From Metabolism to Longevity: Molecular Mechanisms Underlying Metformin’s Anticancer and Anti-Aging Effects
Slavica Vujovic, Svetlana Perovic, Milorad Vlaovic, Andjelka Scepanovic, Stasa Scepanovic

TL;DR
Metformin, a diabetes drug, may slow cancer and aging by altering cell energy processes, but more research is needed to confirm its effectiveness in non-diabetic people.
Contribution
This paper reviews metformin's molecular mechanisms in cancer and aging, highlighting the need for large-scale trials to validate its potential in non-diabetic populations.
Findings
Metformin may slow cancer and aging by modulating AMPK and mTOR pathways.
Preclinical success with metformin does not consistently translate to human clinical trials.
Observational data on metformin's anticancer effects are often confounded by clinical variables.
Abstract
Metformin has stood as the primary clinical tool for type 2 diabetes for decades, yet its potential reach into oncology and gerontology is only now being critically dissected. This review evaluates how metformin might actually pull the levers of cancer progression and biological aging. Evidence from across various models suggests that the drug works by recalibrating cellular energy homeostasis—specifically by triggering AMPK and dampening the mTOR pathway. This signaling shift ripples through downstream processes like autophagy and oxidative stress regulation, theoretically slowing tumor growth and pushing back against cellular senescence. However, our look at the literature from PubMed, Scopus, and Web of Science shows a messy reality where preclinical success often stalls during clinical translation. Even though observational data point toward lower cancer rates in diabetic cohorts,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
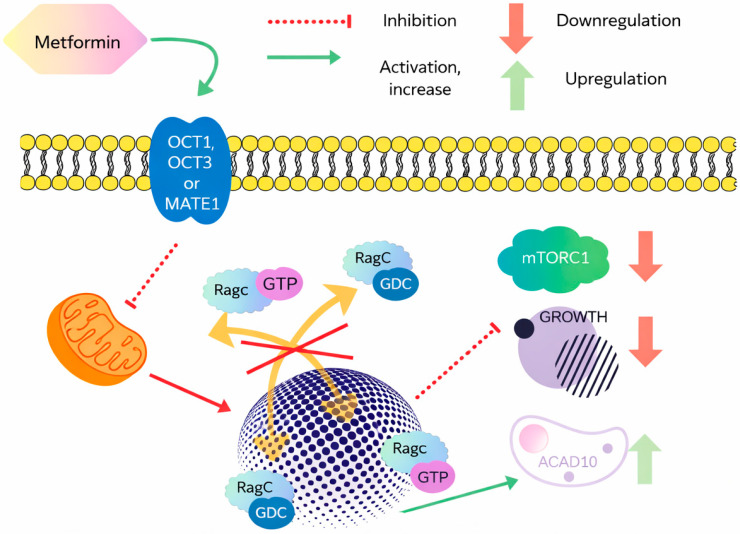 Figure 1
Figure 1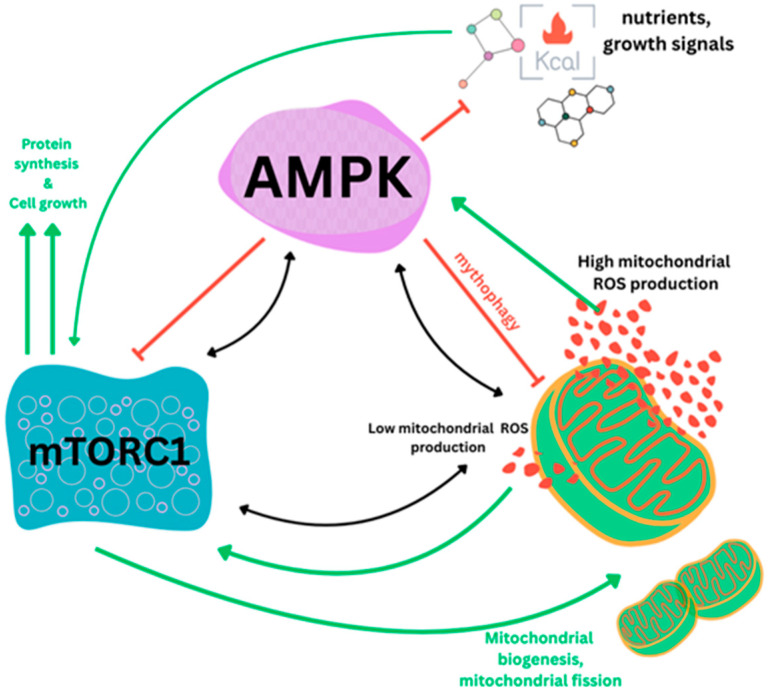 Figure 2
Figure 2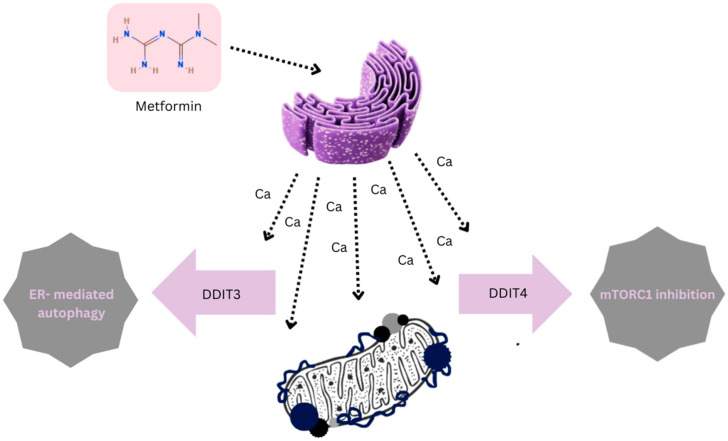 Figure 3
Figure 3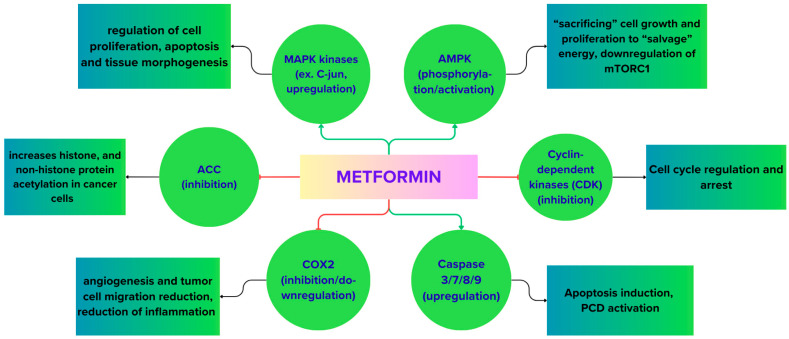 Figure 4
Figure 4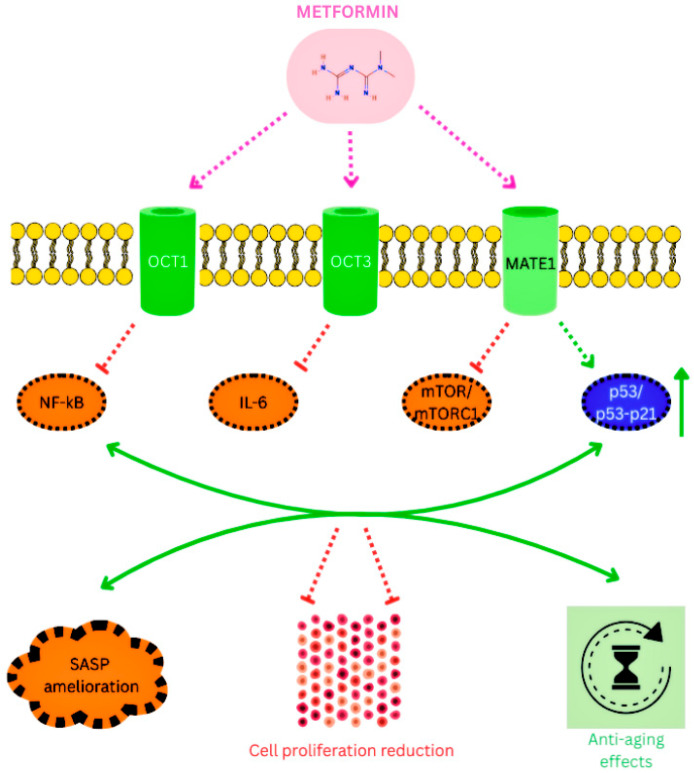 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism, Diabetes, and Cancer · Biological Research and Disease Studies · Cancer Risks and Factors
1. Introduction
Metformin remains the clinical gold standard for managing type 2 diabetes mellitus (T2DM). Since its introduction, it has maintained its position as a first-line therapy, primarily valued for its consistent efficacy in lowering HbA1c and its well-established safety profile [1]. However, in recent years, the focus of metformin research has shifted significantly. While its role in glucose suppression is clear, the underlying molecular landscape remains surprisingly complex and, in several aspects, contentious. Beyond the classic inhibition of hepatic gluconeogenesis, it is now evident that metformin’s influence reaches into skeletal muscle and the intestinal environment, involving a sophisticated interplay between AMPK signaling and intestinal receptors [2,3].
This metabolic versatility has sparked interest in “repurposing” metformin for conditions far beyond diabetes—most notably in oncology and gerontology [4,5]. The last twenty years have seen a surge in data exploring whether metformin can actually lower cancer incidence or slow down the biological clocks of aging [6]. At the cellular level, the drug appears to “reprogram” energy sensing. By activating AMPK and suppressing the mTOR pathway, metformin targets the very mechanisms that drive tumor growth and cellular senescence [7,8]. Consequently, it is no longer viewed just as an antidiabetic agent, but as a potential candidate for broad-spectrum metabolic intervention [6,9].
The first real hint of metformin’s anticancer potential emerged in 2001, when a study on hamsters showed that metformin treatment significantly reduced the development of pancreatic lesions [10]. Since that observation, a mountain of epidemiological data has attempted to link T2DM, obesity, and cancer risk. Observational studies frequently suggest that diabetic patients on metformin have lower rates of various malignancies—including colorectal, liver, and breast cancers—compared to those on other treatments [7,11,12].
However, we must approach these findings with caution. When Evans et al. (2005) first reported a reduced cancer risk in a Scottish cohort, it opened the floodgates for similar research [8], yet the results have been far from uniform. While some meta-analyses point to a 31% reduction in tumor incidence [13], other studies on lung and prostate cancers show almost no survival benefit [14,15,16]. For instance, in colorectal cancer, some data sets even failed to show a statistically significant advantage (HR 1.06) [17]. A massive 2023 meta-analysis of 80 studies recently tried to settle this, suggesting that while breast cancer patients might see the most gain, we still lack the definitive evidence that only large-scale randomized controlled trials (RCTs) can provide [18].
The core of the problem lies in the “noise” of clinical data. We cannot simply equate correlation with causation here. Many of the reported benefits might be skewed by clinical confounders; for example, metformin users often have less severe or “younger” diabetes than those prescribed insulin. The situation is further complicated by the common co-prescription of aspirin or statins, which have pleiotropic effects of their own and make it challenging to pinpoint the precise role of metformin. The data’s ongoing heterogeneity is a result of the frequent inconsistencies in the length of diabetes and the precise glycemic control attained (HbA1c levels) between studies. For this reason, we must go beyond clinical observations and investigate the drug’s direct molecular actions in greater detail.
Mechanistically, metformin operates through several distinct routes. In the gut (enterocytes), it appears to disrupt mitochondrial complex I, which shifts the cellular energy balance and triggers AMPK signaling [2,3]. In the liver, however, it can also act independently of AMPK. By noncompetitively blocking mitochondrial glycerol-3-phosphate dehydrogenase (mG3PD), metformin directly shuts down the conversion of lactate and glycerol into glucose [19]. More recently, an epigenetic layer has been identified: metformin can upregulate microRNAs like let-7, which in turn disrupts the TET3–HNF4α axis to suppress hepatic glucose production [20]. This multi-layered mechanism—spanning from mitochondrial enzymes to microRNA regulation—highlights why metformin remains one of the most intriguing molecules in modern pharmacology.
2. Materials and Methods
2.1. Literature Search and Study Selection
This article was designed as a narrative review of the literature focusing on the molecular mechanisms underlying the anticancer and anti-aging effects of metformin. To ensure transparency and methodological rigor, we employed a structured approach to literature identification and selection.
A literature search was performed using the PubMed, Scopus, and Web of Science databases, covering literature until March 2025. The search terms were combined using the keyword ‘metformin’ and terms related to the key molecular and biological pathways, including AMPK, mTOR signaling, aging, and cancer. Some examples of search terms included “metformin AND AMPK,” “metformin AND mTOR,” “metformin AND aging,” and “metformin AND cancer.”
After combining the search results from all three databases, a total of 3200 articles were found. The duplicates (approximately 1200) were removed before screening. The remaining 2000 articles were screened for relevance to the mechanistic and clinical aspects of the review by title and abstract. At this stage, 1600 articles were removed because they did not have information regarding molecular mechanisms, were not clinically relevant, or were not relevant to the manuscript.
The full-text screening was done for 400 articles. Out of these, 294 articles were excluded because they did not have any mechanistic data, had data related to supra-pharmacologic doses of metformin that are not clinically relevant, or were published in languages other than English.
A total of 106 articles were selected for the qualitative analysis, which is equivalent to the number of references included in the final manuscript.
2.2. Data Synthesis
Relevant data were extracted and qualitatively synthesized to identify key molecular pathways influenced by metformin, including AMPK activation, mTOR inhibition, regulation of mitochondrial metabolism, oxidative stress, and cellular senescence. The findings were integrated into a thematic narrative aimed at highlighting shared and divergent mechanisms underlying metformin’s metabolic, anticancer, and anti-aging effects.
3. Results and Discussion
This review combines the existing knowledge about the molecular pathways through which metformin works as an anticancer and anti-aging compound. The main crucial biological pathways through which metformin works as an anticancer and anti-aging compound are the activation of AMPK, the inhibition of the mTOR pathway, the regulation of oxidative stress levels, and the modulation of the metabolism of mitochondria. Although the final compound has been found to be useful in the treatment of diabetes, its antiproliferative properties against cancer and its anti-aging effect can be employed in various applications. This will be explained in the later sections.
3.1. Metformin-Mediated Inhibition of Mitochondrial Activity, Nuclear Pore Function, and ACAD10 Induction
Here, we focus on the direct, insulin-independent mechanisms of metformin’s anticancer action. Metformin’s effects on tumor cells are multitargeted and can broadly be categorized as direct (insulin-independent) or indirect (insulin-dependent) mechanisms [21]. A key direct target is the mitochondrial respiratory chain. Because cancer cells rely heavily on glucose metabolism (the “Warburg effect”), their elevated glucose uptake can lead to local glucose depletion relative to surrounding normal tissues [22,23].
Wu and colleagues identified two molecular targets linking metformin to antitumor activity within the same pathway: the nuclear pore complex (NPC), responsible for macromolecular trafficking, and acyl-CoA dehydrogenase 10 (ACAD10) [24]. After entering cancer cells via organic cation transporters (OCT1 and OCT3) and the multidrug and tox-in extrusion transporter (MATE1) [25,26], metformin inhibits mitochondrial complex I. This leads to reduced intracellular energy levels, limits transport of the RagA–RagC GTPase heterodimer through the NPC, and ultimately inactivates mTORC1, thereby suppressing tumor cell proliferation (Figure 1). Concurrently, metformin also stimulates the transcriptional upregulation of ACAD10, which regulates β-oxidation and has been shown to extend the longevity of Caenorhabditis elegans [27].
In both human melanoma and pancreatic cancer cells, the restrictions caused by the NPC and the elevation of ACAD10 induced by biguanide drugs have also been noted. However, the abrogation of protective actions through the forced opening of nuclear pores or the genetic ablation of ACAD10 illustrates the importance of functional NPC-ACAD10 signaling in metformin-induced cancer prevention and longevity [27]. Biochemical experiments also showed that the antiproliferative effect in cancer cells at various doses of metformin occurs through the depletion of the TCA cycle due to the reduced activity of the mitochondria complex I [28]. Additionally, the same antiproliferative effect occurs in AMPK-deficient cancer cells, demonstrating that the contribution of AMPK activation to the metformin antiproliferative effect might be unnecessary [29].
Hexokinase II, a glycolytic enzyme that interacts with the outer membrane of mitochondria, has higher levels in cancer cells than in normal cells [30,31]. Metformin directly interacts with glycolysis through binding at the glucose-6-phosphate (G6P) site of Hexokinase II to trigger apoptosis [29]. Another target protein of interest involves the glycerol-3-phosphate dehydrogenase of mitochondria (GPDH). Inhibition of GPDH leads to a diminished transformation of glycerol-3-phosphate to dihydroxyacetone phosphate, hence less lactate and glycerol being used in the production during gluconeogenesis [32]. As GPDH levels are increased in thyroid cancer cells, this tumorigenesis can be targeted through the interaction of metformin and the modulation of oxidative phosphorylation [33].
Apart from the above direct actions, metformin can also indirectly impact cancer cells through the systemic control of the insulin/IGF1 signaling pathways as described below.
3.2. Insulin- and IGF1-Dependent Mechanisms
Because cancer cells rely heavily on glucose, metformin can also act by modifying insulin and IGF1 signaling, especially in patients with type 2 diabetes [23,34]. Cells with impaired glucose metabolism or mitochondrial complex I mutations (mtDNA mutations) are also more susceptible to metformin and its derivative phenformin than wild-type cells when glucose levels are low [29].
Through the Ras/Raf/MEK/ERK and PI3K/AKT/mTORC1 (PAM) pathways, insulin and IGF1 act as growth factors for many tumors, encouraging cell survival, proliferation, and resistance to apoptosis [34,35]. Because insulin-like growth factor-binding proteins (IGFBPs) are reduced by hyperinsulinemia, the IGF1 receptor—a key factor in malignant transformation and tumor survival—is activated, increasing free IGF1 concentrations. By reducing the levels of IGF1 and insulin in the blood and suppressing the expression of their receptors, metformin interferes with this process [34,35].
3.3. AMPK-Dependent Mechanisms
Given that AMPK serves as a metabolic master switch—prioritizing cell survival over proliferation—its activation is widely regarded as a central mechanism behind metformin’s anticancer effects [36]. The process begins with shifts in cellular energy status; specifically, fluctuations in AMP:ATP or ADP:ATP ratios act as the primary trigger for AMPK [32]. Within this framework, Liver Kinase B1 (LKB1) acts as an indispensable mediator by directly phosphorylating AMPK [33,34]. Recent in vitro data further suggest that inositol polyphosphate multikinase (IPMK) interacts with LKB1 to amplify this response in the presence of metformin [37].
Once activated, AMPK targets the tuberous sclerosis complex, phosphorylating both TSC1 (hamartin) and TSC2 (tuberin). This signaling cascade effectively shuts down Rheb GTPase, a key activator of mTORC1. Beyond this direct route, metformin exerts further control over mTORC1 through alternative pathways. For instance, it can induce the hypoxic stress-response protein REDD1 via a p53-dependent mechanism [38] or interfere with Rag GTPases, which usually drive mTORC1 activation in response to nutrient availability [39,40,41]. The interplay between nutrient availability, levels of mitochondrial oxidative stress, AMPK and mTOR1 in carcinogenesis, aging and as potential targets for metformin should therefore not be overlooked (Figure 2) [19,36,37].
Acetyl-CoA carboxylase (ACC) phosphorylation and inhibition in ovarian and prostate cancer cell lines require AMPK activation via LKB1. Gene-expression profiles are changed as a result of increased acetylation of histone and non-histone proteins [42]. Furthermore, fatty acid oxidation is increased and lipogenic enzyme levels are decreased when the ACC gene is inactivated [39].
Metformin also inhibits lipogenesis [39,43], angiogenesis [44], cytokine production [45], and CD8+ tumor-infiltrating lymphocyte infiltration [46], among other AMPK downstream effects.
3.4. AMPK-Independent Mechanisms
3.4.1. Direct Antineoplastic Effects: Beyond the AMPK Paradigm
While AMPK is a central player, it is becoming increasingly clear that metformin’s efficacy does not rely on this pathway alone. The drug appears to operate through a dose-dependent hierarchy; at low concentrations, energy stress triggers AMPK, but at higher doses, alternative mechanisms take the lead. This suggests a redundant, multi-layered system rather than a single point of failure. Crucially, metformin can suppress tumor progression and mitigate aging markers even in cells lacking functional AMPK signaling, indicating that its systemic benefits—such as the reduction in circulating insulin—often bypass the need for local AMPK activation [47,48].
Metformin’s direct influence also extends to the tumor suppressor protein p53. In prostate cancer models, for instance, metformin significantly inhibits growth in p53-wild-type cells, whereas this effect is markedly diminished in p53-deficient environments. However, a major caveat remains: most laboratory findings rely on metformin concentrations far exceeding those found in human patients, leaving the clinical relevance of the p53-axis still open to debate [49]. Furthermore, metformin can inhibit mTORC1 by upregulating the stress-response protein REDD1 in a p53-dependent manner, effectively bypassing traditional nutrient-sensing routes [50]. Beyond these pathways, the drug actively reshapes the tumor microenvironment by downregulating pro-inflammatory and angiogenic factors such as VEGF, HIF-1α, and PDGF-B, thereby hindering the epithelial–mesenchymal transition (EMT) and limiting metastatic potential [51].
3.4.2. Indirect Systemic Mediators
Metformin also combats cancer and aging by fundamentally altering the body’s systemic environment. One of its most potent indirect effects is the suppression of hepatic glucose production. By promoting the accumulation of intracellular AMP, metformin noncompetitively inhibits adenylate cyclase. This leads to a drop in cyclic AMP (cAMP) and a subsequent decrease in protein kinase A (PKA) activity—a process that effectively shuts down glucagon-mediated gluconeogenesis regardless of AMPK status [52].
Perhaps most importantly, metformin’s ability to lower circulating insulin levels remains a cornerstone of its anticancer profile. By improving insulin sensitivity and reducing hyperinsulinemia, the drug starves insulin-dependent tumors of a key growth stimulus [53,54,55]. Far from being mutually exclusive, these AMPK-independent pathways actually reinforce one another. They suggest a multi-layered mechanism where the drug’s impact is heavily dictated by the specific context: the dosage used, the drug’s distribution, and the metabolic state of the model. Because these models are so varied, there is a strong case for future research to be far more transparent. If we are to truly pin down the role of AMPK, it is vital to document intracellular metformin levels alongside the exact status of AMPK signaling—whether through genetic or pharmacological tweaks—and systemic factors like insulin and glucose levels.
3.5. Stress-Induced Effects
Cellular stress is a key factor that shapes the metabolic behavior of cancer cells. Metformin can make cells more susceptible to stress by restricting available energy and worsening already poor vascularization, which becomes especially important under hypoglycemic conditions [38]. When glucose levels drop, the endoplasmic reticulum (ER) activates the unfolded protein response (UPR) to prevent the accumulation of misfolded proteins [56], maintaining this adaptive response until mitochondrial respiration is restored [38,57].
Evidence from tumor cell cultures and xenograft studies shows that severe ER stress can lead to mitochondrial blebbing, largely due to excessive Ca^2+^ influx from the overstressed ER. This, in turn, triggers apoptosis through stress-responsive mediators such as DDIT4 and DDIT3 (Figure 3) [38,58,59]. DDIT4 contributes to mTORC1 inhibition and thereby slows tumor cell proliferation, while DDIT3 plays a central role in initiating ER-mediated apoptotic pathways.
3.6. Autophagy, Apoptosis Induction, and Cell Cycle Arrest
Autophagy is a double-edged sword in cancer biology because, on one side, it offers a survival signal for cancer cells under metabolic stress but, on the other side, it can act as an initiating factor for apoptosis. The differences in these adaptations are mediated by AMPK, which acts as a “thermostat” at the cellular level that detects metabolic stress and triggers the warning system for protective mechanisms [60,61]. The activation of AMPK directly initiates autophagy by regulating the autophagy-start signaling machinery, including mitophagy (Figure 2), which helped in the clearance of damaged mitochondria and consequently led to cell homeostasis. Apoptosis and autophagy are two prominent forms of programmed cell death (PCD) in normal and tumor cells [43,48,59]. Since metformin has the potential to trigger PCD, often through AMPK or enzymatic pathways, it is increasingly being explored as an adjunct in cancer therapy, where the focus would be on encouraging stressed tumor cells towards death rather than survival and enhancing the efficacy of existing treatments [Figure 4].
A clear example comes from the work of Takahashi et al., who found that metformin caused cell cycle arrest at both G1 and G2/M phases in Ishikawa endometrial cancer cells by upregulating the cyclin-dependent kinase inhibitor p21 (CDKN1A) [62]. At higher doses (10 mM), metformin also activated caspase-dependent apoptosis (CASP3/7, -8, -9). Interestingly, the apoptosis response was reduced when the autophagy regulator Beclin1 (BECN1) was inhibited [63].
Variations in the tumor suppressor gene TP53 impact heavily upon tumor characteristics. Approximately 50% of various cancers exhibit irregularities of the p53 protein [63,64]. In the case of p53 proficient colon cancer xenografts, the growth of the tumor was unaffected by metformin, though apoptosis occurred. However, in the case of p53-deficient colon cancer xenografts, the growth of the tumor was inhibited along with apoptosis triggered by metformin [65].
In breast cancer models, Al-Zaidan et al. found that metformin at a concentration of 10 mM resulted in G1 arrest, reduced cell viability, and apoptosis in 72 h [66]. Similarly, Li et al. reported that metformin caused G2/M phase accumulation in osteosarcoma cells both in vitro and in vivo, potentially by reactivating NAC-suppressed JNK/c-Jun signaling. This stress-dependent pathway ties metformin to autophagy, apoptosis, and cell cycle arrest in these cells [67]. However, a critical translational caveat is that these phenotypic changes—such as robust induction of apoptosis and autophagy—are predominantly documented at suprapharmacological concentrations (10 mM). These doses are approximately 100 to 1000 times higher than the steady-state plasma levels of 1–5 μM typically achieved in patients [68]. Consequently, while these studies elucidate the maximal biological signaling potential of metformin, they may overstate its potency as a monotherapy in a clinical setting.
In acute myeloid leukemia SKM-1 cells, metformin induced G0/G1 arrest through an AMPK-dependent mechanism [69]. Treatment decreased CDK4 and Cyclin D1 levels, increased p53 expression, and showed a clear dose-dependent timeline of antiproliferative effects: 1 mM peaked at 72 h, 5–10 mM at 48 h, and 15–20 mM at 24 h. Despite being higher than typical therapeutic concentrations, these doses did not lead to lactic acidosis or other severe adverse effects, which is a promising observation [70,71].
3.7. Metformin in Therapy Combinations and Dose-Dependent Clinical Effects
We are seeing a real shift in how metformin is being tested—specifically, how it behaves at lower, safer doses when paired with other drugs. It turns out that metformin is not just a “stand-alone” treatment. Instead, it seems to act more like a sensitizer, making chemotherapy, hormone therapies, or even DNA-targeting agents work a bit harder [72].
The big takeaway here is that at these low doses, metformin does not actually kill cells on its own. Its real value is in lowering the “apoptotic threshold.” For instance, when combined with hormone treatments, it can actually tinker with gene expression and halt cell proliferation in ways that the drugs could not manage alone. There is even some evidence that it helps shield the heart from the toxic side effects of certain antibiotics. That said, it is not a universal fix; the synergy with drugs like cisplatin, for example, is still pretty hit-or-miss [72,73].
Take the work by Rafaela Erices and her team. They found something quite telling: ovarian cancer cells became far more vulnerable to carboplatin when metformin was added at doses similar to what a diabetic patient would take [73]. On its own, metformin at that level did almost nothing to stop cell growth. This really illustrates the point that metformin’s future in oncology is not as a “miracle pill” on its own, but rather a strategic add-on. By keeping the doses safe and tolerable, we might finally make resistant tumors more responsive to the standard tools we already have.
There are many pathways through which the synergy can occur in metformin. The AMPK/mTOR can be mainly implicated when co-administered in hormone-modulating therapy, while the role of HIF-1, P-gp, and MRP1 approached suppression can be crucial when combined with anti-metabolites [72]. Of late, there has been a study of the synergy of metformin when combined with the phytochemicals quercetin and resveratrol. When co-administered with quercetin, the combination of metformin has been proven to target the reduced viability, migration, and invasiveness of prostate cancer cells (PC-3 and LNCaP). However, the combination of metformin and resveratrol has demonstrated the role of proliferation through PI3K/Akt downregulation, AMPK phosphorylation, and the modulation of mTOR when combined together [74]. The role of nanovectorization has been shown to further enhance the bioavailability and synergistic effects of metformin and phytochemicals.
Berberine, which has antidiabetic and antineoplastic actions similar to metformin’s and is a phytochemical, also has the shared mechanistic targets of AMPK activation and JNK/c-Jun, CDK4, mTOR, and WNT pathways [75,76]. Clinical trials involving the co-administration of the two in metabolic syndrome patients indicate their combined effectiveness over the two drugs separately [77], though there is less information regarding their interaction in cancer research.
The dosing regimen of metformin has a direct impact on its medicinal value. The medication demonstrates varying efficiencies for different types of cancer and other diseases like polycystic ovary syndrome and cardiovascular diseases [77,78]. The dosage of metformin when used to promote longevity and healthspan will be less compared to its dosages in metabolic and cancer treatment applications (Table 1).
Aging has been linked to various biological characteristics: genomic instability, telomere shortening, epigenetics, proteostasis failure, disrupted nutrient sensing pathways, mitochondriopathies, senescent cells, depletion of stem cell pools, dysregulation of intercellular interaction, inflammation, and dysbiosis. Most of the above factors can be traced to the existence of chronic inflammation and oxidative stress in the body, which can cause the development of the tumor microenvironment [79,80,81].
Patients with both T2DM and cancer often share risk factors such as age, sex, obesity, limited physical activity, and suboptimal diet [82]. Hyperglycemia worsens oxidative stress and increases advanced glycation end products (AGEs), adding to genotoxic burden and elevating cancer risk [83,84]. By modulating glucose and insulin metabolism and reducing IGF-1 levels, metformin offers protective benefits—particularly among older individuals with T2DM [85]. In one analysis, the adjusted hazard ratio (HR) for cancer incidence was 0.68 (95% CI 0.51–0.90), although cancer mortality did not change. Interestingly, concurrent daily aspirin use (100 mg) was associated with higher cancer incidence in the same cohort [86].
Systemic inflammation, immunosenescence, and the activation of JNK in the elderly also cause vulnerability to cancer [87,88,89]. Metformin has been found to reduce the production of various factors that cause inflammation, such as IL-6 and NF-Κb [90]. This also reduces the production of PGE2 through the suppression of COX-2 and the expressions of Snail and IL-6 due to its anti-migratory and anti-angiogenic actions [91,92,93,94].
In cancer, among the key factors responsible for the activation of cancer stem cell-like properties, is the dysregulation of Wnt-catenin signaling. The abnormal activation of this signaling pathway is responsible for the continuous proliferation of cells, resistance to treatments, and changes in the tumor microenvironment [95]. For example, it has been shown that metformin, through the activation of AMPK and the subsequent inhibition of mTOR, indirectly inhibits Wnt signaling. Consequently, the transcriptional activity of catenin is inhibited [96,97].
However, it should be noted that most of the evidence for these effects is based on in vitro studies, which are primarily carried out using metformin concentrations that are several times higher than the pharmacologically relevant levels, and thus, not attainable in patients [98]. Hence, the implications of metformin on the Wnt pathway in human cancers are yet to be determined. Moreover, the relationship between Wnt signaling and senescence in cancer is complex. On the one hand, the inhibition of senescence, associated inflammation, and the factors of SASP can inhibit the signals that promote tumors in the microenvironment, but on the other hand, the over-inhibition of senescence can, in theory, inhibit its cancer-inhibiting effects, especially in the very early stages of tumor formation (Figure 5) [99,100]. These points highlight the complexities involved in preclinical research and the need for careful interpretation of the results and their validation through rigorous, context, dependent studies [101,102,103].
Together, these evidence points demonstrate the different biological outcomes of Wnt and SASP pathway modulations in cancer, and therefore these evidence points cannot be merely translated to other biological or pathological processes, especially aging, where the biological roles of these pathways are quite different [95,98,102].
Regarding aging and age-related diseases, the Wnt signaling pathway and cellular senescence are two key elements that clearly determine the pathophysiological process. Therefore, the stem cell population dynamics, the capacity of the tissue to regenerate, and the susceptibility to the degenerative diseases are the common biological effects of the dysfunction of the Wnt pathway, which has been linked to the natural aging process. Under these conditions, it has been proposed that a controlled downregulation of the ab-errant Wnt signaling may favor tissue preservation and ultimately lifespan extension, [96,104] which is something metformin may possibly achieve based on the available data.
Preclinical studies have indicated that metformin could modulate age and age-related pathways primarily by mitigating mitochondrial oxidative stress, enhancing metabolic efficiency, and counteracting chronic low-grade inflammation triggered by the accumulation of senescent cells. The modulation of certain aspects of the Senescence-Associated Secretory Phenotype, therefore, has been hypothesized to be one of the mechanisms by which metformin could mitigate age-related inflammatory issues and slow the onset of functional decline [105]. However, the bulk of the evidence available to date has been obtained from animal and/or cell-based studies, and there is a complete lack of human validation.
In contrast to cancer, where senescence can be either protective or detrimental, depending on the context, reducing persistent SASP (Senescence-Associated Secretory Phenotype) signaling in aged tissues is thought to be, for the most part, a positive process [90,106]. The extent to which this occurs, and when, and in which tissues, is still unclear. To assess the effect of metformin on senescence burden and SASP activity, a long-term human study will be needed to determine if metformin modulates these processes at a clinically relevant dose.
4. Clinical Evidence and Translational Implications
The clinical evidence on the anticancer and anti-aging effects of metformin remains heterogeneous and has been derived largely from observational studies [48,63,66,72,80,81,100]. Despite the fact that a large number of retrospective studies have shown that patients with type 2 diabetes taking metformin have lower incidence of cancer or improved survival rates, these observations should be interpreted cautiously [22,24,26,87]. They could be influenced by confounding by indication, immortal time bias, and marked metabolic heterogeneity (Table 2).
Moreover, the existing evidence indicates a high degree of variability of clinical outcomes depending on the type of cancer. A more consistent positive association has been observed in colorectal, breast, and hepatocellular cancers, whereas the findings in lung, prostate, and pancreatic cancers have been mostly neutral or inconsistent [16,20,33,70,74,93,105]. It is also important to note that the randomized controlled trials conducted so far have not been able to demonstrate a definite anticancer effect of metformin in non-diabetic patients, thus underlining the difficulty of extrapolating preclinical data to the clinical setting [47,50,103].
From a translational perspective, the clinical relevance of metformin appears to be highly context-dependent. For instance, systemic metabolic disorders, drug accumulation in particular tissues, genetic predisposition of tumors, and combination therapies are likely to influence therapeutic responses [27,35]. Another issue is that most of the mechanistic information has been derived from in vitro studies using suprapharmacological concentrations of metformin that are substantially higher than the plasma concentrations that can be achieved clinically [106].
5. Future Directions
Trying to repurpose metformin from a standard diabetes pill into a multi-purpose tool for cancer and aging is not just a matter of running more trials. It actually demands a total rethink of research logic. The singular approach does not promise as much as researching synergism and tailored, individualized doses to specific metabolic phenotypes of a patient.
Perhaps the most persistent issue in the field is the ‘dose-translation gap,’ a discrepancy that undermines much of available in vitro data. Much of in vitro data comes from lab tests using concentrations that are far higher from what you would actually find in human plasma (the typical 10–50 µM range). If our models are ever going to mean anything in a clinic, we have to prioritize doses that actually make sense for the human body. Validating pathways like p53 or Wnt at concentrations humans can actually handle safely is the objectively best way forward.
Additionally, currently available data would suggest that metformin’s real strength is that of a strategic “assistant.” By nudging the “apoptotic threshold” of cancer cells, it could make tumors more vulnerable to standard chemo like Cisplatin—maybe even helping to break through drug resistance. There is also a strong case for its role in immunotherapy, helping to “clean up” the metabolic mess in the tumor microenvironment. Combining it with senolytics or natural compounds like Resveratrol is an exciting frontier, more so if these attempts eventually hold up in human trials.
Also, to reduce or evade side effects like lactic acidosis or stomach issues, we may need a smarter delivery system. Crucially, utilizing nanoparticles as targeted vehicles allows for a more concentrated release of metformin where it is needed most, potentially mitigating the common off-target effects and gastrointestinal distress that currently limit its use. Its effects on B12 levels and VO max (possible reduction) in older adults also warrant further research.
It is important to address the polymorphisms in population which are of interest here. We simply cannot expect uniform results from a population with such diverse metabolic profiles. Future research requires more precision, tailoring and individualization-oriented approach. We need to find the markers—LKB1 status, OCT1 levels, or even gut bacteria—that will “tell us” who will actually respond.
Concerning its life-extending prospective effects: while preclinical data is encouraging, we must acknowledge that murine models are, at best, an approximation of human physiological responses. We desperately need long-term studies like the TAME trial as that is the only proper way to prove if metformin can truly push back age-related diseases and actually extend the human healthspan.
6. Conclusions
It is impossible to overlook how much metformin actually interferes with the core cellular pathways behind both cancer and aging. But we need to stay grounded: moving these lab results into a real clinical setting is a massive uphill battle. We can see the drug hitting targets like inflammation and mitochondria, but we are still quite far from pinning down how metabolic control and cancer onset really talk to each other.
Perhaps the biggest problem facing researchers and the first thing they tend to gloss over is the “dose translation gap.” A massive percentage of our data comes from in vitro studies with unrealistic and massive doses of this stuff. This is not even close to the human body and is completely unrealistic in someone who is not even diabetic in the first place. The “sweet spot” of dosage and timing in a completely healthy human is not just the next step in this process; it is the most pressing issue for the next ten years of study.
If we look at it from a molecular standpoint, the intersection of AMPK and mTOR provides a good lead. In “cleaning up” the chronic inflammation that cancer cells and aging cells feed off of, metformin effectively “cleans up” the mess. Of course, we have to stop referring to it as a “miracle pill,” however. Realistically speaking, it is likely not as effective as a standalone drug. It is more likely to have an impact as a smart, synergistic “add-on” to what we are currently using. To actually get there, we need to move past the idea that one size fits all. We have to have human trials that examine the unique metabolic signature of an individual. It is only then will we be able to tell if the promise we are seeing in a lab setting will translate into a real-life victory.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1UK Prospective Diabetes Study (UKPDS) Group Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34)Lancet 199835285486510.1016/S 0140-6736(98)07037-89742977 · doi ↗ · pubmed ↗
- 2Rena G. Hardie D.G. Pearson E.R. The mechanisms of action of metformin Diabetologia 2017601577158510.1007/s 00125-017-4342-z 28776086 PMC 5552828 · doi ↗ · pubmed ↗
- 3Zake D.M. Kurlovics J. Zaharenko L. Komasilovs V. Klovins J. Stalidzans E. Physiologically based metformin pharmacokinetics model of mice and scale-up to humans for the estimation of concentrations in various tissues P Lo S ONE 202116 e 024959410.1371/journal.pone.024959433826656 PMC 8026019 · doi ↗ · pubmed ↗
- 4Vancura A. Bu P. Bhagwat M. Zeng J. Vancurova I. Metformin as an anticancer agent Trends Pharmacol. Sci.20183986787810.1016/j.tips.2018.07.00630150001 PMC 6153060 · doi ↗ · pubmed ↗
- 5Śliwińska A. Drzewoski J. Molecular action of metformin in hepatocytes: An updated insight Curr. Diabetes Rev.20151117518110.2174/157339981166615032523310825808533 · doi ↗ · pubmed ↗
- 6Chiang G.G. Abraham R.T. Targeting the m TOR signaling network in cancer Trends Mol. Med.20071343344210.1016/j.molmed.2007.08.00117905659 · doi ↗ · pubmed ↗
- 7Yang J. Yang H. Cao L. Yin Y. Shen Y. Zhu W. Prognostic value of metformin in cancers: An updated meta-analysis based on 80 cohort studies Medicine 2022101 e 3179910.1097/MD.000000000003179936626437 PMC 9750609 · doi ↗ · pubmed ↗
- 8Evans J.M.M. Donnelly L.A. Emslie-Smith A.M. Alessi D.R. Morris A.D. Metformin and reduced risk of cancer in diabetic patients BMJ 20053301304130510.1136/bmj.38415.708634.F 715849206 PMC 558205 · doi ↗ · pubmed ↗