Defining Histological Patterns in Inherited Ichthyoses: Toward a Diagnostic Algorithm Based on 66 Confirmed Cases
Kira Süßmuth, Vinzenz Oji, Jacqueline Bodes, Isabelle Jochum, Florian Muhs, Katalin Komlosi, Ingrid Hausser, Matthias Schmuth, Heiko Traupe, Judith Fischer, Dieter Metze

TL;DR
This study identifies six distinct histological patterns in inherited ichthyoses to help doctors diagnose these skin disorders more accurately, especially when genetic testing is not available.
Contribution
The study introduces a pattern-based diagnostic algorithm for ichthyosis subtypes based on histological analysis of 66 confirmed cases.
Findings
Six distinct histological patterns were identified in inherited ichthyosis cases.
A diagnostic algorithm is proposed to improve subtype classification using histological features.
The study provides a valuable tool for settings where genetic testing is limited or inconclusive.
Abstract
Background: Inherited ichthyoses are a heterogeneous group of disorders of cornification caused by mutations in genes encoding epidermal proteins. Clinically, patients with ichthyosis present with erythema, scaling, and occasionally blistering; some subtypes are syndromic. Accurate and timely diagnosis is essential for appropriate management and genetic counseling. Objectives: Diagnosis of ichthyosis typically relies on a combination of clinical features, histopathological and ultrastructural findings, immunohistochemistry, and molecular genetic testing. Dermatopathology can be particularly valuable in three diagnostic scenarios: (i) when the clinical diagnosis of ichthyosis is evident, but the specific subtype remains unclear; (ii) when differential diagnoses such as inflammatory dermatoses need to be excluded; and (iii) when molecular testing is unavailable or yields variants of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
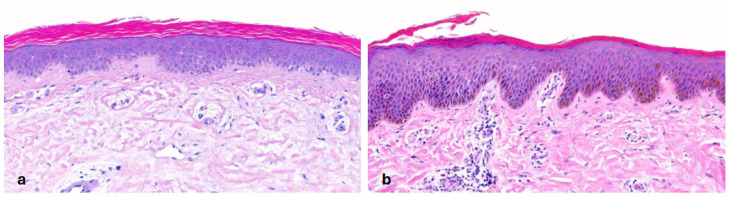 Figure 1
Figure 1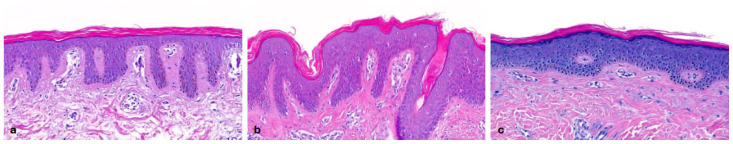 Figure 2
Figure 2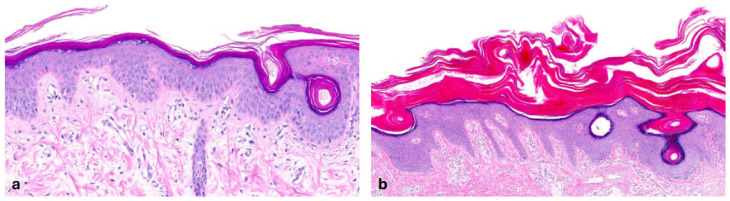 Figure 3
Figure 3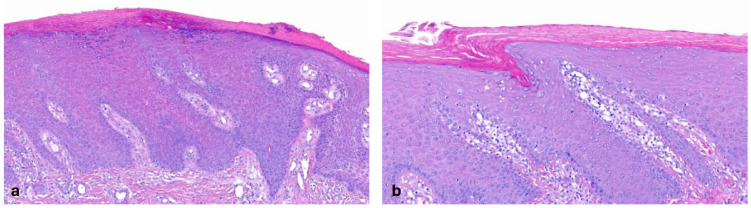 Figure 4
Figure 4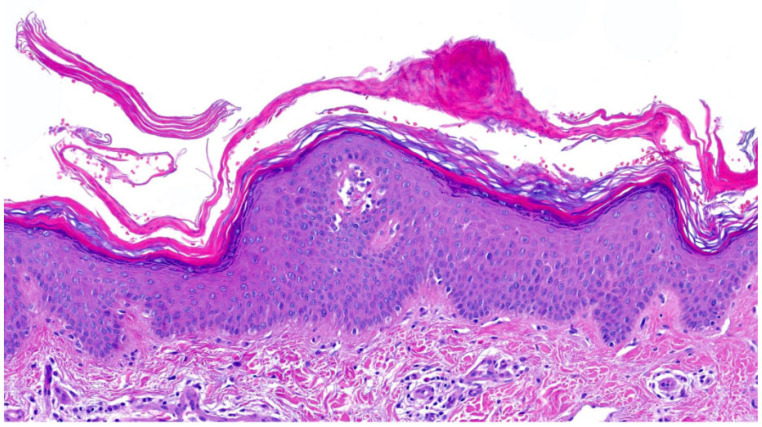 Figure 5
Figure 5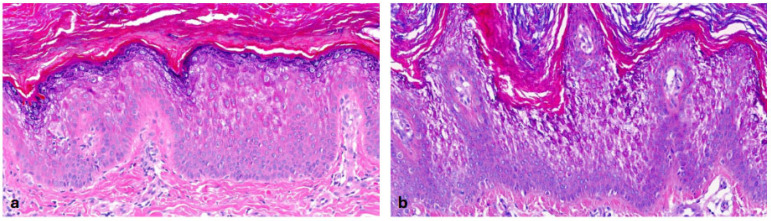 Figure 6
Figure 6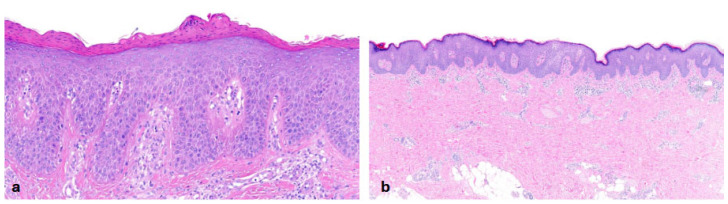 Figure 7
Figure 7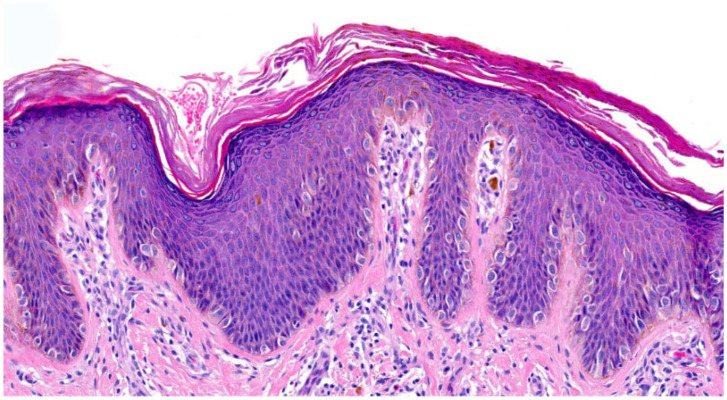 Figure 8
Figure 8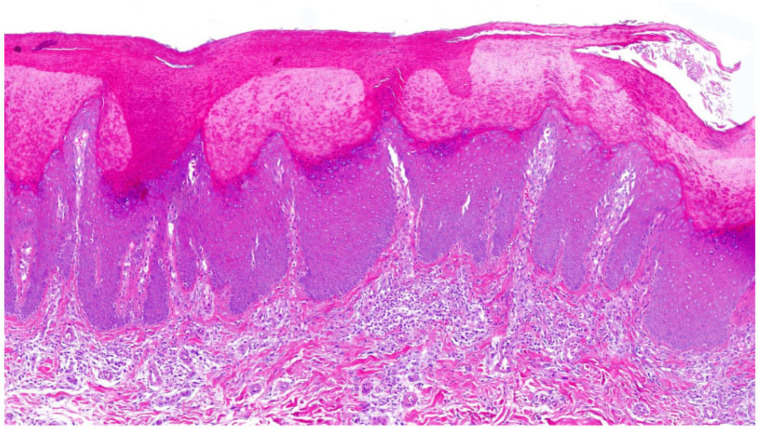 Figure 9
Figure 9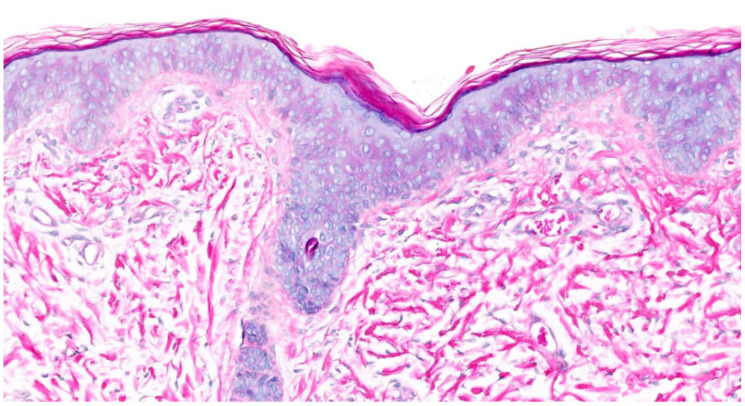 Figure 10
Figure 10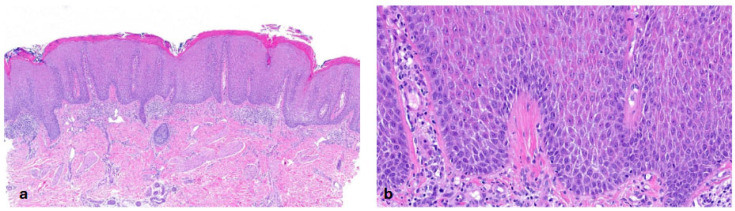 Figure 11
Figure 11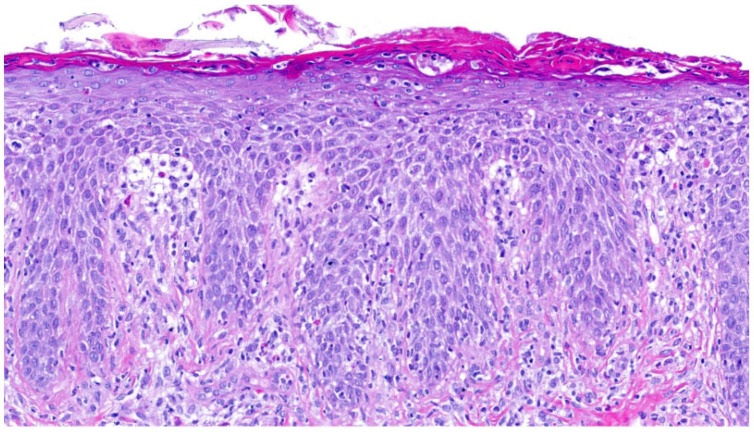 Figure 12
Figure 12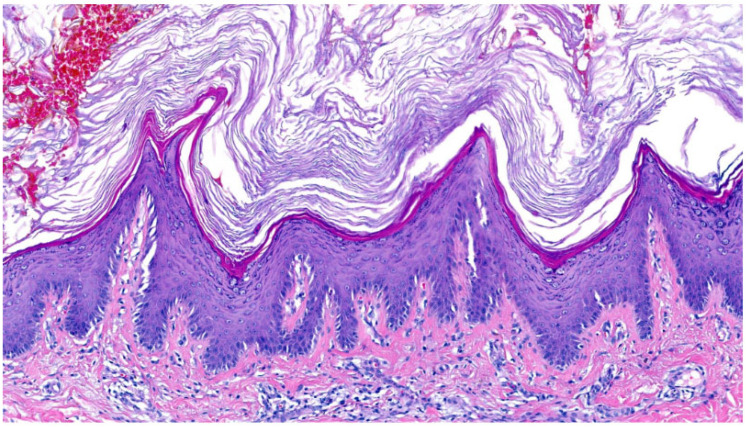 Figure 13
Figure 13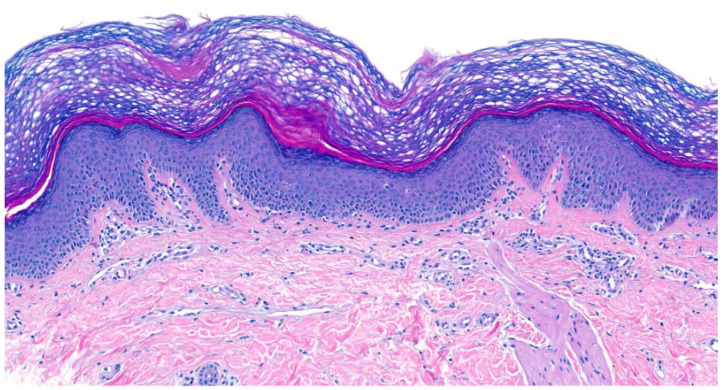 Figure 14
Figure 14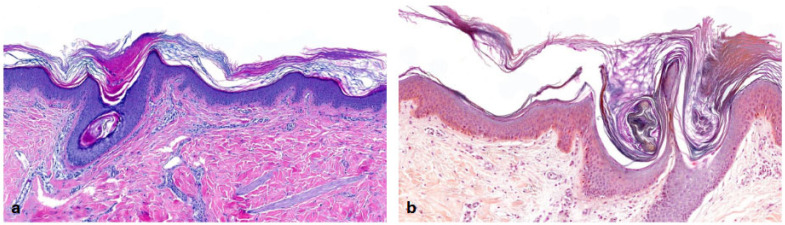 Figure 15
Figure 15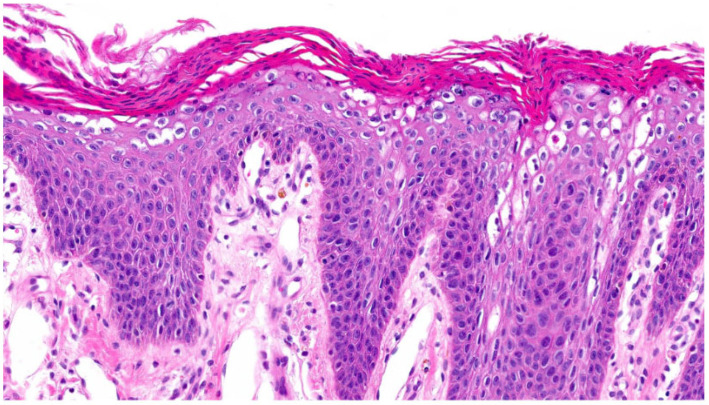 Figure 16
Figure 16| Patient ID | Sex | Age | Biopsy Site |
Diagnosis (Gene); (Mutation);
| Cutaneous and Extracutaneous Symptoms | Histology | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SC | SG | Acanthosis | Inflammation | FHK | AHK | Other Findings | Pattern(s) | ||||||
| 1 | f | 9 | upper limb | IV ( | mild ichthyosis, palmoplantar hyperlinearity, keratosis pilaris, dyshidrosis lamellosa sicca (fingertip), atopic dermatitis | +, OK, lamellar | −− | + | + | FHK | n.a. | I | |
| 2 | m | 11 | upper limb | IV ( | moderate ichthyosis, severe palmoplantar hyperlinearity | +, OK, lamellar | −− | + | focal + | FHK | AHK | I | |
| 3 | m | 71 | forehead | IV ( | ichthyosis, pruritus, comorbidities: basalioma and SCC, lichen planopilaris | +, OK, lamellar | − | − | focal + | FHK | AHK | biopsy captured areas not affected by lichen planopilaris | I |
| 4 | m | 1 month | lower limb | IV ( | mild-to-moderate ichthyosis | +, OK, compact/lamellar | −−/− | − | − | n.a. | n.a. | n.a. | I |
| 5 | f | 50 | lower limb | IV ( | ichthyosis, lymphadenopathy | +, OK, focal PK, compact/lamellar | −/+ | + | + | n.a. | n.a. | I | |
| 6 | m | 12 | 1: right hand; | IV ( | ichthyosis, mild PPK, keratosis pilaris, atopic dermatitis (lower extremities) | 1: +/++, OK, focal PK, lamellar; | 1; 2: −−/− | 1; 2: + | 1; 2: focal + (follicular) | 1; 2: FHK | 1: AHK | 2: focal spongiosis; serum crust | I |
| 7 | m | 60 | lower limb | IV ( | ichthyosis, mild PPK | ++, OK, com-pact/lamellar | −− | + | +/++ | n.a. | AHK | I | |
| 8 | f | 29 | upper limb | IV ( | moderate ichthyosis, atopic diathesis with recurrent eczema, hypohidrosis | +, OK, lamellar | −−/− | + | none | FHK | n.a. | BV: subepidermal dilatated | I |
| 9 | m | 7 | lower limb | XLI ( | mild-to-moderate ichthyosis, atopic dermatitis | +, OK, lamellar | − | + | none | n.a. | AHK | focal spongiosis | I |
| 10 | m | 17 | 1,2: lower limb | XLI ( | n.a. | 1,2: +, OK, lamellar | 1,2: +, focal ++ | 1: +/++, regular | 1,2: + | n.a. | AHK with SG+ | II | |
| 11 | m | 27 | upper limb | XLI ( | mild-to-moderate ichthyosis, no atopic dermatitis | +, OK, compact | + | + | none | n.a. | n.a. | II | |
| 12 | m | 25 | posterior trunk | XLI ( | plaque-like scaling, light brown to dark brown, popliteal fossae with mild scaling, hypohidrosis | +, OK, lamellar | − | +/++, regular | none | n.a. | n.a. | I | |
| 13 | m | 40 | posterior trunk | XLI ( | n.a. | ++, OK, compact | + | + | none | n.a. | AHK with SG+ | II | |
| 14 | m | 9 | posterior trunk | XLI ( | n.a. | +, OK, lamellar | − | + | + | n.a. | AHK with SG + | I | |
| 15 | m | 58 | upper limb | XLI ( | white fine-to-lamellar scaling, atopic diathesis; astigmatismus | +, OK, compact/lamellar | + | + | + | n.a. | n.a. | II | |
| 16 | m | 51 | lateral trunk | XLI ( | mild-to-moderate ichthyosis, keratosis pilaris upper and lower limbs, mild palmoplantar hyperlinearity | +, OK, lamellar | −−/− | + | + | n.a. | n.a. | I | |
| 17 | m | 46 | anterior trunk | XLI ( | severe ichthyosis, no atopy | +, OK, compact | −−/− | + | + | n.a. | n.a. | I | |
| 18 | m | 7 | upper limb | XLI ( | severe fine-to-mild lamellar ichthyosis, atopic diathesis, palmoplantar hyperlinearity | +/++, OK, com-pact/lamellar | −−/− | ++ | + | n.a. | n.a. | I | |
| 19 | m | 50 | trunk | ARCI ( | CIE, mild phenotype; palmoplantar hyperlinearity; comorbidities: SCC, lichen planopilaris | ++, OK, basket-weave-like/lamellar | −/+ | ++, regular | + | n.a. | AHK | II | |
| 20 | f | 3 days | upper limb | ACRI ( | mild generalized, white scaling | +/++, OK, basket-weave-like/compact/lamellar | +/++ | ++, regular | + | n.a. | n.a. | II | |
| 21 | f | 29 | forehead | ARCI ( | extreme hypohidrosis, fine lamellar scaling, kinking ear, many papillomatous nevi | +, OK, lamellar | + | +/++ | + | n.a. | n.a. | BV: dilatated, kissing vessels | II |
| 22 | f | 47 | upper limb | ARCI ( | lichenification (hyperkeratotic), fine-to-mild lamellar scaling, face included, mild PPK with mild hyperlinearity | +, OK, compact/lamellar | +/++ | ++, regular | + | n.a. | AHK | BV: dilatated | II |
| 23 | f | 25 | 1: posterior trunk; | ARCI ( | CIE, very high IgE levels, milk allergy | 1: +, OK/PK, lamellar; | 1: +/++; | 1: ++; regular, no neutrophils | 1: ++; | n.a. | n.a. | 1; 2: BV: dilated; | II |
| 24 | f | 3 | lower limb | ARCI ( | lamellar ichthyosis | +, OK, compact/lamellar | +/++ | ++, regular | none | n.a. | AHK | BV: dilatated | II |
| 25 | f | 13 | gluteal | ARCI ( | CIE, mild lamellar dark scaling | +, OK, lamellar | + | ++, regular | + | FHK | AHK | II | |
| 26 | f | 11 months | lower limb | ARCI ( | lamellar ichthyosis, focal yellowish PPK | ++/+++, OK, lamellar | ++ | ++, regular | +/++ | FHK | AHK | BV: dilated; | II |
| 27 | f | 30 | posterior trunk | ARCI ( | lamellar ichthyosis; comorbidity: actinic keratosis | +, OK, lamellar/compact | + | +/++, regular | + | FHK | n.a. | II | |
| 28 | f | 50 | shoulder | ARCI ( | CIE | ++, OK, focal PK, com-pact/lamellar | ++ | ++, regular | + | n.a. | n.a. | BV: dilated; focal spongiosis | II |
| 29 | m | 43 | upper limb | ARCI ( | lamellar ichthyosis, collodion baby, hypohidrosis | +/++, OK, lamellar/basket-weave-like | + | +/++, regular | + | n.a. | n.a. | BV: dilated | II |
| 30 | m | 13 | upper limb | ARCI | lamellar ichthyosis with gray and bright-brown scaling, PPK | ++, OK, compact > lamellar | + | ++, regular | + | n.a. | n.a. | II | |
| 31 | m | 56 | upper limb | ARCI | lamellar ichthyosis | ++, OK, compact/lamellar | + | +/++, regular | + | FHK | n.a. | BV: dilated | II |
| 32 | f | n.k. | n.k. | ARCI, bathing suit ichthyosis | extreme scaling of abdomen (cranial of umbilicus), yellowish PPK; growth retardation; | ++, OK, compact/lamellar | + | +/++, regular | + | FHK | AHK | BV: dilated | II |
| 33 | f | 12 | gluteal | ARCI | lamellar ichthyosis | +++, OK, focal PK, compact/lamellar | ++ | ++, regular | + | FHK | n.a. | BV: dilated, kissing vessels | II |
| 34 | m | 2 months | upper limb | ARCI ( | lamellar ichthyosis, focal erythema | +, OK, lamellar/compact | + | + | + | FHK | n.a. | II | |
| 35 | f | 44 | posterior trunk | ARCI ( | white mild lamellar scaling, mild erythroderma, tinea plantar | +, OK, lamellar/basket-weave-like | −/+ | +/++, regular | + | n.a. | n.a. | II | |
| 36 | m | 15 | upper limb | harlequin ichthyosis, overlap lamellar ichthyosis | severe erythroderma and scaling; clinical overlap of lamellar ichthyosis with harlequin ichthyosis | +, OK, focal PK, compact | + | ++/+++, regular | ++ | n.a. | normal | neutrophils in SC; BV: dilatated, kissing vessels | II |
| 37 | m | 20 | upper limb | harlequin ichthyosis | severe erythroderma and scaling | +, OK, focal PK, compact | −− | ++, regular | ++ | n.a. | n.a. | neutrophils in SC; BV: dilatated, kissing vessels, | I |
| 38 | f | n.k. | n.k. | ADLI ( | mild lamellar scaling, mild diffuse PPK | +/++; OK/PK, lamellar | + | +, irregular | + | n.a. | n.a. | III | |
| 39 | f | n.k. | n.k. | ADLI ( | mild lamellar scaling, mild diffuse PPK | +/++; OK/PK, lamellar | + | +, irregular | none | n.a. | n.a. | III | |
| 40 | m | 3 months | n.k. | EI (n.a.) | nevoid epidermolytic ichthyosis, neck and joints accentuated, face involved, no PPK; hypohidrosis | ++/+++, OK, | + | + | + | n.a. | n.a. | EHK continuous, suprabasal | IV |
| 41 | m | 39 | trunk | EI ( | generalized mild scaling, PPK; comorbidity: urticaria | −, OK, | + | + | ++/+++ | n.a. | n.a. | EHK continuous, suprabasal | IV |
| 42 | m | 15 | upper limb | EI ( | hyperkeratosis and erosions over joints, PPK | ++, OK, | + | ++, regular | + | n.a. | n.a. | EHK almost continuous, suprabasal, papillomatosis | IV |
| 43 | m | 68 | knee | EI ( | ichthyotic scaling lesions with blistering and erosions, yellowish PPK | +++, OK, compact/lamellar | + | ++, regular | + | n.a. | n.a. | EHK, continuous, suprabasal, suprapapillar epidermis thin; | IV |
| 44 | f | 16 | knee | EI ( | clinical similarities to SEI, brown hyperkeratosis and scaling | +++, OK, compact/lamellar | + | ++, regular | + | n.a. | n.a. | EHK non-continuous, suprabasal, papillomatosis | IV |
| 45 | f | 21 | neck | NTS ( | generalized ichthyosiform erythroderma, severe pruritus | +, OK, focal PK, lamellar | +/++ | ++, regular | ++ | n.a. | n.a. | BV: dilatated, kissing vessels, corneocytes detached (“flying birds”), suprapapillar epidermis thick | VI |
| 46 | f | 7 | gluteal | NTS ( | ichthyosiform erythroderma with aspects of ichthyosis linearis circumflexa; severe pruritus, vitamin d deficiency | 1: +, OK, focal PK, basket-weave-like; neutrophils and lymphocytes; | 1: −−/+; | 1: ++, regular; | 1,2: ++ with eosinophils in 2 | FHK | n.a. | BV: dilatated, kissing vessels, suprapapillar epidermis normal/thin, | VI |
| 47 | f | 4 months | n.k. | NTS ( | erythroderma, scaling, hypotrichosis, bacterial ( | 1: +, OK, focal PK; | 1: −−/+; | 1: ++, regular; | ++ | FHK | n.a. | 1; 2: BV: dilatated, kissing vessels, suprapapillar epidermis thin, spongiosis; | VI |
| 48 | m | 1 | gluteal | NTS ( | erythroderma with few scales | +, PK | −− | ++, regular | ++ | n.a. | n.a. | BV: dilatated, kissing vessels, | VI |
| 49 | m | 18 | upper limb | PSD ( | severe atopic dermatitis (EASI 41.5), diffuse peeling and inflammation of the skin (esp. on extremities), severe pruritus | ++, OK/PK, subcorneal clefting | −−/++ | +++, regular | ++ | n.a. | n.a. | suprapapillar epidermis thin, clumsy rete ridges | VI |
| 50 | m | 7 | trunk | PSD ( | mild phenotype, localized inflammation, multiple nevi, multiple allergies | 1–3, 8–14: +, OK, basket-weave-like and lamellar; | 1, 2, 4, 6, 9–14: ++, single acantholytic cells; | 1, 6, 9, 10, 12, 13: +; | 1; 2; 4–5; 9; 11–13: +; | n.a. | n.a. | 1: suprapapillar epidermis thin, BV: dilated, kissing vessels, subcorneal clefting; | VI |
| 51 | f | 9/13/16 | trunk | PSD ( | diffuse peeling and inflammation of the skin, severe pruritus, multiple nevi, verrucae vulgaris, vitamin D deficiency | 1: ++, PK, focal OK | 1: −−, | 1: +++, irregular | 1; 3: ++ | FHK | AHK | 1: suprapapillar epidermis thin, | VI |
| 52 | f | 51 | trunk. | mono-symptomatic CHILD (CHILD nevus) ( | right body side with erythema and scaling | +++, OK, focal PK, compact | +, focal missing | +++, irregular | +/++ | n.a. | n.a. | no neutrophils in SC, | VI |
| 53 | f | n.k. | n.k. | CHILD | unilateral inflammation and scaling, scoliosis | ++, OK, focal PK, compact | +, focal −− | ++, irregular | ++ | n.a. | n.a. | neutrophils in SC, BV: dilatated, kissing vessels, clumsy rete ridges, few xanthoma cells | VI |
| 54 | m | n.k. | n.k. | IFAP syndrome | hyperkeratosis on back, dry skin, no erythema, no collodion baby, | +, OK, lamellar/basket-weave-like | − | + | none | n.a. | n.a. | hair follicle rudimentary/atrophic, BV: dilatated, club-shaped rete ridges | I |
| 55 | f | 33 | upper limb | IFAP syndrome | n.a. | ++/+++, OK, basket-weave-like/compact | + | − | none | FHK | n.a. | I | |
| 56 | m | n.k. | n.k. | SAM syndrome ( | severe erythroderma, PPK, severe life-threatening infections, dilated cardiomyopathy | +, PK | −− | +++, regular | ++ | n.a. | n.a. | neutrophils in SC, | VI |
| 57 | f | 11 months | n.k. | MALT1 deficiency ( | dry and scaling skin, severe infections, brain atrophy, syndactyly | +, OK, focal PK | −− | ++, regular | +++ | − | − | neutrophils in SC, | VI |
| 58 | m | n.k. | n.k. | KID | massive hyperkeratosis, multiple SCC | +++, OK, lamellar | ++ | + | ++ | n.a. | n.a. | BV: dilatated, kissing vessels, | II |
| 59 | f | 39 | posterior trunk | KID | multiple hyperkeratoses | +++, OK, lamellar | −−/− | + | + | FHK | AHK | long and thin rete ridges, suprapapillar thinning | I |
| 60 | f | n.k. | n.k. | KID | n.a. | +++, OK, lamellar/basket-weave-like | −/+ | + | ++ | FHK | n.a. | BV: dilatated, kissing vessels, papillomatosis, hyperplastic sweat glands | II |
| 61 | f | 21 | trunk | KID ( | multiple follicular hyperkeratoses | +++, OK, lamellar | −/+ | + | + | FHK | AHK | II | |
| 62 | f | 5 | gluteal | SLS | fine scaling on trunk and extremities, cobblestone scaling in neck area, severe pruritus | ++, OK, compact/basket-weave-like | ++ | + | + | FHK | n.a. | BV: normal | II |
| 63 | f | 13 | posterior trunk | SLS | generalized lichenification and fine lamellar scaling | +, OK, basket-weave | − | + | + | n.a. | n.a. | I | |
| 64 | m | 22 | upper limb | Chanarin–Dorfman syndrome | generalized fine-to-midlamellar scaling | +, OK, focal PK, compact/lamellar | + | + | + | FHK | n.a. | discrete vacuolization of basal keratinozytes | II |
| 65 | f | 14 | lower limb | Conradi–Hünermann–Happle syndrome/X-linked dominant chondrodysplasia punctata ( | brown lamellar scaling, follicular hyperkeratosis, scarring alopecia | 1 (2 years old): +, OK; | 1: −−/−; | 1: +; | 1: +; | 1: FHK; | 1: AHK; | 1: club-shaped and broadened rete ridges, | I |
| 66 | m | 3 months | trunk | KIDAR syndrome (mutation found in brother: | n.a. | +, OK, compact/basket-weave-like | ++ | + | + | FHK | AHK | BV: dilatated, kissing vessels, | II |
| Pattern | Clinical Feature | Diagnosis |
|---|---|---|
| Pattern I | Palmoplantar hyperlinearity, atopy | Ichthyosis vulgaris |
| Male patient with gray-to-brown coarse lamellar scaling, flexures spared, improvement in summer, no palmoplantar hyperlinearity; maldescensus testis, hyperactivity, autism, protracted birth; with/without atopy | X-linked ichthyosis | |
| Late onset of ichthyosis, comorbidities | Acquired ichthyosis | |
| Conradi–Hünermann–Happle syndrome | ||
| Erythroderma, ectropium, eclabium | Harlequin ichthyosis | |
| Ichthyosis follicularis with alopecia (atrichia) and photophobia (IFAP) syndrome | ||
| Brownish hyperkeratosis | Keratitis ichthyosis deafness (KID) syndrome | |
| Cobblestone hyperkeratosis, severe pruritus | Sjögren–Larsson Syndrome | |
| Pattern II | Fine scaling, mild erythroderma | Autosomal recessive congenital ichthyosis (erythrodermic) |
| Coarse dark and brown scaling | Autosomal recessive congenital ichthyosis (non-erythrodermic) | |
| Coarse dark and brown scaling in warmer areas of the body | Bathing suit ichthyosis | |
| Erythroderma, ectropium, eclabium | Harlequin ichthyosis | |
| Male patient with gray-to-brown coarse lamellar scaling, flexures spared, improvement in summer, no palmoplantar hyperlinearity; maldescensus testis, hyperactivity, autism, protracted birth; with/without atopy | X-linked ichthyosis | |
| Cobblestone hyperkeratosis, severe pruritus | Sjögren–Larsson syndrome | |
| Ichthyosis follicularis with alopecia (atrichia) and photophobia (IFAP) syndrome | ||
| Brownish hyperkeratosis | Keratitis ichthyosis deafness (KID) syndrome | |
| Chanarin–Dorfman syndrome | ||
| Autosomal recessive keratitis–ichthyosis–deafness (KIDAR) syndrome | ||
| Pattern III | Scaly erythema, palmoplantar keratoderma, no systemic manifestation | Erythrokeratoderma variabilis (EKV) |
| Keratitis ichthyosis deafness (KID) syndrome | ||
| Dark, coarse scales, lichenification of the dorsum of the hands/feet, knees and plantar hyperkeratosis | Autosomal dominant lamellar ichthyosis | |
| Pattern IV | With palmoplantar keratoderma | Epidermolytic ichthyosis with |
| Without palmoplantar keratoderma | Epidermolytic ichthyosis with | |
| Superficial epidermolytic ichthyosis | ||
| Annular epidermolytic ichthyosis | ||
| Pattern V | Ichthyosis with confetti-like normal skin | Congenital reticular ichthyosiform erythroderma (CRIE) |
| Spiny hyperkeratosis | Ichthyosis hystrix of Curth–Macklin | |
| Pattern VI | Ichthyosiform erythroderma, trichorrhexis invaginata, predisposition to infections, food allergies, hypereosinophilia, elevated IgE, failure to thrive | Netherton syndrome |
| Erythema, peeling of the skin, allergies, pruritus, hypereosinophilia, elevated IgE, normal body size and weight | Peeling skin disease | |
| Congenital hemidyplasia with ichthyosiform nevus and limb defects (CHILD) syndrome | ||
| Ichthyosiform erythroderma, alopecia, allergic symptoms, hypereosinophilia, elevated IgE, failure to thrive | Severe dermatitis, multiple allergies, metabolic wasting (SAM) syndrome | |
| Predisposition to infections, failure to thrive, periodontal disease, enteropathy | Malt 1 deficiency |
| Follicular Hyperkeratosis in Ichthyoses |
|---|
| Ichthyosis vulgaris with follicular keratosis |
| AR-congenital ichthyosis |
| Sjögren–Larsson syndrome |
| Ichthyosis follicularis with alopecia and photophobia |
| Hystrix-like ichthyosis with deafness (HID) and keratitis ichthyosis deafness (KID) syndromes |
| Other ichthyoses (see |
|
|
| Pachyonychia congenita |
- —“Innovative Medical Research” fund of the University of Münster Medical School
- —German Dermatological Society (“Deutsche Dermatologische Gesellschaft (DDG)”)
- —Arbeitsgemeinschaft Dermatologische Forschung (ADF)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Dermatological and Skeletal Disorders · Genetic and rare skin diseases.
1. Introduction
Ichthyoses are a heterogeneous group of inherited cornification disorders (Mendelian Disorders of Cornification, MeDOCs; new nomenclature: non-syndromic epidermal differentiation disorders (nEDDs) and syndromic epidermal differentiation disorders (sEDDs)) caused by gene variants affecting epidermal barrier function [1,2]. Patients with ichthyosis present with scaling and dry skin but may also have erythroderma and blistering. Some types of ichthyoses have syndromic manifestations (e.g., immunodeficiency, deafness, skeletal anomalies, neurological symptoms) and some are potentially life-threatening. The prevalence ranges from 1:200 to less than 1:1,000,000 [3,4,5,6].
Nowadays, the diagnosis of ichthyoses is based on clinical and histological findings and can be supported by electron microscopy and immunohistochemistry. For most dermatologists, clinical assignment to a subtype is very difficult or even impossible. However, the most reliable way of confirming ichthyosis is through molecular genetic analysis.
Histology is a fast, cost-effective and well-established method of dermatodiagnosis (although patients and physicians are often reluctant to perform a (minimally) invasive biopsy). In clinical practice, there are three scenarios in the diagnosis of ichthyosis where histology may be involved: (i) ichthyosis can be diagnosed with clinical certainty, but the subtype remains obscure; (ii) it is not clear whether the patient suffers from ichthyosis or an inflammatory dermatosis—a typical situation, for instance, in erythroderma in infants; (iii) molecular genetic diagnostics are not possible or detect variants with unclear significance.
To date, only a limited number of characteristic histological and ultrastructural findings in cornification disorders have been well documented in the literature [7,8,9,10,11]. Epidermolytic hyperkeratosis is the only well-defined pattern and is associated with keratin defects due to pathogenic variants in KRT1, KRT2 and KRT10. Ross et al. characterized the histological features of keratinopathic ichthyosis and nevi in terms of epidermal changes and correlated these findings with the phenotype. Histological distinction between KRT2 and KRT1 or KRT10 mutations was not always possible [7].
As previously shown, dermatopathology can be useful in the differential diagnosis of neonatal and infantile erythroderma [12,13]. In a cohort of 51 patients with erythodermic ichthyosis, histological analysis was useful in half of the patients, excluding those with Netherton syndrome [12].
To date, there is no valid definition of patterns in ichthyosiform skin disorders that allows for an algorithmic analysis of inflammatory skin diseases as described by Ackerman et al. [14]. Systematic analyses of larger cohorts are rare, and many publications are monographs, case reports or publications without genetic confirmation of these rare diagnoses [8,9,10,11].
Working in the Reference Center for Ichthyoses and Palmoplantar Keratoderma (ReCIP) in Münster collaborating with the Center for Cornification Disorders in Freiburg, we see many patients with hereditary cornification disorders in our consultations and carry out all the necessary analyses to obtain a diagnosis. With years of clinical experience, molecular genetic analysis, modern immunohistological staining methods and enzyme activity measurements, we could establish exact diagnoses in the majority of our patients, especially in recent years.
We systematically analyzed 87 biopsies from 66 patients with inherited ichthyosis. We then correlated the histological findings with clinical and genetic data, and we were able to define six histological patterns. On this basis, we developed a diagnostic algorithm that enables the histological classification of patients into specific ichthyosis subgroups and helps narrow down differential diagnoses.
2. Materials and Methods
2.1. Materials
In total, we identified 87 biopsies from 66 patients in whom the diagnosis was confirmed by clear clinical features and various diagnostic techniques. Genetic analysis was performed in 89.4% of patients.
It is noteworthy that the clinical evaluation, particularly the systematic screening for phenotypic findings, was performed by dermatologists from our centers with many years of experience in ichthyosis and keratinization disorders. All patients were phenotyped using a standardized questionnaire. As a member of the European Reference Network (ERN-Skin, Ichthyosis and Palmoplantar Keratoderma (IPK) subgroup), we have been involved in drafting the current version of a standardized questionnaire for ichthyosis and palmoplantar keratoderma. In addition, all patients have been discussed within our regular case conferences across specialized centers. Regarding clinical manifestations, we examined the presence and severity of ichthyosis (mild to moderate), palmoplantar hyperlinearity, palmoplantar keratoderma, erythroderma, presence of a collodion membrane at birth, (skin) malignancies, allergies, atopic dermatitis/diathesis and other dermatoses. In cases without records of disease severity, we collected data on the type of scaling. This study adhered to the Declaration of Helsinki and was approved by the local ethics committees (XTrau1, AN 2016-0260, 368/4.22, 454/AM1).
2.2. Histological Analysis
Histological analyses were performed by one of the authors, who is an experienced dermatopathologist (DM). For histology, 4 mm punch biopsies were taken, fixed in 4% formaldehyde, embedded in paraffin, sectioned and stained with hematoxylin and eosin [9,10].
The slides were evaluated semi-quantitatively using a scoring system (regarding the Stratum granulosum—absent: −−; reduced: −; normal: +; prominent: ++—and inflammation—no inflammation: −; mild: +; moderate: ++; severe: +++). In addition, the mean thickness of the stratum corneum (SC) hyperkeratosis and the vital epidermis was measured quantitatively (no hyperkeratosis: −; mild hyperkeratosis ≤ 0.1 mm: +; moderate hyperkeratosis > 0.1–0.2 mm: ++; severe hyperkeratosis > 0.2 mm: ++; no acanthosis ≤ 0.1 mm: −; acanthosis > 0.1–0.2 mm: +; acanthosis > 0.2–0.3 mm: ++; acanthosis > 0.3 mm: +++).
Psoriasiform hyperplasia was defined as regular hyperplasia with or without suprapapillary thinning. The presence of suprapapillary thinning was included under “other findings” in Table 1. For hyperplasia less than 0.1 mm, we did not differentiate between irregular and regular hyperplasia, because the assessment of suprapapillary epidermis seemed ambiguous. We performed a thorough histological evaluation of follicular and acrosyringeal hyperkeratosis. We also examined all biopsies for dilated capillaries in the papillary body and analyzed whether they were in close contact with the basal membrane (“kissing phenomenon”). Abnormal blood vessel findings were listed under other findings.
2.3. Genetic Analysis
In 59 patients, the clinical diagnosis was confirmed by genetic analysis. In all patients, genomic DNA was isolated from peripheral blood lymphocytes, followed by either Sanger sequencing or NGS methods (e.g., gene panel sequencing or whole-exome sequencing) [15].
Further diagnostics were performed via immunohistochemistry (n = 3) and in situ monitoring of transglutaminase 1 activity (n = 4) (Table 1).
Statistical analysis was carried out with IBM SPSS Statistics Version 28.1.1 (2021, IBM).
2.4. Cohort Demographics and Diagnostic Spectrum
We studied a total of 87 biopsies in 66 patients altogether, including 32 female and 34 male patients with the following diagnoses: ichthyosis vulgaris (new nomenclature for non-syndromic epidermal differentiation disorders (nEDDs) and syndromic epidermal differentiation disorders (sEDDs)) (FLG-nEDD) (n = 8), X-linked ichthyosis (STS-sEDD) (n = 10), autosomal recessive congenital ichthyosis (TGM1-nEDD; SDR9C7-nEDD; ALOXE3-nEDD; ALOX12B-nEDD; NIPAL4-nEDD; ABCA12-nEDD) (n = 19), autosomal dominant lamellar ichthyosis (ASPRV1-nEDD) (n = 2), epidermolytic ichthyosis (KRT1-nEDD; KRT2-nEDD; KRT10-nEDD) (n = 5), Netherton syndrome (SPINK5-sEDD) (n = 4), peeling skin disease (CDSN-nEDD) (n = 3), and rare (syndromic) subtypes (n = 15). Rare syndromic subtypes included keratitis–ichthyosis–deafness (KID) syndrome (GJB2-sEDD-KID); congenital hemidysplasia with ichthyosiform nevus and limb defects (CHILD) syndrome (NSDHL-sEDD-CHILD); ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome (MBTPS2-sEDD-IFAP); severe dermatitis, multiple allergies and metabolic wasting (SAM) syndrome (DSG1-sEDD); MALT1 deficiency; autosomal recessive keratitis–ichthyosis–deafness (KIDAR) syndrome (AP1B1-sEDD-KID); and neutral lipid storage disease with ichthyosis and Sjögren–Larsson syndrome (ALDH3A2-sEDD). The median age at biopsy was 17 years (minimum: 3 days; maximum: 68 years). We had information on the biopsy site in 55 cases. All biopsies were taken from affected skin, with the majority from the extremities (Table 1).
Detailed information regarding whether biopsies from the extremities were obtained from ventral or dorsal sites was not available. However, when biopsies were taken from anatomical locations with characteristically thicker skin, such as over joints, knees, elbows or from the palms or soles, this information was documented and explicitly reported.
3. Results
3.1. Histological Analysis of Different Subtypes of Ichthyosis
3.1.1. Common Ichthyoses: Ichthyosis Vulgaris (IV) and X-Linked Ichthyosis (XLI)
In IV, due to filaggrin (FLG) mutations, all histologies showed orthohyperkeratosis with a reduced or absent stratum granulosum (SG). SC was lamellar or compact. Mild acanthosis was observed in all patients except for two patients with only one FLG mutation. Notably, mild inflammation was common (75%), but only one patient had clinical erythema. A history of atopic dermatitis (AD) was reported in three patients, one of whom had histologically confirmed spongiosis and serum crust. Interestingly, prominent hyperkeratosis and absent SG were not significantly more common in patients with biallelic FLG mutations compared to those with one mutation (p = 0.464; p = 0.464). Follicular or acrosyringial hyperkeratosis was observed in six patients (Figure 1).
In the steroid sulfatase deficiency group causing XLI, 6 out of 10 patients showed orthohyperkeratosis with a reduced or absent SG; the rest had orthohyperkeratosis and a well-developed SG. Of note, three patients had additional filaggrin mutations with and without reduced SG. The differences in reduced or normal SG between the groups with and without additional FLG mutations, respectively, were not significant (p = 0.52). The SC was compact or lamellar. All patients showed at least mild acanthosis, and two patients showed moderate regular hyperplasia. Inflammation was present in six patients. Acrosyringeal hyperkeratosis was observed in four patients (three without additional FLG mutation), with the formation of an SG in the acrosyringium in three patients (Figure 2). Hair follicles could not be assessed.
3.1.2. Autosomal Recessive Congenital Ichthyosis (ARCI), Including Harlequin Ichthyosis
In the heterogeneous subset of ARCI, we recruited 19 patients. Three of them had ALOXE3 mutations, two had ALOX12B mutations, three had SDR9C7 mutations and four had NIPAL4 mutations in genetic testing [15]. We also included five patients with TGM1 mutations and two patients with harlequin ichthyosis due to ABCA12 mutations. Almost all patients showed orthohyperkeratosis and a well-developed stratum granulosum (Figure 3). Mild-to-moderate hyperkeratosis presented as lamellar to compact. One patient with transglutaminase 1 (TG1) deficiency showed severe orthohyperkeratosis and one patient with a NIPAL4 mutation showed moderate-to-severe orthohyperkeratosis. Of note, one patient with an SDR9C7 mutation had a reduced-to-normal SG, possibly due to an additional FLG mutation detected during routine genetic analysis. Mild-to-moderate epidermal acanthosis was observed in the majority of patients. Mild inflammation was common (n = 14), and moderate inflammation was rare (n = 2; ALOX12B, NIPAL4). The remaining patients had biopsies with different patterns of inflammation (mild to moderate, or one biopsy with moderate and one with mild-to-moderate inflammation). Follicular hyperkeratosis (FHK) and acrosyringial hyperkeratosis (AHK) were described in all patients, whenever hair and sweat glands could be assessed. In contrast to the other ARCI patients, the two patients with harlequin ichthyosis showed compact orthohyperkeratosis with focal parakeratosis, moderate-to-severe acanthosis, moderate inflammation and a stratum corneum with neutrophils. The SG was absent in the patient with isolated harlequin ichthyosis. In one patient with clinical overlap with lamellar ichthyosis (ID 36), the SG was regular (Figure 4).
3.1.3. Autosomal Dominant Lamellar Ichthyosis
Autosomal dominant lamellar ichthyosis was first described in 1984 by Traupe et al. [16] and genetically confirmed in 2020 by Boyden et al. [17], who reported a gene defect involved in filaggrin degradation. Both patients included in our study with a heterozygous mutation in the ASPRV1 gene showed lamellar hyperkeratosis with ortho- and parakeratosis with a preserved stratum granulosum. The epidermis showed irregular acanthosis and one patient had mild inflammation (Figure 5). Clinically, these patients also presented with a diffuse PPK. Although the pattern of inheritance is different from ARCI, clinical differentiation between these two subtypes can be challenging.
3.1.4. Keratinopathic Ichthyosis
In the group of keratinopathic ichthyoses, we included patients with mutations in keratin (KRT)1 (n = 2) and KRT10 (n = 2). One patient did not have a genetic report but had clear clinical and histological features of keratinopathic ichthyosis. All patients had orthohyperkeratosis. Acanthosis was mild to moderate and regular. All patients exhibited a pattern of suprabasal epidermolytic hyperkeratosis characterized by irregular keratohyalin granules and vacuolated keratinocytes with hypereosinophilic granules (granular degeneration) (Figure 6); it was non-discontinuous in one patient and continuous in all remaining cases. Hair follicles and acrosyringia were not assessable. Moderate-to-severe inflammation was observed only in one patient with a KRT1 mutation who had a comorbidity of urticaria. The skin biopsy was taken from a non-urticaria area. Inflammation was mild in the other patients. One patient with a KRT10 mutation presented PPK, which is typically seen in patients with a KRT1 mutation.
3.1.5. Netherton Syndrome (NTS)
Netherton patients carry biallelic mutations in SPINK5. All patients (n = 4) with NTS showed a psoriasis-like pattern with orthohyperkeratosis, focal parakeratosis and regular moderate hyperplasia of the epidermis (Figure 7) [2]. In one case, neutrophils were found in the SC. One patient had a normal-to-prominent SG. All other patients had at least a partial absence of SG. Spongiosis was present in two cases. Inflammation was moderate in all patients, with one biopsy showing eosinophils. Blood vessels were dilated, showing the kissing phenomenon (n = 4) (vessels close to basal keratinozytes), and the suprapapillary epidermis was thin (n = 3). Follicular hyperkeratosis was present in two patients. The acrosyringium was not assessable.
3.1.6. Peeling Skin Disease
Peeling skin disease caused by biallelic variants in corneodesmosin shows some clinical overlaps with Netherton syndrome [18]. The patients in our cohort had a psoriasis-like pattern similar to Netherton syndrome. Of note, we had 18 biopsies from three patients. Two patients had multiple dark pigmented nevi, which was the reason for multiple biopsies in order to exclude malignancy. The extent of acanthosis varied compared to NTS. We had one biopsy with no acanthosis, six biopsies with mild acanthosis, nine biopsies with moderate acanthosis and two biopsies with severe acanthosis, including eight biopsies with regular hyperplasia and three biopsies with irregular hyperplasia. Subcorneal clefts or acantholytic cells due to corneodesmosin deficiency were prominent in peeling skin disease but were not found in all patients (Figure 8).
3.1.7. Other Syndromic Subtypes
Congenital hemidysplasia with ichthyosiform nevus and limb defects (CHILD) syndrome is due to nonsense or missense mutations in the NSDHL gene. It is inherited in an X-chromosomal dominant pattern and is therefore usually lethal to males in utero [2]. In our cohort, we found a psoriasis-like pattern in our two patients with moderate-to-severe orthohyperkeratosis, focal parakeratosis with neutrophils (n = 1) and a compact SC. The SG was normal or partially absent. Epidermal hyperplasia was moderate and irregular (Figure 9). Blood vessels were dilated with kissing vessels in one case. Of note, xanthoma cells were present in the papillary dermis in only one case.
Ichthyosis follicularis, alopecia and photophobia (IFAP) is an X-linked recessive disorder caused by mutations in the MBTPS2 gene involved in the cholesterol pathway [19]. In our two patients, orthohyperkeratosis was lamellar with a basket weave pattern. The SG was normal in one case and absent in the other patient. Inflammation was absent. The patients had follicular hyperkeratosis (Figure 10). In one patient, the hair follicles were rudimentary or atrophic.
Severe dermatitis–multiple allergies–metabolic wasting (SAM) syndrome is caused by mutations in desmoglein 1 (DSG1) or desmoplakin (DSP). Patients with DSP mutations may have cardiac involvement [20]. Histology shows hyperkeratosis, focal parakeratosis with neutrophils, absent SG, severe regular hyperplasia of the epidermis and typical dehiscence of keratinocytes accompanied by moderate inflammation (Figure 11a,b).
The patient with Mucosa-Associated Lymphoid Tissue Lymphoma Translocation Protein 1 (MALT1) deficiency showed orthohyperkeratosis with focal parakeratosis, few neutrophils, absence of SG and regular epidermal hyperplasia. There were few dyskeratotic keratinocytes and moderate spongiosis. Inflammation was severe with many lymphocytes, erythrocytes and neutrophils, including inflammation of the hair follicles. Blood vessels were dilated without showing a kissing phenomenon (Figure 12).
In keratitis–ichthyosis–deafness (KID) syndrome caused by connexin 26 (GJB2) mutations, all patients showed massive orthohyperkeratosis, mostly lamellar [2]. SG thickness varied from absent to prominent. Other findings included mild acanthosis, papillomatosis and long and thin rete ridges. Inflammation was mild to moderate. Follicular hyperkeratosis and acrosyringial hyperkeratosis were common. Blood vessels were dilated in two cases. Hyperplastic sweat glands were observed in one patient (Figure 13).
In Sjögren–Larsson syndrome, a neurocutaneous disease due to fatty aldehyde dehydrogenase deficiency (FALDH), hyperkeratosis with orthokeratosis was observed [2,21]. The SG was prominent (in one case thinned), and we saw mild acanthosis, papillomatosis (in one patient steeple-shaped appearance) and inflammation (Figure 14). Follicular hyperkeratosis was present.
Conradi–Hünermann–Happle syndrome is caused by mutations in the emopamil-binding protein (EBP) gene [2,22]. In our patient, we observed orthohyperkeratosis with a reduced or absent SG. Acanthosis was mild to moderate (Figure 15a). Inflammation was mild, and FHK and AHK were present in one of two biopsies. Of note, calcification of the horny layer of the epidermis and the hair follicles can be demonstrated by von Kossa staining (Figure 15b) [22].
Our patient with neutral lipid storage disease with ichthyosis (Chanarin–Dorfman syndrome) showed lamellar-to-compact orthohyperkeratosis with focal parakeratosis and a normal SG [2]. Acanthosis and inflammation were mild. Basal keratinozytes were slightly vacuolated.
A patient with suspected keratitis–ichthyosis–deafness (KIDAR) syndrome, characterized by deafness, ichthyosis and erythroderma, showed mild orthohyperkeratosis, a prominent SG, mild acanthosis and mild inflammation [23]. Follicular hyperkeratosis and acrosyringial hyperkeratosis were present.
4. Discussion
4.1. Definition of Six Histological Patterns Including an Algorithm for the Diagnosis of Ichthyosis
Ichthyoses are a very heterogeneous group of cornification disorders with high variability in clinical presentation. There are over 36 entities [4]. Our histological study of 66 patients with well-defined ichthyoses identified six histological patterns that may help to narrow the differential diagnosis (Table 1).
The first pattern is defined by “orthohyperkeratosis with a reduced or absent stratum granulosum”, which occurred in IV, XLI, Conradi–Hünermann–Happle syndrome, Sjögren–Larsson syndrome, IFAP syndrome, harlequin ichthyosis, KID syndrome and acquired ichthyosis-like condition (Figure 1, Figure 2, Figure 4, Figure 10 and Figure 15). The latter was not part of our cohort. Acquired ichthyosis-like conditions typically manifest in adulthood and may represent paraneoplastic manifestations or be due to, for example, extreme diets and drug interactions. Some cases are associated with an underlying disease such as venous insufficiency of the leg and mycosis fungoides [24].
Interestingly, the stratum granulosum findings in our sample did not correlate with the number of FLG mutations. Therefore, additional immunohistochemistry may be useful in the histology of ichthyosis vulgaris. Keratosis follicularis as a clinical finding may be present in histology and, like palmoplantar hyperlinearity, may be a clinical diagnostic clue. In addition, FLG deficiency in IV can be associated with moderate alterations in epidermal barrier function [25]. Therefore, spongiosis and acanthosis may be present. As follicular hyperkeratosis was common in our overall cohort, clinical features and supportive methods may help to further differentiate between ichthyosis subtypes with follicular hyperkeratosis belonging to the same pattern (Table 3). Notably, inflammatory cells were frequently observed in these non-inflammatory subtypes. Regarding the differentiation between XLI and IV, a reduced SG does not necessarily seem to be a diagnostic clue for IV. Even in some cases with a reduced SG in XLI, we could not find an additional FLG mutation implying modifying genetic factors. As an acquired ichthyosis-like condition may also present with massive orthohyperkeratosis with absent or thin stratum granulosum, it cannot be distinguished histologically from inherited forms of ichthyoses (personal observation, DM).
The second pattern is classified by “orthohyperkeratosis and a well-developed stratum granulosum”. Our cohort represented a very heterogeneous group of ARCIs, including bathing suit ichthyosis, harlequin ichthyosis, XLI, Sjögren–Larsson syndrome, IFAP, KID, Chanarin–Dorfman syndrome and KIDAR syndrome (Figure 2, Figure 3, Figure 4, Figure 13 and Figure 14). Of note, inflammatory conditions such as lichen simplex chronicus share similarities in the epidermal pattern but differ in the fibrosis of the papillary dermis. Clinical and histological differentiation between XLI and ARCI can be difficult. Clinical features, associated symptoms and electron microscopy may be helpful (Table 2). XLI shows a retention hyperkeratosis which is characterized by the persistence of corneodesmosomes in electron microscopy. Of note, ARCI often showed dilated vessels and the kissing phenomenon even in patients without erythroderma. This correlation should be investigated separately in a larger cohort of ARCI.
The third pattern, “hyperkeratosis with ortho- and parakeratosis with preserved or prominent stratum granulosum”, was seen in KID syndrome (personal observation, DM) and autosomal dominant lamellar ichthyosis (Figure 5). However, inflammatory skin diseases in general may show a similar pattern, e.g., lichen simplex chronicus and pityriasis rubra pilaris.
KID syndrome shares some features with erythrokeratodermia (EKV): pseudoepitheliomatous, verruciform or psoriasiform hyperplasia with dyskeratosis and rarely perinuclear vacuoles (“bird’s eye”) [9,26]. The orthohyperkeratotic horny layers show focal parakeratosis with focal small roundish nuclear remnants (“shadow nuclei”). Keratotic plugging of hair follicle openings and atrophic sweat ducts are typical. In areas of alopecia, hair follicles are absent or atrophic. Eccrine sweat glands may be reduced in number and/or be atrophic. In general, connexin mutations result in a broad spectrum of histological changes, making a specific diagnosis difficult. Immunohistochemical staining for the expression pattern of connexins may be helpful [26].
The fourth pattern is characterized by “epidermolytic hyperkeratosis” (EHK), which occurs in KPI due to mutations in KRT1, KRT10 or KRT2, as well as in epidermal nevi, acanthomas, leukoplakia or epidermolytic palmoplantar keratoses [27]. In our cohort, three out of five patients with KPI had non-continuous and suprabasal EHK (Figure 6). Two patients had non-continuous or almost continuous EHK. We could not deduce a specific mutation status from the histological findings, as in superficial epidermolytic ichthyosis, a mutation in KRT2 may also lead to signs of EHK in the lower parts of the epidermis (personal observation, DM). The patient with clinical findings of superficial epidermolytic ichthyosis carried a KRT10 mutation. The EHK pattern can also be seen in epidermal nevi, epidermolytic acanthoma, and incidentally in actinic damaged skin, actinic keratosis, invasive squamous cell carcinoma, basal cell carcinoma, epidermal and pilar cysts and various inflammatory diseases [28,29].
Notably, one patient with EI (ID 41) did not show EHK. As this patient had only a mild-to-moderate phenotype, we can speculate that the biopsy was taken from a mildly affected body site.
Annular epidermolytic ichthyosis (KRT10-nEDD-annular) is caused by a dinucleotide mutation in KRT10 and presents with polycyclic, hyperkeratotic plaques predominantly on the trunk and the proximal extremities [28]. With its histology showing epidermolytic hyperkeratosis, it belongs to the group of keratinopathic ichthyoses [28,29]. There are also annular forms with KRT1 mutations [1].
The fifth pattern with “perinuclear vacuoles and binucleated keratinocytes” is seen in congenital reticular ichthyosiform erythroderma (CRIE) and ichthyosis hystrix of Curth–Macklin (KRT1-nEDD-spiny) [29,30]. KRT10 mutations in CRIE result in almost complete loss of keratin filaments in the perinuclear cytoplasm and vacuolization of suprabasal keratinocytes, some of which are binucleated without eosinophilic intracytoplasmic inclusions. The SG is almost absent, and parakeratotic corneocytes manifest with large nuclei representing transitional cells (Figure 16, unpublished case). Patients with CRIE show spots of normal skin that increase in size over time and appear to be surrounded by erythrokeratotic and hyperpigmented areas in a reticular pattern (“confetti”) during childhood. Notably, the spots of normal skin do not show any pathological changes. Hyperpigmentation of keratinocytes is possible, as is a perivascular lymphocytic infiltrate with melanophages. Histologically, pagetoid dyskeratosis is an important differential diagnosis, defined as an incidental histological finding of no clinical relevance due to friction and maceration with pyknotic nuclei, clear halo, rim of stippled cytoplasm and eosinophilic granules [27,29]. In contrast, ichthyosis hystrix of Curth–Macklin has a KRT1 mutation in the tail domain that disrupts supramolecular keratin organization, resulting in a shell-like structure of loosely arranged tonofilaments, but unlike CRIE, it forms an SG and an orthohyperkeratotic horny layer [27,30].
The sixth pattern is characterized by “psoriasis-like features”, which in some cases are indistinguishable from psoriasis vulgaris (Figure 7, Figure 8, Figure 9, Figure 11 and Figure 12) [31]. This pattern contributes to an entirely new understanding of the overlap between different inflammatory skin conditions. The histology of Netherton syndrome (NS) can be similar to that of psoriasis vulgaris, except NS has a very thin stratum corneum in early life. However, clinical aspects (Table 2) are not usually characteristic of psoriasis and provide additional clues [31]. Interestingly, the histological spectrum of NS is broad and includes atopic dermatitis-like features with spongiosis (Figure 7) [8,31]. Immunostaining for LEKTI allows a reliable specific diagnosis of NS by showing loss of LEKTI in the epidermis and hair follicle epithelia [8]. Other psoriasiform dermatoses may exhibit reduced LEKTI expression in the epidermis, while hair follicles remain distinctly immunoreactive.
Peeling skin disease (PSD) is characterized by lifelong patchy erythema with peeling of the skin, as with NS patients, who are at high risk of food allergy, pruritus and asthma. Histologically, the subcorneal clefts in psoriasiform dermatitis have been difficult to recognize (Figure 8). Sometimes step sections are helpful. We observed erythrocytes in the subcorneal clefts, derived from the biopsy procedure, which may serve as a diagnostic clue to exclude artificial clefting after tissue processing. Immunohistochemical staining for corneodesmosin may also help to differentiate PSD from other ichthyoses.
In CHILD syndrome, xanthomatous cells in the papillary dermis, if present, are a helpful histological clue when present in association with a psoriasiform pattern (Figure 9) [32]. In addition, immunostaining for adipophilin can highlight lipid inclusions in fibroblasts.
Severe dermatitis–allergies–metabolic wasting (SAM) syndrome presents with congenital erythroderma reminiscent of Netherton syndrome. Food allergies, recurrent skin and respiratory infections, failure to thrive and growth retardation may cause severe problems. Skin biopsy shows psoriasiform dermatitis with additional histological evidence. Mutations in desmosomal proteins can lead to the histological finding of “desmosomal-type acantholysis”, defined by hypereosinophilic epidermal keratinocytes, widening of intercellular spaces and partial loss of intercellular bridges in the suprabasal layers (Figure 11) [33]. Immunostaining for desmoglein or desmoplakin may help to rapidly establish the diagnosis (personal observation).
MALT1 deficiency also presents with postpartum periodic erythroderma with scaling and recurrent systemic infections and severe internal complications. Histology is characterized by psoriasiform and spongioform dermatitis, which can be easily differentiated from NS by immunohistochemistry for LEKTI (Figure 12).
As a result of our cohort analysis, we suggest that follicular hyperkeratosis not be defined as an independent pattern, as it may be associated with all subtypes of ichthyosis (Table 1 and Table 2). Furthermore, pachyonychia congenita and ectodermal dysplasia may show aspects of follicular hyperkeratosis (Table 3). Hyperkeratosis of the acrosyringeum is also a variable feature in many ichthyoses and is thought to lead to impaired sweating. However, we were not able to systematically analyze this criterion, as the sections did not show a significant number of sweat gland orifices.
In summary, the value of histological patterns in diagnosing inherited ichthyoses is underestimated. Here, we describe a practically relevant algorithm consisting of six histological patterns. In settings lacking access to genotyping, the analysis of skin biopsy sections by light microscopy can aid in narrowing down the differential diagnosis. If available, molecular findings and other laboratory values combined with clinical clues and histological examination not only support the diagnostic process but also increase our understanding of the consequences of the genotype for the phenotype (ranging from molecule-level effects to tissue morphology).
4.2. Limitations
We could not include all of the diagnoses listed in the consensus nomenclatures for epidermal differentiation disorders, previously referred to as ichthyotic skin diseases [1,2,6]. This was due, among other things, to the lack of histological examination in confirmed cases or the lack of genetic results and other diagnostic methods. Therefore, we deliberately did not include erythrokeratodermia (EKV). Recently, an EKV-like phenotype has also been shown in kallikrein (KLK)11 mutations, which were initially not yet part of the panel diagnosis [34]. For the sake of completeness, we have included some diagnoses that were missing in our cohort in our diagnostic algorithm. The criteria for these diagnoses are based on experience with consultation cases of ichthyosis and findings in the literature that provide convincing histological descriptions.
Moreover, due to the rare-to-ultrarare nature of the diseases investigated, the number of available patients is limited, and subtype representation may be uneven. Nevertheless, the overall number of patients and the inclusion of a well-characterized cohort from specialized reference settings provides meaningful insights into conditions for which comprehensive datasets are otherwise scarce.
Notably, some diagnoses present with variable patterns, making differential diagnosis difficult. As we usually only look for distinct mutations in genetic analysis, we cannot completely exclude that patients have other modifying genetic factors outside of the known genes for genodermatoses. Further studies correlating the phenotype, mutational status and histological patterns in larger cohorts of patients are needed to understand the clinical and histological heterogeneity of cornification disorders. The importance of immunological mechanisms and modifier genes, as well as environmental factors, contributes to this heterogeneity. Recently, a highly variable response to biologic agents has been demonstrated, underscoring this heterogeneity [35]. In addition, variations in histology may be related to the biopsy site, age of the lesion or pretreatment.
5. Conclusions
In this study, we propose histological patterns and diagnostic pathways that may help to improve the diagnosis of ichthyoses, some of which are rare, syndromic and life-threatening.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Akiyama M. Choate K. Hernández-MartínÁ. Aldwin-Easton M. Bodemer C. Gostyński A. Hovnanian A. Ishida-Yamamoto A. Malovitski K. O’Toole E.A. Nonsyndromic epidermal differentiation disorders: A new classification toward pathogenesis-based therapy Br. J. Dermatol.202519361964110.1093/bjd/ljaf 15440308026 · doi ↗ · pubmed ↗
- 2Paller A.S. Teng J. Mazereeuw-Hautier J. Hernández-MartínÁ. Granier Tournier C. Hovnanian A. Aldwin-Easton M. Tadini G. Schwartz J. Sprecher E. Syndromic epidermal differentiation disorders: A new classification toward pathogenesis-based therapy Br. J. Dermatol.202519359261810.1093/bjd/ljaf 12340184496 · doi ↗ · pubmed ↗
- 3Almendra N.V. Duran L.A. Ictiosis hereditaria: Desafío diagnóstico y terapéutico. [Hereditary ichthyosis: A diagnostic and therapeutic challenge]Rev. Chil. Pediatr.20168721322310.1016/j.rchipe.2015.07.02526471314 · doi ↗ · pubmed ↗
- 4Gutiérrez-Cerrajero C. Sprecher E. Paller A.S. Akiyama M. Mazereeuw-Hautier J. Hernández-Martín A. González-Sarmiento R. Ichthyosis Nat. Rev. Dis. Primers 20239210.1038/s 41572-022-00412-336658199 · doi ↗ · pubmed ↗
- 5Joosten M.D.W. Clabbers J.M.K. Jonca N. Mazereeuw-Hautier J. Gostyński A.H. New developments in the molecular treatment of ichthyosis: Review of the literature Orphanet J. Rare Dis.20221726910.1186/s 13023-022-02430-635840979 PMC 9287901 · doi ↗ · pubmed ↗
- 6Oji V. Tadini G. Akiyama M. Blanchet Bardon C. Bodemer C. Bourrat E. Coudiere P. Di Giovanna J.J. Elias P. Fischer J. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009 J. Am. Acad. Dermatol.20106360764110.1016/j.jaad.2009.11.02020643494 · doi ↗ · pubmed ↗
- 7Ross R. Di Giovanna J.J. Capaldi L. Argenyi Z. Fleckman P. Robinson-Bostom L. Histopathologic characterization of epidermolytic hyperkeratosis: A systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders J. Am. Acad. Dermatol.200859869010.1016/j.jaad.2008.02.03118571597 PMC 2517215 · doi ↗ · pubmed ↗
- 8Metze D. Traupe H. Süßmuth K. Ichthyoses—A Clinical and Pathological Spectrum from Heterogeneous Cornification Disorders to Inflammation Dermatopathology 2021810712310.3390/dermatopathology 802001734066992 PMC 8161842 · doi ↗ · pubmed ↗