancreatic transcription factors (Pdx1, MafA, and Ngn3; PMN factors) [5,7,8,9,10]. Despite these promising approaches, limitations such as low conversion efficiency and potential adverse effects underscore the need for alternative and potentially simpler reprogramming strategies [3,11].
Paired Box 4 (Pax4) is a transcription factor critical for pancreatic β-cell and gut endocrine cell development, promoting lineage specification [12,13,14]. Beyond its developmental role, Pax4 has garnered significant attention for its potency as a reprogramming factor for generating insulin^+^ cells from non-β sources [12,15,16,17,18]. Notably, ectopic expression of Pax4 is sufficient to convert pancreatic glucagon-producing (glucagon^+^) α-cells into functional β-like cells capable of ameliorating toxin-induced diabetes in vivo [18]. Interestingly, gut endocrine glucagon^+^ L-cells share key molecular and developmental features with pancreatic α-cells, including Ngn3-dependent differentiation as well as glucose-sensing and hormone secretion machinery [19,20,21,22].
Based on this rationale, we investigated whether ectopic Pax4 expression could also induce the formation of insulin^+^ cells in the gut in vivo. Our findings demonstrate that Pax4 alone is sufficient to generate gut-derived β-like cells expressing key β-cell markers and glucose sensing components. These cells are functionally capable of responding to glucose, supporting the concept that the GI epithelium may serve as a renewable source of self-sustaining β-like cells with potential applications for glycemic control.
2. Materials and Methods
2.1. Animal Manipulations
To induce targeted Pax4 ectopic expression in glucagon^+^ L-cells, Gcg-Cre^ERT2^::Pax4-OE mice were generated by crossing Gcg-Cre^ERT2^ mice [23], expressing a tamoxifen (TAM)-inducible Cre^ERT2^ recombinase knocked into the endogenous pre-proglucagon gene, with the previously described Pax4-OE mouse line [17].
For validation of Cre^ERT2^ recombination efficiency and specificity, Gcg-Cre^ERT2^ mice were also crossed with the Rosa26-β-gal reporter line [24], which carries a loxP-flanked transcriptional STOP cassette, including a neomycin resistance gene (NEO^R^), upstream of the β-galactosidase cDNA under the control of the ubiquitous Rosa26 promoter.
Animals were genotyped via GFP fluorescence and PCR for Cre^ERT2^ and β-galactosidase genes. Recombination was induced in 8-week-old male mice by intraperitoneal injection of TAM (20 mg/mL; Sigma–Aldrich, St. Louis, MO, USA) daily for five consecutive days, combined with oral administration in drinking water (50 mg/L; Biogaran, Colombes, France), freshly prepared weekly.
2.2. Isolation of Murine Pancreatic Islets of Langerhans
Murine pancreatic islets were isolated from 8-week-old TAM-treated Gcg-Cre^ERT2^::Pax4-OE mice and control littermates by perfusing a collagenase solution (1 mg/mL; Sigma–Aldrich) diluted in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) into the main pancreatic duct. Perfused pancreata were incubated at 37 °C for 12 min to allow enzymatic digestion, and the reaction was subsequently stopped by adding high-glucose DMEM containing 10% fetal bovine serum (FBS; Gibco). Following several centrifugation steps, islets were purified by density gradient separation using Histopaque (10771 and 11191; Merck, Darmstadt, Germany). Purified islets were then cultured in RPMI-1640 medium (Merck) supplemented with 10% FBS and 1% penicillin/streptomycin (10,000 U/mL; Gibco) in a humidified incubator at 37 °C with 5% CO_2_. After an overnight recovery period, islets were hand-picked under a stereomicroscope, and 300 islets per sample were collected into clean Eppendorf tubes for subsequent RNA extraction.
2.3. Immunohistochemistry
GI and pancreatic tissues were fixed in 4% paraformaldehyde (Microm Microtech, Francheville, France) for 30 min at 4 °C, embedded in paraffin, and sectioned at 5 μm. Sections were processed as previously described [25], with DAPI counterstaining.
Primary antibodies included: guinea pig anti-insulin (1:500; Dako, Glostrup, Denmark), mouse anti-glucagon (1:500; Sigma–Aldrich), rabbit anti-PC1/3 (1:500; Millipore, Burlington, MA, USA), anti-chromogranin A (CgA, 1:500; Abcam, Cambridge, UK), anti-GLUT-2 (1:500; Millipore), anti-glucose-dependent insulinotropic polypeptide (GIP, 1:100; Abcam), anti-Kir6.2 (1:500; Abcam), rat anti-C-peptide (1:500; Phoenix Pharmaceuticals, Burlingame, CA, USA), and chicken anti-β-galactosidase (1:500; Abcam). Secondary antibodies were Alexa Fluor 488, 594, and 647 conjugates (1:1000; Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
Images were acquired using an Axioimager Z1 (Zeiss, Oberkochen, Germany) with appropriate filter sets and processed with Axiovision Rel. 4.8. For quantification of insulin^+^, glucagon^+^, and GIP^+^ cells, 100 CgA^+^ cells per animal were analyzed across at least 10 randomly selected sections from five animals per genotype.
Colonoids were stained following a modified protocol [26]. Organoid cultures were seeded in 4-well Nunc Lab-Tek chamber slides (Thermo Fisher Scientific, Waltham, MA, USA). Primary and secondary antibodies were used at 1:200 and 1:250, respectively. Images were captured using a Nikon Confocal Spinning Disk TiE W1 (Nikon, Tokyo, Japan) and processed with ImageJ 1.52n (National Institutes of Health, Bethesda, MD, USA).
2.4. Gene Expression Analysis
Total RNA was extracted from GI segments (fundus, corpus, antrum, duodenum, jejunum, ileum, and colon) using the RNeasy Mini Kit (Qiagen, Hilden, Germany), and from isolated pancreatic islets and colonoids using the RNeasy Micro Kit (Qiagen), according to the manufacturer’s instructions. RNA quantity and quality were assessed using a NanoDrop One spectrophotometer (Thermo Fisher Scientific).
Reverse transcription was performed with Superscript (Invitrogen), and quantitative PCR (qPCR) analyses were carried out using the QuantiTect SYBR Green Kit (Thermo Fisher Scientific) with validated mouse-specific primers (Qiagen). Gene expression levels were normalized to GAPDH and are reported as relative mRNA levels (fold-change).
2.5. Glucose Tolerance Tests and Blood Glucose Measurement
Mice were fasted for 6 h, then administered glucose (2 g/kg of body weight; Sigma-Aldrich) either orally (oGTT) or intraperitoneally (ipGTT). Blood glucose was measured at indicated intervals using an ONETOUCH Verio glucometer (LifeScan, Inc., Malvern, PA, USA).
2.6. Biochemical Analyses
Blood samples were collected at baseline, 10, and 15 min post-glucose administration. Plasma was isolated by cold centrifugation and stored at −80 °C. Plasma GLP-1 and GIP levels were measured using ELISA kits (CC-81508 and CC-81527; Crystal Chem, Itasca, IL, USA) following the manufacturer’s instructions.
2.7. Colonoid Generation, Maintenance, and 4-Hydroxytamoxifen Treatment
Colonoids were generated from colon crypts isolated from 8-week-old Gcg-Cre^ERT2^::Pax4-OE mice as previously described [10,27], with minor modifications. Crypts were embedded in Matrigel (Corning, Corning, NY, USA) and cultured in IntestiCult™ Organoid Growth Medium (STEMCELL Technologies, Vancouver, BC, Canada) supplemented with primocin (100 μg/mL; InvivoGen, San Diego, CA, USA) and penicillin/streptomycin (Gibco).
Colonoids were maintained at 37 °C, 5% CO_2_, and split 1:2 every 10–14 days. Two days after seeding, 10 μM 4-hydroxytamoxifen (4-OHT; Sigma–Aldrich) was applied for 72 h to induce recombination. Medium was then replaced with 4-OHT–free medium, and colonoids were harvested four days later for analysis.
2.8. Glucose-Stimulated Insulin Secretion (GSIS) Assay
Twenty to thirty colonoids were incubated in Krebs-Ringer buffer containing 2.8 mM glucose, 16.7 mM glucose, or 35 mM KCl. Secreted insulin was quantified using Mouse Ultrasensitive ELISA Kit (10-1243-01; Mercodia, Uppsala, Sweden) according to the manufacturer’s instructions.
2.9. Data Analysis
Data are presented as mean ± SEM from at least three independent animals. Normality was assessed with the D’Agostino–Pearson omnibus test. Statistical analyses were performed using GraphPad Prism 10 (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant at p < 0.05 (), p < 0.01 (), p < 0.001 (), and p < 0.0001 (****).
3. Results
3.1. Generation and Characterization of Gcg-CreERT2::Pax4-OE Animals
To determine whether Pax4 ectopic expression in gut glucagon^+^ L-cells could affect cell phenotype and identity in vivo, we first validated the specificity and efficiency of the Gcg-Cre^ERT2^ system. Gcg-Cre^ERT2^ mice [23], harboring a TAM-inducible Cre^ERT2^ recombinase knocked into the endogenous pre-proglucagon locus, were crossed with the Rosa26-β-gal reporter line [24], which carries a loxP-flanked transcriptional STOP cassette—including the neomycin resistance gene—upstream of the β-galactosidase cDNA under the control of the ubiquitous Rosa26 promoter (Figure 1A).
Recombination efficiency was assessed in the colon, the GI segment displaying the highest density of L-cells [28]. Immunohistochemical analyses of colonic sections from TAM-treated Gcg-Cre^ERT2^::Rosa26-β-gal mice confirmed Cre^ERT2^ activity exclusively in glucagon^+^ L-cells (Figure 1C). Quantitative analyses revealed recombination at the Rosa26 locus in 41 ± 6.5% of L-cells (Figure 1D). Importantly, no glucagon^−^/β-galactosidase^+^ cells were detected, further confirming the specificity of Cre^ERT2^-mediated recombination to the L-cell lineage (Figure 1C).
Having validated the recombination efficiency and specificity of the model, Gcg-Cre^ERT2^ mice were subsequently crossed with Pax4-OE animals [17]. In this line, the transgene consists of the ubiquitous CAG promoter driving a loxP-flanked GFP-STOP cassette followed by the Pax4 cDNA linked to an IRES-galactosidase reporter (Figure 1B). Because no reliable antibodies are currently available to specifically detect Pax4, its expression in GI tissues was assessed by RT-qPCR. Following TAM administration, Pax4 mRNA expression was significantly increased in Gcg-Cre^ERT2^::Pax4-OE mice compared to control littermates, reaching up to approximately 3.5-fold induction depending on the GI region analyzed (Figure 1E; Figure S1A).
Importantly, Gcg-Cre^ERT2^::Pax4-OE mice were viable and fertile, with no evidence of premature mortality or overt health abnormalities.
3.2. Pax4 Misexpression in L-Cells Promotes Their Loss in the Gut
L-cells are classically distributed along the GI tract, from the upper intestine to the colon, with their density increasing towards the distal segments [28]. Using the Swiss-roll method to enable comprehensive scanning of the entire GI tract [29], we evaluated the impact of ectopic Pax4 expression on L-cell populations and distribution.
Immunohistochemical analyses confirmed the presence of L-cells throughout the tract in both control and TAM-treated Gcg-Cre^ERT2^::Pax4-OE animals (Figure 2A,B).
Consistent with previous reports, quantitative analyses of glucagon^+^/chromogranin A^+^ cells (CgA, a common marker of mature gut endocrine cells) revealed higher L-cell counts in the ileum and colon compared with other intestinal segments [19] (Figure 2C).
Importantly, ectopic Pax4 expression was associated with a significant reduction in L-cell numbers, with up to a 48 ± 8.2% decrease observed in the colon. This reduction was further supported by gene expression analyses, which showed a marked reduction in glucagon mRNA levels in transgenic tissues relative to controls (Figure 2D). Notably, Pax4 misexpression did not significantly alter the expression of other gut hormones (Figure S2).
Together, these results indicate that Pax4 misexpression in L-cells promotes their substantial loss throughout the GI tract.
3.3. Pax4 Ectopic Expression Drives the Conversion of Gut L-Cells into Insulin+ Cells
To determine whether the decrease in glucagon expression observed in Pax4-misexpressing gut tissues was accompanied by the acquisition of a β-like cell phenotype, as previously reported in the pancreas [17,18], additional quantitative immunohistochemical analyses were performed.
As expected, no insulin^+^ cells were detected along the GI tract in control animals (Figure 3A). In contrast, following TAM treatment of Gcg-Cre^ERT2^::Pax4-OE animals, numerous insulin^+^ cells were observed throughout the entire GI tract, with their numbers progressively increasing from the fundus to the colon (Figure 3B,C and Figure S3A–F).
In agreement with these observations, RT-qPCR analysis revealed a substantial increase in insulin mRNA levels in transgenic tissues compared to controls, reaching up to a 65.35 ± 3.55-fold induction in the colon (Figure 3D).
Among Gcg-Cre^ERT2^::Pax4-OE GI tissues, the colon appeared to represent the largest reservoir of insulin^+^ cells (Figure 3C,D). Accordingly, subsequent analyses focused on this region. Interestingly, a small subset of bihormonal cells co-expressing glucagon and insulin was detected exclusively in transgenic tissues (Figure S4A,B), suggesting the existence of a transitional state during the conversion of L-cells into insulin^+^ cells. Importantly, no co-expression of insulin with other gut hormones was observed.
To further investigate the fate of Pax4-misexpressing L-cells, β-galactosidase lineage tracing was performed by comparing TAM-treated Gcg-Cre^ERT2^::Rosa26-β-gal control tissues with TAM-treated Gcg-Cre^ERT2^::Pax4-OE tissues. In Gcg-Cre^ERT2^::Rosa26-β-gal sections, β-galactosidase^+^ cells were, as expected, scattered throughout the epithelium (Figure S4C), and none co-expressed insulin.
In contrast, in TAM-treated Gcg-Cre^ERT2^::Pax4-OE sections, approximately 20.6% of β-galactosidase^+^ cells expressed insulin (Figure S4D,E), in agreement with previous quantifications (Figure 3C). Notably, all insulin^+^ cells were consistently labeled with the β-galactosidase tracer, confirming their origin from glucagon-expressing L-cells. Although overall recombination efficiency reached approximately 41 ± 6.5% (Figure 1D), insulin expression was detected in a smaller proportion of lineage-traced cells in such 5-day-long analysis, consistent with a progressive acquisition of insulin expression following Pax4 ectopic expression.
Collectively, these data demonstrate that Pax4 misexpression alone induces the conversion of gut L-cells into insulin^+^ cells.
3.4. Gut-Derived Insulin+ Cells Display a β-like Cell Phenotype
To further characterize the molecular identity of the newly generated insulin^+^ cells, we performed comprehensive immunohistochemical analyses to assess the expression of bona fide β-cell markers in colonic sections from TAM-treated Gcg-Cre^ERT2^::Pax4-OE animals.
Our analyses revealed that all colonic insulin^+^ cells uniformly expressed key β-cell markers, including PC1/3 (required for insulin maturation; Figure 4A), GLUT2 (a glucose transporter involved in glucose sensing; Figure 4B), Kir6.2 (the principal pore-forming subunit for the K_ATP_ channel involved in glucose-stimulated insulin secretion (GSIS); Figure 4C), and C-peptide (a byproduct of insulin biosynthesis; Figure 4D).
To further substantiate these findings and overcome limitations related to antibody availability, we analyzed the expression of a broad panel of β-cell-associated genes by RT-qPCR. These analyses revealed a significant upregulation of β-cell-specific mRNA levels in transgenic tissues relative to controls (Figure 4E). Notably, critical regulators of β-cell development and function—including Nkx6.1, NeuroD1, Isl1, Rfx6, Insm1, Sur1, and Gck—were significantly enriched in Gcg-Cre^ERT2^::Pax4-OE tissues, supporting the acquisition of a β-like cell phenotype by gut-derived insulin^+^ cells.
Concomitantly, we observed a marked decrease in the expression of multiple L-cell-associated markers [30], including Arx, Prox1, Hoxb1, Trim35, Etv1, and Hopx, in Pax4-misexpressing tissues compared with controls (Figure 4F).
Collectively, these results indicate that Pax4-misexpressing L-cells progressively lose their original identity while acquiring a β-like cell phenotype.
3.5. Improved Glucose Tolerance in Pax4-Misexpressing Mice Is Associated with Functional Gut Insulin+ Cells and Compensatory K-Cell Expansion
To determine whether gut-derived insulin^+^ cells were functionally competent and capable of improving glucose handling, we performed an ipGTT on TAM-treated Gcg-Cre^ERT2^::Pax4-OE animals and matched controls.
Analysis of glycemic curves revealed a significant improvement in glucose tolerance in transgenic mice, characterized by a reduced glycemic peak and a faster return to fasting glycemia compared to controls (Figure 5A), suggesting an increase in functional β-cell mass and/or enhanced insulin-secreting capacity.
Because L-cells play a critical role in glucose homeostasis through the secretion of glucagon-like peptide 1 (GLP-1), an intestinal hormone that enhances GSIS, we next examined whether their conversion into insulin^+^ cells affected GLP-1 production. Consistent with the reduction in L-cell numbers (Figure 2C), glucose-stimulated GLP-1 levels were significantly decreased in transgenic animals compared to controls (Figure S5A). However, this reduction had limited metabolic impact.
Indeed, during an oGTT, Gcg-Cre^ERT2^::Pax4-OE mice again displayed significantly improved glucose tolerance, with no substantial difference in ΔAUC compared to ipGTT results (Figure 5B,C). These findings suggest the involvement of compensatory mechanisms, potentially involving K-cells, which secrete the incretin hormone glucose-dependent insulinotropic polypeptide (GIP) and may help preserve glucose homeostasis.
To investigate this possibility, we first examined the distribution of GIP^+^ K-cells and confirmed their presence in both control and transgenic tissues (Figure 6A–H).
Notably, we observed a marked increase in K-cell numbers in Gcg-Cre^ERT2^::Pax4-OE intestines, reaching up to approximately 87.5% depending on the intestinal segment analyzed (Figure 6I).
Consistent with this observation, plasma GIP levels following the oral glucose challenge were increased in transgenic mice compared to controls, reaching statistical significance at 10 min and showing a trend toward significance at 15 min (p = 0.1431) (Figure 6J). Together, these results indicate that compensatory expansion of K-cells occurs in transgenic animals, likely counterbalancing L-cell loss and contributing to preserved incretin responsiveness.
Importantly, the improved glucose tolerance observed in Gcg-Cre^ERT2^::Pax4-OE mice cannot solely be attributed to gut-derived insulin^+^ cells and compensatory K-cells. This model also induces the conversion of pancreatic glucagon^+^ α-cells into insulin^+^ cells [17,18], which may additionally contribute to enhanced glucose control.
3.6. Bioengineered Pax4-Misexpressing Mini-Guts Release Insulin upon Glucose Stimulation
To circumvent the intrinsic limitations of the in vivo murine model and directly assess the functional competence of gut-derived insulin^+^ cells, we implemented an ex vivo approach using bioengineered murine mini-guts. Colon-derived organoids (colonoids) were generated from Gcg-Cre^ERT2^::Pax4-OE mice and corresponding controls.
We first determined whether ex vivo Pax4 misexpression in L-cells could recapitulate the phenotype observed in vivo. Immunohistochemical analyses confirmed the presence of glucagon^+^ L-cells (Figure 7A) and revealed insulin^+^ cells exclusively in 4-OHT-treated Gcg-Cre^ERT2^::Pax4-OE colonoids (Figure 7B).
Consistent with these observations, RT-qPCR analyses demonstrated a significant reduction in glucagon mRNA levels (~41.24%; Figure 7C) together with a marked increase in insulin transcripts (79.67 ± 9.67-fold; Figure 7D) in transgenic colonoids compared to controls.
Further characterization identified bihormonal (glucagon^+^/insulin^+^) cells exclusively in Pax4-misexpressing colonoids (Figure S6A), suggesting an intermediate conversion state. Lineage-tracing experiments confirmed that insulin^+^ cells originated from L-cells ex vivo (Figure S6B). Moreover, expression analyses of canonical β-cell markers demonstrated that these insulin^+^ cells exhibited a bona fide β-cell molecular signature (Figure S6C–E). Collectively, these findings validate Pax4-misexpressing colonoids as a robust ex vivo platform to investigate the functionality of gut-derived insulin^+^ cells.
To directly evaluate their secretory competence, we performed GSIS assays on colonoids derived from control and Gcg-Cre^ERT2^::Pax4-OE animals. Exposure to low glucose (2.8 mM) did not induce detectable insulin secretion in either group (Figure 7E). In contrast, high glucose stimulation (16.7 mM) induced robust insulin release exclusively in Pax4-misexpressing colonoids, corresponding to an approximately 4.36-fold increase compared to basal conditions. Additional validation was obtained using 35 mM KCl, a well-established depolarizing agent that induces insulin vesicle exocytosis [31,32], which similarly resulted in a significant increase in insulin release in transgenic colonoids (Figure 7E).
Together, these results demonstrate that gut-derived insulin^+^ cells generated through Pax4 misexpression are not only capable of producing insulin but also exhibit glucose-regulated secretory activity characteristic of functional β-like cells.
4. Discussion
In this study, we demonstrate that gut endocrine cells represent a potential source for cellular reprogramming to replace lost pancreatic β-cells in T1D. Our data show that ectopic Pax4 expression is sufficient to convert gut L-cells into insulin^+^ cells exhibiting key molecular and functional hallmarks of authentic β-cells. Using bioengineered Pax4-misexpressing mini-guts, we further confirmed their functionality ex vivo, providing compelling evidence that patient-specific gut-derived β-like cells may represent a promising strategy to restore normoglycemia in T1D.
4.1. Ectopic Pax4 Expression Converts Gut L-Cells into Insulin+ Cells
Previous studies demonstrated that ectopic Pax4 expression in pancreatic glucagon^+^ α-cells is sufficient to convert them into β-like cells [17,18]. Given the abundance of glucagon^+^ L-cells in the gut and their molecular similarities to pancreatic α-cells, we investigated whether a comparable reprogramming event could be induced in the GI epithelium. Unlike earlier studies in which the minimal glucagon promoter restricted Pax4 expression primarily to pancreatic α-cells [17,18], we used a Gcg-Cre^ERT2^ mouse line with Cre^ERT2^ inserted into the endogenous pre-proglucagon locus [23], thereby enabling targeted Pax4 misexpression in gut L-cells.
This strategy resulted in the generation of insulin^+^ cells throughout the GI tract, accompanied by a partial reduction in L-cell numbers. Importantly, the spatial distribution of newly generated insulin^+^ cells closely paralleled the endogenous distribution of L-cells, with the highest conversion efficiency observed in the distal intestine and colon. This observation is consistent with the higher density of L-cells reported in these regions [19].
Although the loss of gut endocrine cells has been associated with metabolic disturbances, including impaired lipid absorption and altered glucose homeostasis [33], Pax4-misexpressing animals displayed no overt physiological abnormalities. This likely reflects the remarkable regenerative capacity of the GI epithelium, which continuously replenishes endocrine populations through intestinal stem cell activity, as well as the partial Cre^ERT2^ recombination efficiency observed in our model.
Following five consecutive days of TAM treatment, insulin^+^ cells were detected throughout the GI tract. Compared with shorter treatment regimens, this protocol proved to be the most efficient, as it encompassed a broader temporal window including both differentiating and mature L-cells susceptible to Pax4-mediated reprogramming. Notably, prolonged TAM exposure did not further increase the number of insulin^+^ cells, suggesting a cellular turnover rate comparable to that of native gut endocrine cells, estimated at approximately five to seven days [34,35]. Consistent with this interpretation, withdrawal of TAM resulted in the disappearance of intestinal insulin^+^ cells within one week, correlating with a partial decline in glycemic control (Figure S7A,B).
Lineage tracing analyses provided strong evidence that L-cells gradually transition toward a β-like identity. Specifically, L-cells initially adopt a transient glucagon^+^/insulin^+^ bihormonal phenotype before acquiring a more complete β-like transcriptional program. Such intermediate states have been observed in multiple endocrine reprogramming models and likely reflect the progressive remodeling of transcriptional networks required to stabilize the new cellular identity [17,18,36].
Importantly, only a subset of lineage-traced cells expressed insulin this 5-day-long analysis, reflecting the progressive nature of the conversion process. Further quantitative studies would therefore be required to determine the full efficiency of the reprogramming process and to reconcile the discrepancy between the proportion of recombined cells and the fraction of insulin^+^ cells detected in gut tissues.
Molecular characterization revealed that gut-derived insulin^+^ cells expressed key β-cell markers involved in insulin synthesis, processing, and secretion. In parallel, we observed a marked downregulation of several L-cell-associated genes, including Arx. This observation is consistent with the well-established antagonistic relationship between Pax4 and Arx during endocrine lineage specification. As previously reported [14], Arx expression is largely confined to nascent hormone-producing cells rather than fully differentiated ones, supporting the hypothesis that Pax4-mediated reprogramming may primarily target early differentiating L-cells.
As an initial step toward elucidating the molecular mechanisms underlying this conversion, we examined the expression of Cdx-2, a transcription factor that acts as a master regulator of intestinal identity and has previously been shown to impede β-cell reprogramming [8,37]. Interestingly, Cdx2 expression was markedly reduced in colonic tissues from Pax4-misexpressing animals. These findings raise the possibility that Pax4, or some of its downstream targets, may partially suppress intestinal lineage programs, thereby facilitating the acquisition of β-cell characteristics. Nevertheless, future genome-wide transcriptional and epigenomic analyses will be required to fully elucidate the regulatory networks governing Pax4-driven reprogramming in gut endocrine cells.
A limitation of the present study relates to the genetic model used to induce Pax4 misexpression. As the Gcg-Cre^ERT2^ mouse line drives recombination in all glucagon-expressing cells [23], Pax4 overexpression is not solely restricted to gut L-cells and may also occur in pancreatic α-cells. Additional analyses of pancreatic sections confirmed recombination in peripheral α-cells but did not reveal detectable α-to-β cell conversion during the short induction period used in this study (Figure S8A). Consistently, analysis of isolated pancreatic islets revealed a significant increase in Pax4 mRNA levels without detectable changes in insulin expression (Figure S8B). One likely explanation is that the relatively brief TAM exposure may induce Pax4 expression without allowing sufficient time for complete cellular conversion in the pancreas. In contrast, the rapid turnover and regenerative dynamics of the intestinal epithelium may facilitate faster cellular reprogramming events [38], potentially explaining the preferential detection of insulin^+^ cells in gut tissues.
4.2. Pax4-Misexpressing Animals Display Improved Glucose Metabolism
Functional analyses demonstrated that Pax4-misexpressing animals exhibited improved glucose tolerance. Despite a reduction in L-cell numbers and decreased glucose-stimulated GLP-1 secretion, transgenic mice maintained enhanced glycemic control. This observation indicates that newly generated insulin^+^ cells are functionally competent and may contribute to improved glucose handling.
Interestingly, the loss of L-cells was accompanied by a significant increase in K-cell counts and enhanced GIP secretion. These findings suggest the presence of compensatory mechanisms within the entero-insular axis. Similar adaptive responses have been reported in GLP-1 receptor knockout models, where increased GIP signaling contributes to maintaining glucose homeostasis [39,40]. Our observations therefore highlight the remarkable plasticity of intestinal endocrine populations and suggest that alterations in one enteroendocrine lineage may trigger compensatory expansion of others.
In addition, pancreatic GLP-1 and glucagon may function as local gluco-incretins capable of sustaining insulin secretion when intestinal GLP-1 levels are reduced [40,41,42]. Whether these mechanisms operate synergistically with gut-derived insulin^+^ cells to improve glycemic control remains an important question for future investigation.
4.3. Pax4-Misexpressing Mini-Guts Demonstrate Functional Insulin+ Cells
Given the potential physiological consequences of L-cell conversion within the intestinal epithelium [13,33,43], we developed bioengineered mini-guts as an ex vivo platform to study Pax4-mediated reprogramming. Organoid analyses confirmed that gut-derived insulin^+^ cells express canonical β-cell-specific markers and respond to glucose stimulation by secreting insulin.
These findings provide direct evidence that Pax4-mediated reprogramming generates functionally competent β-like cells capable of regulated insulin secretion. Importantly, the organoid system offers a powerful experimental platform to dissect the molecular mechanisms of gut endocrine reprogramming and to evaluate potential therapeutic applications.
Future transplantation experiments into chemically induced diabetic NOD-scid IL2Rg^null^ (NSG) mice will be essential to determine whether gut-derived β-like cells can restore normoglycemia in vivo [44,45].
Compared with previously described reprogramming approaches, including PMN factors overexpression or Foxo1 deletion [8,10], our strategy presents several advantages: (i) ectopic expression of a single factor, Pax4, is sufficient to induce a physiologically relevant β-cell phenotype; (ii) conversion occurs throughout the GI tract, providing a potentially abundant source of reprogrammable cells; and (iii) Foxo1 function remains intact, preserving its tumor-suppressive and β-cell-protective roles [11,46,47,48,49]. Taken together, these features suggest that Pax4-mediated reprogramming may represent a simpler and potentially safer strategy for generating β-like cells.
4.4. Conclusions and Perspectives
Our study demonstrates that Pax4 misexpression enables the generation of insulin^+^ cells in the GI epithelium and highlights the remarkable plasticity of gut endocrine populations. These findings suggest that gut-derived β-like cells could represent a promising cellular source for β-cell replacement therapies.
Importantly, the gastrointestinal tract represents an accessible tissue that can be sampled through minimally invasive procedures such as endoscopic biopsies. This raises the possibility of generating patient-specific β-like cells using autologous gut tissue.
Future work would therefore be required to determine whether similar reprogramming events can be induced in human intestinal organoids and whether small molecules capable of mimicking Pax4 activity could be identified. Another critical question would be whether gut-derived β-like cells could evade autoimmune destruction in the context of T1D. Addressing these challenges would be essential to putatively translate Pax4-mediated reprogramming strategies into clinically relevant therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kalo E. Read S. Ahlenstiel G. Reprogramming—Evolving Path to Functional Surrogate beta-Cells Cells 202211281310.3390/cells 1118281336139388 PMC 9496933 · doi ↗ · pubmed ↗
- 2Wei R. Hong T. Lineage Reprogramming: A Promising Road for Pancreatic beta Cell Regeneration Trends Endocrinol. Metab.20162716317610.1016/j.tem.2016.01.00226811208 · doi ↗ · pubmed ↗
- 3Mc Kimpson W.M. Accili D. Reprogramming Cells to Make Insulin J. Endocr. Soc.201931214122610.1210/js.2019-0004031187080 PMC 6546342 · doi ↗ · pubmed ↗
- 4Basile G. Qadir M.M.F. Mauvais-Jarvis F. Vetere A. Shoba V. Modell A.E. Pastori R.L. Russ H.A. Wagner B.K. Dominguez-Bendala J. Emerging diabetes therapies: Bringing back the beta-cells Mol. Metab.20226010147710.1016/j.molmet.2022.10147735331962 PMC 8987999 · doi ↗ · pubmed ↗
- 5Baafi K. March J.C. Harnessing gut cells for functional insulin production: Strategies and challenges Biotechnol. Notes 2023471310.1016/j.biotno.2022.11.00539416909 PMC 11446352 · doi ↗ · pubmed ↗
- 6Helander H.F. Fandriks L. Surface area of the digestive tract-revisited Scand. J. Gastroenterol.20144968168910.3109/00365521.2014.89832624694282 · doi ↗ · pubmed ↗
- 7Chen Y.J. Finkbeiner S.R. Weinblatt D. Emmett M.J. Tameire F. Yousefi M. Yang C. Maehr R. Zhou Q. Shemer R. De novo formation of insulin-producing “neo-beta cell islets” from intestinal crypts Cell Rep.201461046105810.1016/j.celrep.2014.02.01324613355 PMC 4245054 · doi ↗ · pubmed ↗
- 8Ariyachet C. Tovaglieri A. Xiang G. Lu J. Shah M.S. Richmond C.A. Verbeke C. Melton D.A. Stanger B.Z. Mooney D. Reprogrammed Stomach Tissue as a Renewable Source of Functional beta Cells for Blood Glucose Regulation Cell Stem Cell 20161841042110.1016/j.stem.2016.01.00326908146 PMC 4779391 · doi ↗ · pubmed ↗