Genomic Biomarkers and Mutational Landscape of Nonsyndromic Hearing Loss (NSHL) in the Singaporean Population: Clinical Translational Implications
Che Kang Lim, Mei Shuang Cheng, Gerard Low, Joyce Zhi’en Tang, Jia Hui Ng, Ni Gin Ong, Pei Shan Leem, Su Ann Lim, Jiun Fong Thong, Vanessa Yee Jueen Tan

TL;DR
This study identifies genetic biomarkers for nonsyndromic hearing loss in Singaporeans, revealing key genes and variants linked to different hearing loss severities.
Contribution
The study expands the mutational spectrum of NSHL in Singaporeans and identifies novel variants with clinical implications.
Findings
A molecular diagnosis was achieved in 57% of NSHL cases with high pathogenic variant classification.
Common and novel genes like GJB2, SLC26A4, and MYO6 were linked to specific hearing loss severities.
The findings support improved diagnosis and precision medicine approaches for hearing loss in Singapore.
Abstract
Nonsyndromic hearing loss (NSHL) is a highly prevalent, genetically heterogeneous condition, yet its molecular basis in the Singaporean population remains underexplored. We performed whole-exome sequencing and integrative bioinformatics analysis in 115 patients with NSHL to define population-specific genetic biomarkers. A molecular diagnosis was achieved in 57% of cases, with 76% of identified variants classified as pathogenic or likely pathogenic and 24% exhibiting high pathogenic potential. Common East Asian NSHL genes, including GJB2, SLC26A4, and OTOF, were frequently detected alongside less prevalent genes such as ACTG1, CEACAM16, COL11A2, DIAPH1, KCQN4, MYH14, MYO6, MYO7A, MYO15A, SLC17A8, SMPX, STRC, TJP2, TMC1, TMPRSS3, highlighting extensive genetic heterogeneity. Notably, multiple novel variants, including MYO6 c.554-2A>G, and TNC p.N750Y, were identified, expanding the known…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
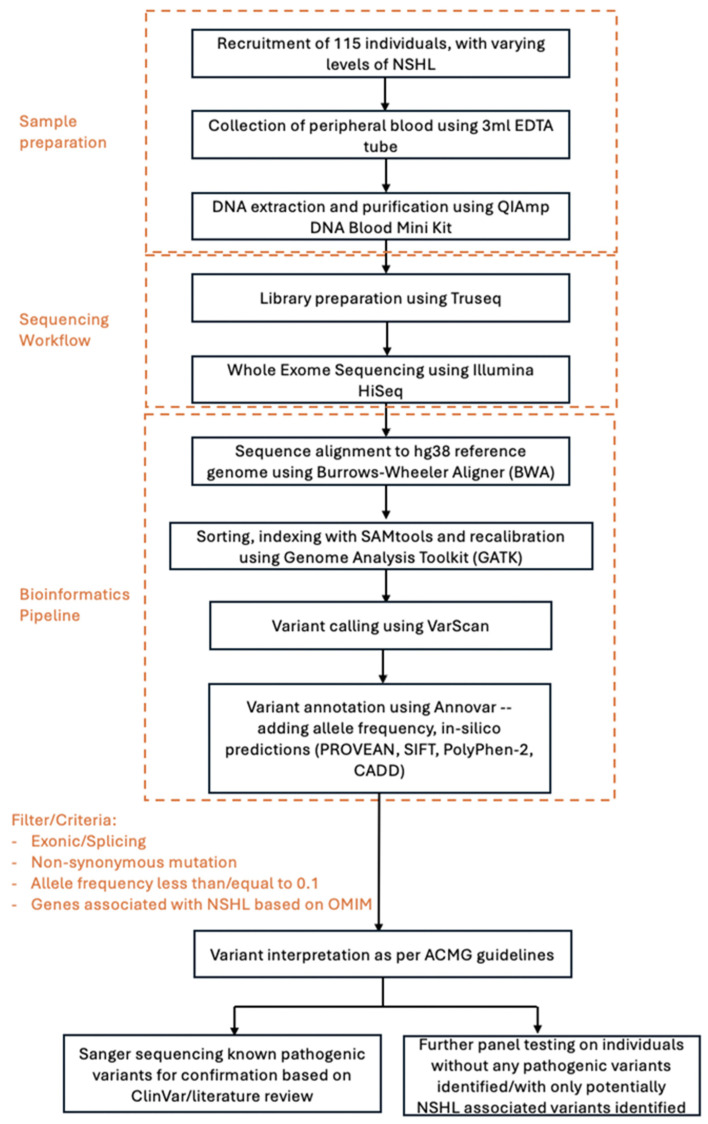 Figure 1
Figure 1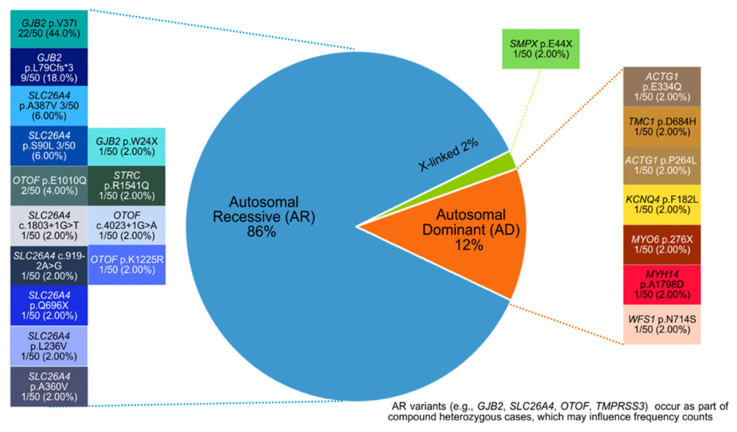 Figure 2
Figure 2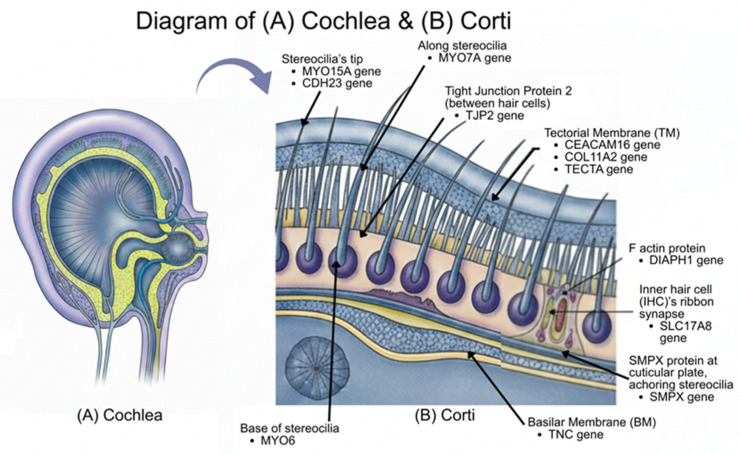 Figure 3
Figure 3- —SGH Health Development Fund
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHearing, Cochlea, Tinnitus, Genetics · Genomics and Rare Diseases · Vestibular and auditory disorders
1. Introduction
Hearing loss represents one of the world’s most pervasive sensory impairments, impacting approximately one in five people globally. According to the World Health Organization (WHO), over 1.5 billion people currently live with some degree of hearing loss, and over 5% of the world’s population (430 million people), require rehabilitation to address their disabling hearing loss [1]. This burden is projected to grow substantially, with over 700 million people expected to require hearing rehabilitation by 2050. The unaddressed cost to the global economy is estimated by the WHO at USD 980 billion annually [1]. On a regional level, the challenge is equally pronounced. In Singapore, four out of every 1000 newborns are identified with a significant hearing impairment [2], underscoring the substantial clinical and developmental urgency of early detection and intervention within the local population.
Amongst the myriad of known causes, genetic factors are recognized as the leading known etiology of hearing loss, accounting for approximately 50% of all cases. Approximately 70% of hereditary hearing loss cases are classified as non-syndromic hearing loss (NSHL) [3,4]. Non-syndromic hearing loss (NSHL) is a partial or complete loss of hearing that occurs without any other associated signs or symptoms. One common classification method of NSHL is by the condition’s pattern of inheritance: autosomal dominant (DFNA), autosomal recessive (DFNB), X-linked (DFNX), or mitochondrial (no special designation). Most cases of NSHL are sensorineural, resulting from disruption of essential cellular function in the inner ear. Rarely, cases are described as conductive, resulting from defects in the development of middle ear structures [5].
Early detection and accurate etiological diagnosis are crucial, as untreated hearing impairment can affect language development, education, and social integration [6]. However, there is difficulty in identifying NSHL-associated variants present in individuals due to the highly heterogenous genetic etiology of NSHL. To date, over 150 distinct genes have been identified and the number continues to grow [7,8,9]. In addition, the distribution of NSHL-related genes varies across populations and ethnic groups [10]. This limits the applicability of existing gene panels and leads to complications when interpreting variants of uncertain significance despite the advancement in sequencing technology. While Northeast Asian populations, including Chinese and Japanese cohorts, have been extensively studied [11,12,13], there is a notable lack of Southeast Asian-specific, particularly adult-focused, genetic research. Hence, we highlight a need for targeted, population-specific studies in the Singaporean context.
Biomarker discovery helps confirm the cause of hearing loss, reducing the need for other costly and extensive investigations. It can also help predict the severity and likely progression of the condition, inform regarding recurrence risks for family planning, and holds potential for advancing precision medicine through matching patients to optimal treatment, thereby improving patient outcomes. Different types of hearing loss-associated variants implicate appropriate disease management as well as therapeutic approach. For example, both SLC26A4 and *GJB2-*associated hearing loss are different in their severity and progressive nature, possibly implicating the timing of rehabilitative interventions such as cochlear implants [14]. In addition, identification of a genetic cause can possibly pave the way for future gene therapies. In fact, clinical studies on gene therapy for OTOF-related hearing loss are underway and early results have shown promising outcomes with dramatic improvements in hearing in some patients [15]. Hence, data from this study can provide crucial insights for diagnosis, intervention, and genetic counselling for underrepresented patients with NSHL.
This study aims to identify genetic variants associated with NSHL in a Singaporean cohort and to evaluate molecular biomarkers for diagnosis and clinical management. By integrating variant annotation, pathogenicity assessment, and frequency data, this work seeks to expand on the understanding of hereditary hearing loss and support precision medicine initiatives in Southeast Asia.
2. Materials and Methods
2.1. Participants
2.1.1. Eligibility Criteria
Eligible participants included patients with NSHL in Singapore and their immediate family members (parents or siblings) who carried mutations in genes associated with hearing impairment. The inclusion of family members enables the study of inheritance modes of identified genes, and facilitates phase resolution of potential compound heterozygous variants.
2.1.2. Screening Process
All potential participants underwent standardized screening using a structured questionnaire. Information collected included demographic data (age, sex, ethnicity), perinatal history, age of onset, laterality and symmetry of impairment, and type of loss (sensorineural or conductive). To prioritize cases with likely genetic causes, individuals with a history of syndromic, conductive, or non-genetic prenatal/perinatal hearing impairment risk factors (such as intrauterine infections or specific medication exposures) were excluded. The criteria further excluded those with acquired hearing issues like acoustic neuroma, head trauma, meningitis, Meniere’s disease, otitis media, ototoxic medication exposure, noise exposure, and viral or bacterial labyrinthitis.
2.1.3. Ethical Considerations
Written informed consent was obtained from all patients and their participating family members. The study was approved by the SingHealth Centralised Institutional Review Board under approval protocol 2019-2337-CIRB on 2 July 2019.
In overall, a total of 115 individuals (probands) with varying levels of NSHL from mild, moderate, severe to profound was recruited. Participants’ age ranged 16–81 years and comprised all major ethnicity in Singapore population, including 88.7% Chinese, 6.1% Malay, 3.5% Indian and 1.7% other Southeast Asian. This ensured the study reflects the nation’s ethnically diverse population to the best it could.
2.2. Sample Preparation and Sequencing Workflow
Peripheral blood collection was carried out utilising 5 mL EDTA tubes. The DNA extraction and purification were performed efficiently using Qiagen’s QIAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany). A quality check for the purity of the extracted DNA (between AD260/AD280 of 1.8 to 2.0) was performed before proceeding with sequencing steps.
Whole-Exome Sequencing
Library preparation was performed using the Agilent SureSelectXT Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions. In brief, genomic DNA was randomly sheared into short fragments with the size of 180–280 bp. Subsequently, fragments were end repaired, A-tailed, and further ligated with the adapters. Adapter-ligated fragments were PCR-amplified, size-selected, and purified. The prepared libraries were hybridized with biotin-labeled probes, and streptavidin-coated magnetic beads were used to capture targeted exons. After washing away non-hybridized fragments and digesting the probes, the captured libraries were further enriched by PCR amplification.
The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries were pooled and then paired-end sequenced using a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA).
2.3. Bioinformatics Pipeline
Sequencing reads were aligned to the GRCh38/hg38 human reference genome using BWA (Burrows–Wheeler Aligner v0.7.17). The resulting SAM files were converted to BAM format, sorted, and indexed using SAMtools. Duplicate reads were removed and base quality score recalibration was performed with the Genome Analysis Toolkit (GATK, Broad Institute) prior to variant calling as well as to improve variant calling accuracy. Variants were then identified with GATK’s HaplotypeCaller, focusing on single-nucleotide variants (SNVs) and small insertions/deletions (indels). Stringent quality control filters were applied to exclude low-confidence calls. Only variants meeting established thresholds for read depth (≥20), mapping quality (≥30), and base quality (≥30) were retained for further analysis. Variants passing quality filters were annotated using ANNOVAR (v2020Jun07) [16] to obtain functional information including predicted coding effect (e.g., synonymous/nonsynonymous, stop-gain etc.), and integrated population allele frequencies from public databases such as gnomAD, ExAC and the 1000Genomes Project. Additional annotation included gene-based information, regulatory region mapping, and conservation scores, enabling comprehensive characterization and prioritization of variants with potential clinical relevance.
Variants were first filtered by hearing loss-associated genes recorded in the OMIM (Online Mendelian Inheritance in Man) [17] database. Then, variants with a minor allele frequency (MAF) ≤ 1% in any population database were retained. The clinical significance of the remaining variants was interpreted using ClinVar [18], dbSNP [19], gnomAD [20] and Deafness Variation Databases [21,22], following the classification ACMG-AMP 2015 guidelines of “Pathogenic”, “Likely pathogenic”, “Benign”, “Likely benign” and “Uncertain significance”.
To assess potential pathogenicity, amino acid substitutions were further evaluated using multiple in silico prediction tools. PROVEAN and SIFT predicted the effect of missense variants on protein function based on sequence homology and amino acid properties. PolyPhen-2 estimated the likelihood of structural or functional disruption of the encoded protein, and CADD (Combined Annotation Dependent Depletion) provided an integrative score reflecting the deleteriousness of each variant across multiple annotations. Through this method, potentially pathogenic variants whose allele frequency was low and which in silico prediction pointed towards being potentially deleterious were also identified.
2.4. Variant Validation Using Sanger Sequencing
Disease-causing variants identified through next-generation sequencing (NGS) were confirmed by Sanger sequencing. Briefly, regions of interest were amplified using polymerase chain reaction (PCR) to generate specific DNA fragments. The PCR products were then purified and subjected to Sanger sequencing to verify the presence, zygosity, and accuracy of the identified variants. This confirmation step ensured that only validated genetic variants were reported, minimizing the risk of false positives from high-throughput sequencing.
The workflow of the methodology, spanning from sample recruitment to variant validation, is presented in Figure 1.
3. Results
3.1. Summary of Demographic and Clinical Characteristic of NSHL Patients
A total of 115 probands were included in the study. Of these, 56.5% were male, and all major ethnic groups in the Singaporean population were included. The patients’ ethnicity percentages were 88.7% Chinese, 6.1% Malay, 3.5% Indian and 1.7% other Southeast Asian (Indonesian and Filipino). Participants ranged in age from 16 to 81 years (age of onset/at diagnosis from newborn to 76 years old). Overall, 37.4% experienced onset of hearing loss before the age of 20, including 17.4% with congenital onset and 10.4% with prelingual onset. In contrast, only 7.0% reported onset after the age of 60.
Regarding laterality, majority of participants had bilateral hearing loss (88.7%). The predominant audiometric configuration was down-sloping (62.6%). In terms of severity, 51.3% had worse than moderatesevere hearing loss, while the remaining 48.7 near-normal to moderate hearing loss(Table 1). All hearing loss was sensorineural in nature.
3.2. Overall Diagnostic Yield
Of the 115 samples analyzed, around 57% (66/115) were found to harbor causative pathogenic variants, likely pathogenic variants, or variants with high potential pathogenicity. Within this subset of diagnosed patients, the majority, around 76% (50/66), carried causative pathogenic or likely pathogenic variants, while approximately 24% (16/66) exhibited variants classified as having high potential for pathogenicity. Notably, almost two-thirds (41/66) of the diagnosed samples underwent orthogonal validation by Sanger sequencing, confirming the accuracy of the variant calls.
This distribution underscores the diagnostic yield of the WES pipeline, demonstrating that a meaningful proportion of clinically relevant variants can be identified and validated with high confidence.
3.3. Genomic Biomarkers Analysis and Identification
In our cohort with documented pathogenic mutations, 86% of patients were from families with autosomal recessive inheritance, 12% from those with autosomal dominant inheritance, and 2% from those with X-linked inheritance, as shown in Figure 2. GJB2 emerged as the most frequently mutated gene in our cohort (27.8%, 32/115), with the p.V37I and p. L79Cfs*3 variants identified in 31 of 66 diagnosed patients (47%) (Figure 2 and Supplementary Table S1). Most GJB2 variants occurred in the homozygous state; however, four patients carried both variants as compound heterozygotes. The second most frequently mutated gene was SLC26A4, detected in approximately 20% (13 of 66) patients with identified causative mutations, where a diverse spectrum of variants was observed (c.1803+1G>T, c.919-2A>G, p.A360V, p.A387V, p.S90L, p.L236V). In addition, several autosomal dominant variants were identified in gene variants (ACTG1, KCQN4, MYO6, TMC1, MYH14), while autosomal recessive variants were identified in genes (OTOF, STRC, TMPRSS3). An X-linked variant in SMPX was also detected.
Autosomal dominant biomarkers are shown in orange, autosomal recessive biomarkers in blue, and X-linked biomarkers in green. Causative variants corresponding to each inheritance category are displayed in text boxes adjacent to their respective regions, with each text box shaded in a color matching or closely approximating the category color.
Overall, we identified three novel mutations and fourteen very rare, damaging mutations (Table 2). 70% of these highly potential pathogenic variants followed an autosomal dominant inheritance mode, including CEACAM16, COL11A2, DIAPH1, MYH14, MYO6, MYO7A, SLC17A8, TECTA, TJP2 and TNC, while only 24% followed an autosomal recessive inheritance mode, including CDH23, MYO15A and TJP2. Interestingly, 3 of the variants identified were novel variants, including TNC p.750Y, CDH23 p.N2063K and MYO6 c.554-2A>G. For MYO6 c.554-2A>G, a nearby mutation, MYO6 c.554-4A>G was recently confirmed to be associated with hearing loss, strongly supporting the potential pathogenicity of the MYO6 c.554-2A>G variant [23]. Most of the genes encode stereocilia-associated proteins (e.g., CDH23 p.N2063K, CDH23 p.Q2227P [24], DIAPH1 p.R243W [25], MYH14 p.A1798D, MYH14 p.D1023Y, MYH14 p.R1777C [26], MYO6 c.554-2A>G [23], MYO15A c.4143-1G>A, MYO15A p.G2177R [27], MYO7A p.G2177R [28], SLC17A8 p.G246A [29], TJP2 p.A116T [30]) where disruption of their respective encoded protein function is known to affect the transduction of auditory signals and hence result in sensorineural hearing loss (SNHL).
3.4. Genotype and Clinical Phenotype Manifestation/Association
As shown in Table 3, within our Singaporean cohort, GJB2 demonstrated the clearest genotype–phenotype relationship in relation to the severity of NSHL. Homozygous missense GJB2 variants such as p.V37I were typically associated with bilateral mild to moderate SNHL and often had a later onset. This genotype–phenotype link was observed in 90% (18 out of 20) of patients carrying this GJB2 point mutation. On the other hand, homozygous truncating GJB2 variants, such as p. L79Cfs*3 or p.W24X, were associated with early-onset moderate to profound SNHL. This was observed in all six patients carrying the mutations.
In comparison, all eight patients with SLC26A4 mutations had congenital or early-onset severe to profound SNHL. Most of these patients had Incomplete Partition type II (IP-II) inner ear malformation as diagnosed on computed tomography (CT) scans. The dominant genes detected, such as ACTG1, TMC1, MYH14 and MYO6, were mostly found in patients with adult-onset bilateral hearing loss of varying severity (observed in four-fifths of patients). Furthermore, the X-linked SMPX gene detected was associated with severe to profound hearing loss detected around 5–6 years old while the recessive STRC gene was associated with congenital moderate SNHL. However, due to the small sample size for these biomarkers, further validation for the phenotypic correlation is required.
3.5. Similarities and Differences to Southeast Asia, China Regions and USA
In comparison to regional studies, including Thailand, Indonesia, USA and parts of China, similar and different genes were detected across all five regions (Table 4). Among shared biomarkers, GJB2 and SLC26A4 were detected across five countries. In addition, other genes like ACTG1, OTOF and STRC could be noted as detected in at least one other country as well, supporting regional consistency in major genes implicated in NSHL across East and Southeast Asian populations. Reports from heterogeneous populations such as the USA further support their broader clinical relevance [31,32,33,34,35,36].
4. Discussion
This study, the first comprehensive genetics investigation of non-syndromic hearing loss in Singaporean cohort, highlights the value of key molecular biomarkers such as GJB2, SLC26A4, and OTOF, which were consistently detected at high frequencies across the East Asian region. Notably, the recurrent identification of variants as GJB2 c.235delc and p.V37I, previously recognised as East Asian founder alleles [37], demonstrates their broader applicability within Singaporean individuals and reinforces their relevance in local diagnostic efforts.
The GJB2 gene codes gap junction protein beta 2, more commonly known as connexin 26. Connexin proteins form channels called gap junctions that allow the movement of potassium ions between cells. Hearing requires the conversion of sound to electrical impulses, which involves ionic movement between cells. The high frequency and consistent detection of these variants strongly supports the incorporation of focused genetic panels as a first-tier diagnostic tool, enabling earlier, more accurate identification and reducing the diagnostic burden for affected individuals.
Additionally, in our cohort, 90% of homozygous missense GJB2 p.V37I were observed as late-onset and associated with mild to moderate SNHL; while homozygous truncating GJB2 variants, p. L79Cfs*3 or p.W24X, were linked with moderate to profound phenotypes that are early onset.
These findings underscore the importance and clinical utility of genetic testing not only in juvenile but also in adult hearing loss patients, supporting accurate diagnosis, guiding clinical follow-up and prognostic assessment, as well as provide crucial information for family planning.
Our study expands the spectrum of causative genes for NSHL, reporting three novel mutations and fourteen very rare damaging mutations. Among the genes identified as high potential pathogenicity, most have not been previously confirmed as causative in the context of NSHL, except for MYH14. This highlights a crucial gap in clinical knowledge about these genes, particularly within our Singaporean cohort, and emphasizes the need for further investigation. Our approach, which integrates allele frequency analysis, in silico pathogenicity prediction, and genomic context including conservation, proximity to known pathogenic residues, and gene-specific mechanisms, allowed us to identify variants that extend beyond those with established clinical significance. By focusing on variants with an allele frequency of less than 0.03, we ensured that pathogenic variants like GJB2 were preserved while minimizing the inclusion of common, tolerated variants. Furthermore, SIFT as well as PolyPhen predictions, alongside CADD scores above 12, provide strong support for the potential deleterious nature of the identified variants. We also examined whether the affected sites are conserved or if nearby mutations with known pathogenicity could be used as reference points.
In addition, we confirmed that all the shortlisted genes encode proteins that are involved in cochlear physiology or play a role in the noise signal transduction pathway in the inner ear, reinforcing their relevance in NSHL (Figure 3). Notably, the MYO15A exon9: c.4143-1G>A splice site variant stands out as a highly supported pathogenic variant, as canonical ±1 or 2 splice site mutations often lead to exon skipping or intronic material inclusion. Such variants, especially when previously reported in the same exon, typically result in pathogenic outcomes, particularly given the gene’s established role in hearing mechanisms [38]. Similarly, variants like CEACAM16 p.V338L, COL11A2 p.G353W, and TECTA p.H1400Y are associated with the tectorial membrane (TM), which is crucial for hearing. CEACAM16 plays a key role in TM integrity and its adhesion to stereocilia for mechanical amplification [39], while mutations in COL11A2 disrupt collagen protein structure, causing malformations in the TM’s ultrastructure [40]. TECTA encoding alpha-tectorin is critical for the normal formation of the TM, thereby affecting hearing thresholds [41] (Figure 3B). On the other hand, TNC encoding Tenascin-C, a glycoprotein found in the basement membrane of the basilar membrane (BM), is significant in auditory development and repair of hearing injuries [42]. These findings underscore the significance of these genes in cochlear function and support their candidacy as high potential pathogenic variants for NSHL.
Our general diagnostic yield of approximately 57% falls at the higher end of what has been reported for NSHL, where most studies typically achieve yields of 30–45% [31,43,44,45]. This elevated yield demonstrates the effectiveness of our sequencing and variant-interpretation strategy in uncovering both known and novel pathogenic variants within this cohort, highlighting the potential for improving NSHL diagnosis in populations with limited genetic data.
One limitation of our study is the overrepresentation of Chinese ethnic individuals, reflecting the national demographic distribution, while the smaller sample sizes of other ethnic groups limit ethnicity-specific comparisons and reduce the representativeness of findings for non-Chinese populations. Therefore, the analysis was conducted at the level of the Singaporean population as a whole to provide results that are more applicable at the national clinical level. In our data, 17% of cases were unknown onset, which may introduce some bias in the clinical phenotype–genotype correlation. This limitation should be considered when interpreting the results. Future studies with more precise data collection regarding the onset of disease could help clarify the impact of this variable and provide a more accurate analysis. Additionally, the lack of consanguineous marriage data is also a limitation in our study. Consanguineous marriage rates are known to be higher in certain regions, including parts of Asia, the Middle East, and Africa, which could potentially influence genetic analyses. However, in our study, the effect of consanguinity is likely minimal due to low prevalence in Singapore [46]. We acknowledge this limitation and recommend that future research should collect consanguinity data to better assess its impact on genetic analysis on NSHL. In addition, some variants were classified as of uncertain significance, and challenges in confirming the genetic basis for certain samples limited our detection rate. This reinforces the need for further investigation of variants unique to the Singaporean population to enhance diagnostic accuracy. Furthermore, environmental and epigenetic factors are likely to influence penetrance and expressivity, suggesting that further validation studies are essential to confirm genotype-phenotype associations in this cohort.
While our study contributes to an expanded framework for interpreting NSHL variants, functional validation of the identified variants is crucial for definitively establishing their pathogenicity. Furthermore, this work highlights the importance of integrating in silico prediction tools and clinical phenotypes with empirical data to support genomic interpretation, particularly in settings where reference data remain scarce.
5. Conclusions
In summary, our study provides important insights into the genetic architecture of NSHL within Singaporean populations. By identifying both known and novel pathogenic variants, we demonstrate an improved yield in molecular diagnosis and highlight the value of genetic testing in guiding clinical management decisions. The discovery of new mutations further broadens the diagnostic spectrum, underscoring the complexity and diversity of NSHL in the region.
Overall, these findings support the clinical utility of targeted genetic testing in NSHL and provide a framework for improved diagnostic strategies and genetic counseling tailored to the region. Furthermore, by enabling more precise molecular classification, this work contributes to the implementation of precision medicine approaches in clinical genetics and hearing loss management.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization: WHO Deafness and Hearing Loss 2025 Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss(accessed on 1 December 2025)
- 2Tang J.Z.T. Ng P.G. Loo J.H.Y. Do all infants with congenital hearing loss meet the 1-3-6 criteria? A study of a 10-year cohort from a universal newborn hearing screening programme in Singapore Int. J. Audiol.20236279580410.1080/14992027.2022.209553735830492 · doi ↗ · pubmed ↗
- 3Shearer A.E. Hildebrand M.S. Odell A.M. Smith R.J.H. Genetic Hearing Loss Overview Gene Reviews((R)) Adam M.P. Bick S. Mirzaa G.M. Pagon R.A. Wallace S.E. Amemiya A. University of Washington Seattle, WA, USA 199320301607 · pubmed ↗
- 4Asaad M. Mahfood M. Al Mutery A. Tlili A. Loss-of-function mutations in MYO 15A and OTOF cause non-syndromic hearing loss in two Yemeni families Hum. Genom.2023174210.1186/s 40246-023-00489-1PMC 1018680937189200 · doi ↗ · pubmed ↗
- 5Medline Plus National Library of Medicine (US) Nonsyndromic Hearing Loss 2016 Available online: https://medlineplus.gov/genetics/condition/nonsyndromic-hearing-loss/(accessed on 1 December 2025)
- 6Verhaert N. Willems M. Van Kerschaver E. Desloovere C. Impact of early hearing screening and treatment on language development and education level: Evaluation of 6 years of universal newborn hearing screening (ALGO) in Flanders, Belgium Int. J. Pediatr. Otorhinolaryngol.20087259960810.1016/j.ijporl.2008.01.01218295908 · doi ↗ · pubmed ↗
- 7Aboagye E.T. Adadey S.M. Wonkam-Tingang E. Amenga-Etego L. Awandare G.A. Wonkam A. Global Distribution of Founder Variants Associated with Non-Syndromic Hearing Impairment Genes 20231439910.3390/genes 1402039936833326 PMC 9957346 · doi ↗ · pubmed ↗
- 8Lee N.K. Uhler K.M. Yoon P.J. Santos-Cortez R.L.P. Clinical Genetic Testing for Hearing Loss: Implications for Genetic Counseling and Gene-Based Therapies Biomedicines 202412142710.3390/biomedicines 1207142739062005 PMC 11274279 · doi ↗ · pubmed ↗