Targeting T-Cells for Cancer Treatment: Current Clinical Strategies and Challenges
Anand Rotte, Mentor Sopjani, Madhuri Bhandaru

TL;DR
This paper reviews how T-cells are being used in cancer treatment, focusing on current strategies, challenges, and new developments in immunotherapy.
Contribution
The paper provides a comprehensive overview of the latest advancements and challenges in T-cell-directed cancer immunotherapies.
Findings
Bispecific T-cell engagers and next-generation CAR T-cells are being developed to improve treatment of solid tumors.
Challenges include resistance, limited efficacy in solid tumors, and manufacturing difficulties.
Evolution from monoclonal to bispecific antibodies is a key trend in immunotherapy development.
Abstract
Modulation of immune response to target tumor cells has been shown to be a successful strategy for cancer treatment. Over the past years immunotherapy has been integrated into cancer treatment and PD-1 blockers have become the backbone of treatment regimens for multiple cancer types. Several classes of immunotherapies, such as immune checkpoint blockers, bispecific antibodies, chimeric antigen receptor (CAR) T-cells, and tumor-infiltrating lymphocytes (TILs), were approved by the US FDA in the last decade and many more are in clinical trials. Research on redirecting effector T-cells to treat cancer has been aimed at addressing the limited responses in solid tumors, emergence of resistance, treatment-limiting adverse events and logistical challenges. Bispecific immune checkpoint blockers, developed to simplify the combination therapies; bispecific T-cell engagers, developed to connect…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
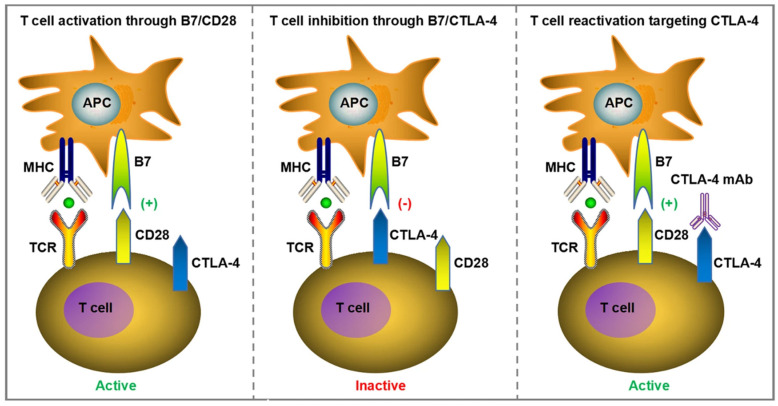 Figure 1
Figure 1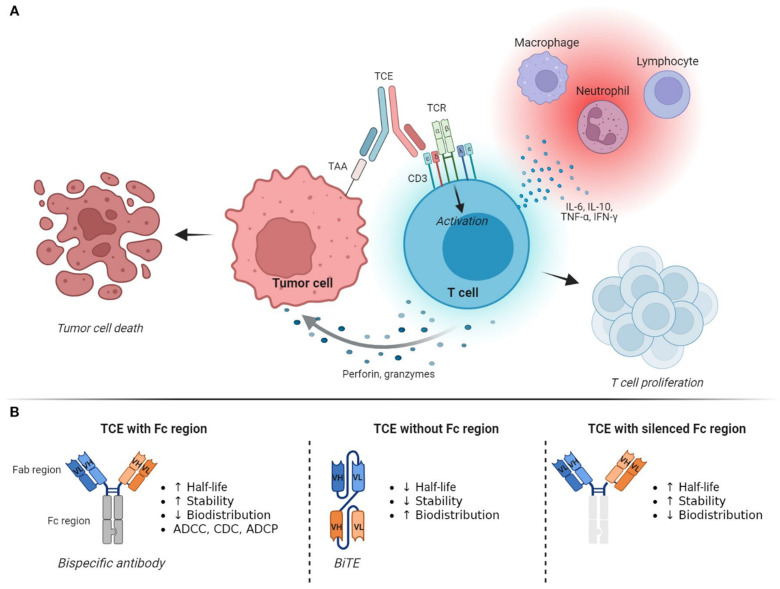 Figure 3
Figure 3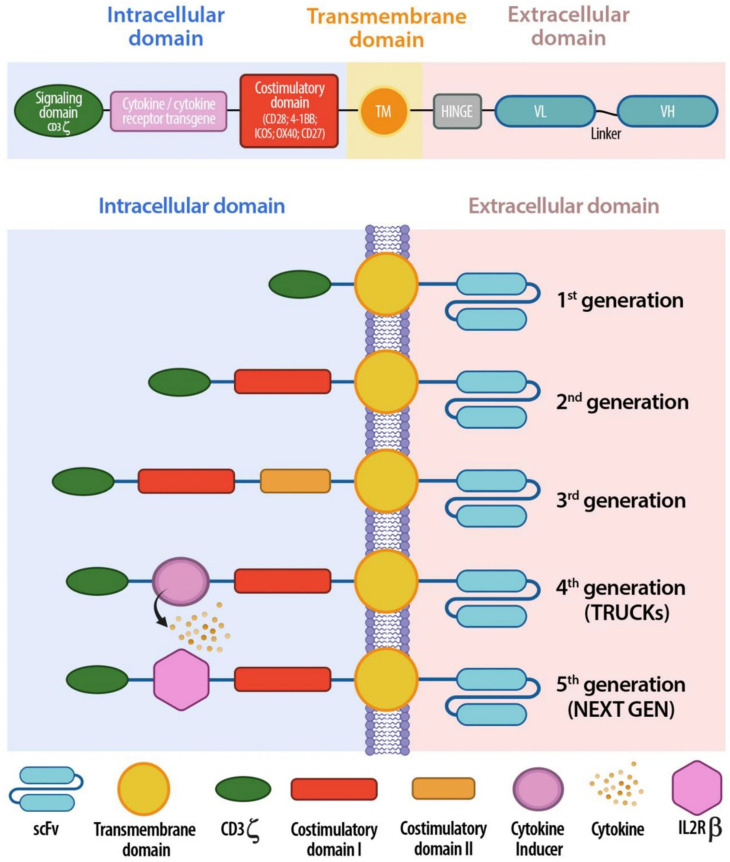 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCAR-T cell therapy research · Monoclonal and Polyclonal Antibodies Research · Cancer Immunotherapy and Biomarkers
1. Introduction
The therapeutic modulation of the immune system has emerged as one of the most significant advances in cancer treatment over the past two decades. Unlike conventional cytotoxic therapies, immunotherapies aim to harness and amplify endogenous immune mechanisms to recognize and eliminate malignant cells, leading in some cases to durable clinical responses and long-term survival benefits [1]. Several classes of immunotherapies, including cytokines, cancer vaccines, immune checkpoint blockers (ICBs), bispecific antibodies, chimeric antigen receptor (CAR) T-cells, and tumor-infiltrating lymphocytes (TILs), have been approved by the US FDA for the treatment of cancer, and a majority of the approvals have happened in the past 10 years. Table 1 includes the list of immunotherapy sub-classes that were introduced in the clinic along with the earliest drug approvals. Though theoretically most of the immunomodulatory drugs can target multiple immune cells, including sub-types of T-cells, natural killer (NK) cells, macrophages and dendritic cells, the effector T-cells are generally considered to be the focus for modulation. T-cell-based immunotherapies can be broadly classified into five major categories: (i) cytokine therapies that promote T-cell proliferation (e.g., interleukin-2 [IL-2]); (ii) cancer vaccines that modulate T-cell priming, activation and differentiation; (iii) immune modulators that enhance T-cell activation or reverse T-cell exhaustion, such as immune checkpoint inhibitors (ICIs); (iv) T-cell-redirecting agents, including bispecific T-cell engagers (BiTEs); and (v) adoptive cellular therapies, such as chimeric antigen receptor (CAR) T-cell therapy and tumor-infiltrating lymphocyte (TIL) therapy.
Immunostimulatory cytokines such as interleukin-2 showed the earliest signs of clinical potential for the treatment of cancer, as seen by the approval of IL-2 for the treatment of unresectable metastatic melanoma (Table 1) [2]. Though the overall response rate for IL-2 treatment was relatively low, the responses were durable in the responding patients, and the adverse events were manageable compared to chemotherapy [3,4]. However, the widespread application of cancer immunotherapy was limited until the development of immune checkpoint blocker, until ipilimumab, the anti-Cytotoxic T-Lymphocyte-Associated protein 4 (CTLA-4) monoclonal antibody belonging to the class of ICBs got approved for the treatment of unresectable metastatic melanoma [5]. The approval of ipilimumab was followed by the approval of the next sub-class of ICBs, the anti-Programmed Cell Death Protein 1 (PD-1) monoclonal antibodies nivolumab and pembrolizumab, in 2014 [6]. Anti-PD-1 monoclonal antibodies significantly improved the clinical outcomes in cancer patients and dramatically changed the treatment landscape for cancer, resulting in an explosion of research in immunotherapy. The transformative impact of immune checkpoint blockades on cancer treatment was recognized by the Nobel committee, and Dr. James Allison and Dr. Tasuku Honjo were awarded the Nobel Prize in medicine for their research on immune checkpoints in 2018 [7].
To further improve the responses and survival in patients treated with immunotherapy, combination therapy, including the combination of PD-1/PD-L1 blockers with CTLA-4 blockers, with chemotherapy and with vascular endothelial growth factor (VEGF) receptor blockers were developed. Research in the structure and functions of monoclonal antibodies resulted in the development of bispecific antibodies that were designed to target two different antigens instead of single antigen. The ability to target two different antigens by bispecific antibodies resulted in the development of bispecific T-cell engagers (BiTEs) that mainly worked by binding to both T-cells, through CD3 binding and the tumor cells, through respective target antigen binding, and facilitating the elimination of tumor cells by T-cells [8]. Blinatumomab was the first bispecific antibody targeting CD3 on T-cells and CD19 on malignant B-cells, which was approved for certain hematological cancers [9]. In addition to BiTEs, bispecific antibodies are also aimed at simplifying combination immunotherapy by targeting two different pathways, such as PD-1 and VEGF or PD-1 and CTLA-4.
The early concepts of adoptive cell therapy (ACT), involving the transfer of ex vivo cultured and expanded T-cells, was proposed in the late 1980s [10,11,12,13,14] and the research on the clinical application of ACT continued along with research on immune checkpoints [15]. However, the initial results from clinical studies were not satisfactory and the evidence for clinical efficacy was seen only after the development of second-generation CAR T-cells. CAR T-cells were first approved in 2017 for pediatric and young adult patients with acute lymphoblastic leukemia [15] and TILs were approved for metastatic melanoma in 2024 [16].
Despite these advancements, there are still significant challenges to solve. CAR T-cell and BiTE therapies are often linked to treatment-related toxicities, such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), necessitating specialized monitoring and management strategies. Additionally, the use of T-cell-based therapies in solid tumors has been constrained by tumor antigen heterogeneity, limited T-cell trafficking, and the immunosuppressive tumor microenvironment [17,18,19,20,21,22]. Manufacturing complexity, supply chain constraints, and high treatment costs further restrict broad clinical implementation of adoptive cellular therapies [23,24,25,26].
Many reviews have addressed different types of T-cell-based immunotherapies but have often focused exclusively on either ICBs or CAR T-cell therapy [23,27,28]. There are limited to no reviews offering a comprehensive synthesis encompassing monoclonal antibody-based T-cell modulation, bispecific and multispecific antibody platforms, and adoptive cellular therapies while rigorously analyzing their common mechanisms, constraints, and translational obstacles. This narrative review aims to describe the different types of T-cell-targeting cancer immunotherapies that were successfully introduced into the treatment paradigm, including ICBs, BiTEs, CAR T-cells and TILs, and briefly summarize the development history, clinical success of the class and latest ongoing clinical developments. This review intends to offer a thorough and current framework for comprehending the current status and future trajectories of T-cell-based cancer immunotherapy by integrating clinical and translational viewpoints across various therapeutic platforms. As the review did not aim to address any specific clinical- or nonclinical-related questions due to its narrative nature, literature was not collected based on pre-defined search criteria, including search terms and time period, which is required for a systematic literature review. Studies that are well-known for their impact on the research area were selected for discussion and data from publicly available sites such as FDA and clinicaltrials.gov were collected to summarize in the tables. The most recent package inserts approved by the US FDA for the respective approved drugs were used to review the current status, approved indications, dosage and regimen, safety and efficacy. Where needed simple keywords, such as bispecifics, CAR T-cells, TILs or the generic name of the drug, were used to search and obtain information.
Drugs that act by stimulating T-cell activity and proliferation and by inhibiting T-cell exhaustion, such as ICBs and bispecifics, are discussed first in the review, followed by T-cell-redirecting therapies, such as bispecific T-cell engagers, and then by adoptive cell therapies, such as CAR T-cells and TILs.
2. Modulators of T-Cell Activation and Exhaustion
2.1. ICBs
T-cells are thought to play a central role in the anti-tumor immune response, and their activity is regulated at multiple levels to prevent harming healthy internal organs [29]. Naïve T-cells are primed and activated in the lymph nodes and the activated T-cells are trafficked to the tumor microenvironment, where they initiate anti-tumor immune response upon interaction with tumor antigens. Activation of T-cells requires two signals, one from the presentation of an antigen peptide on the major histocompatibility complex (MHC) on the antigen-presenting cell (APC) to the T-cell receptor, and the second from the binding of a costimulatory receptor such as CD28 with B7 ligands present on APCs (Figure 1). The requirement of a second activation signal in addition to antigen presentation is an important regulatory feature of the immune response. In the absence of a second stimulatory signal, T-cells turn anergic and apoptotic, preventing the activation of T-cells in response to autoantigens [29].
Uncontrolled activation of T-cells is further regulated by the expression of inhibitory molecules on the surface of activated T-cells. The receptors and their respective ligands that are shown to regulate the immune response are commonly referred to as ‘immune checkpoints’ and the antibodies that are developed to block the immune checkpoints are referred to as ‘immune checkpoint blockers’. The discovery in the mid-1980s of pathways regulating immune response and the molecules, including receptors and their ligands, involved in fine-tuning immune cell activity dramatically changed the treatment landscape of cancer [7].
Especially, the characterization of the first immune checkpoint receptor, CTLA-4, expressed on activated T-cells, the identification of its ligands B7-H1 (CD80) and B7-H2 (CD86), expressed on APCs, and the elucidation of its role in inhibiting the costimulatory signals of T-cell activation are key milestones in tumor immunotherapy (Figure 1). The interaction between CTLA-4 and its ligands is understood to inhibit T-cell proliferation and induce anergy in the T-cells [27,28,30,31,32,33]. The discovery of CTLA-4 was closely followed by the discovery of another immune checkpoint, PD-1, and the elucidation of its role in the dampening of immune response [34,35,36]. Research on CTLA-4 and PD-1 resulted in the development of respective monoclonal antibodies blocking the inhibitory pathways to unlock anti-tumor immune responses, and the researchers Dr. James P Allison and Dr. Tasuku Honjo were awarded the Nobel Prize in medicine in 2018 for their pioneering research on CTLA-4 and PD-1 respectively [7].
T-cell activation and CTLA-4 pathway (simplified). Reproduced from an article by Meng et al. [37]. Copyright © Meng et al. Published by Cell Death and Disease [37] under Creative Commons License CC.BY NC 4.0.
The PD-1 pathway has emerged as a key pathway in the regulation of peripheral immune response. PD-1 receptors are expressed on activated T-cells and its ligand PD-L1 is broadly expressed on various cell types (Figure 2). The PD-1 pathway mainly limits uncontrolled activation of immune response and aims to protect healthy tissue from the harmful effects of immune cell activity [27,28,33]. Anti-PD-1 antibodies significantly improved the survival outcomes in cancer patients and became the backbone of treatment algorithms for a majority of cancers [37,38,39,40]. The clinical success of anti-PD-1/PD-L1 antibodies resulted in an explosion of research in immunotherapy and the clinical evaluation of multiple molecular targets for the treatment of cancer [41]. Multiple companies initiated the development of anti-PD-1 and PD-L1 antibodies as a monotherapy or as a combination therapy together with chemotherapy, other checkpoint inhibitors or targeted therapies. The number of registered clinical trials studying the efficacy and safety of anti-PD-1/PD-L1 alone or in combination therapy was estimated to be over 5000 in 2021, indicating the interest in the anti-PD-1/PD-L1 clinical development programs [42]. To date, seven anti-PD-1 and three anti-PD-L1 monoclonal antibodies have been approved by the US FDA as monotherapy or combination therapy for the treatment of various cancer sub-types (Table 2). Anti-PD-1 monoclonal antibodies have received approvals for a broad range of indications, including both solid tumors as well as hematological cancers. Key approved indications for pembrolizumab, an anti-PD-1 monoclonal antibody and its clinical outcomes are summarized in Supplementary Information Table S1. As research has progressed more cancer types have been added to the approved list of indications, and newer molecules targeting PD-1 and PD-L1 with comparatively higher response rates have been developed. A recent study in rectal carcinoma patients reported 100% complete clinical response following the treatment with the anti-PD-1 antibody dostarlimab, and the molecule was granted ‘breakthrough designation’ by the US FDA for the treatment of patients with locally advanced mismatch repair deficient (dMMR)/microsatellite instability-high (MSI-H) rectal cancer [43,44].
Biomarkers have played a significant role in the success of anti-PD-1/PD-L1 monoclonal antibodies and helped in identifying the patients with a high likelihood of response [45,46,47]. The biomarkers that were approved as companion diagnostics for use with anti-PD-1/PD-L1 monoclonal antibodies by the US FDA include PD-L1, dMMR, MSI-H, TMB, microsatellite stable (MMS or MSI-not high), EGFR and BRAF [48]. However, the impact of the biomarkers in explicitly identifying responders for therapy has been limited by factors such as tumor heterogeneity, lack of standard cut-off values, the labor-intensive nature of the techniques, extrapolation of results based on a small sample or small genomic space and the dynamic nature of the tumor [46,47]. Research is ongoing to improve the accuracy of the biomarkers and to simplify the use of diagnostic tools.
While ICBs, particularly anti-PD-1 monoclonal antibodies, have been extremely successful in the treatment of cancer, limitations to the therapy have also been reported. Resistance to ICB treatment due to the loss of tumor antigens, defects in antigen presentation, T-cell exhaustion and mutations in interferon signaling pathways resulting in the recurrence of cancer were commonly reported, along with the occurrence of autoimmunity-related side effects [49,50,51,52,53]. Lack of reliable biomarkers to predict the treatment response and challenges in the optimization of anti-PD-1 combinations with other ICBs or other molecules such as anti-vascular endothelial growth factor (VEGF) monoclonal antibodies have limited the utilization of ICBs and prompted further research.
In patients with disease progression after initial treatment with ICBs, retreatment with ICBs, termed ‘immunotherapy rechallenge’, has been proposed. While clinical evidence supporting the application of immunotherapy rechallenge is limited, early results support the use of immunotherapy rechallenge in patients who tolerated the initial therapy well and showed response [54,55,56,57]. The efficacy of immunotherapy rechallenge was found to be influenced by multiple factors, including the characteristics of patients, PD-L1 expression, cancer sub-type, monotherapy versus combination therapy and the treatment interval between initial treatment and rechallenge. Patients who had a response during initial treatment were found to likely have response during rechallenge. Similarly, patients who experienced grade 3 or higher adverse events during initial treatment were likely to have severe adverse reactions during rechallenge [57,58,59]. Careful screening of patients is recommended prior to rechallenge and confirmatory studies in larger cohorts of patients may be needed to determine the benefits of immunotherapy rechallenge in cancer patients.
To address the concerns of patient compliance and treatment logistics associated with immunotherapy combinations, the use of bispecific and trispecific antibodies was proposed. Bispecific antibodies targeting PD-1 and a second immune checkpoint, such as CTLA-4, and PD-1 and cell surface molecules involved in cancer progression, such as VEGF, have shown promising efficacy in clinical trials and are discussed in the following section.
2.2. Bispecific Antibodies
Monoclonal antibodies were tested and successfully introduced into clinics for the treatment of cancer long before the application of ICBs [60,61]. Monoclonal antibodies against tumor-associated antigens, such as Her2 and epidermal growth factor receptor (EGFR), and cancer-promoting cytokines, such as vascular endothelial growth factor (VEGF-A), have been approved for the treatment of multiple cancer types [61]. In addition, antibody–drug conjugates combining monoclonal antibodies with cytotoxic compounds to selectively deliver the cytotoxic compound to cancer cells via monoclonal antibodies have also been approved for the treatment of multiple cancer types [61]. The overall success of monoclonal antibodies in targeting tumor cells resulted in the evaluation of bispecific antibodies for the treatment of cancer [62,63]. Bispecific antibodies can target two different immune checkpoints (e.g., PD-1 and CTLA-4), target an immune checkpoint (e.g., PD-1) and a tumor-associated antigen (e.g., EGFR) or cytokine (e.g., VEGF), or target the T-cell receptor component CD3 and a tumor-associated antigen. The first two types of bispecific antibodies are discussed in this section, and the third category of bispecific antibodies, which mainly act by forming a link between effector T-cells and tumor cells, termed BiTEs, are discussed separately in the following section.
Bispecific antibodies designed to block two tumorigenic signaling pathways or two immune checkpoints, or to block an immune checkpoint and a tumorigenic signaling pathway, address the concerns of combination therapies, such as the logistics of drug administration and patient compliance. The list of bispecific antibodies approved by the US FDA for the treatment of cancer is presented in Table 3. Amivantamab-vmjw, a bispecific antibody against EGFR and mesenchymal epithelial transition (MET) receptor, has been approved for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations [64]. Other combinations of tumorigenic signaling blockades under advanced stages (phase 2 or above) of evaluation include anti-EGFR and anti-Human Epidermal growth factor Receptor (HER) 3, and anti-EGFR [65] and anti-Leucine-rich repeat-containing G-protein-coupled receptor (LGR) 5 (Table 4) [66]. Anti-PD-1 and anti-CTLA-4 bispecific antibodies are among the most commonly evaluated dual immune checkpoint blockers in advanced stages of development [67,68,69,70]. Other dual immune checkpoint blockers include anti-PD-1 and anti-4-1BB blockers, anti-PD-1 and anti-TIGIT blockers, anti-PD-1 and anti-LAG-3 blockers, and anti-PD-1 and anti-Tim-3 blockers [63].
The combination of anti-PD-1 and anti-VEGF therapies has been shown to improve treatment outcomes in cancer patients and has been approved by US FDA for the treatment of renal cell carcinoma, NSCLC, hepatocellular carcinoma and endometrial carcinoma [71,72,73,74]. The success of the anti-PD-1/PD-L1 and anti-VEGF therapy combination encouraged the development of bispecific antibodies targeting PD-1/PD-L1 and VEGF [75,76,77,78,79,80]. Early clinical studies promising the efficacy of anti-PD-1/PD-L1 and anti-VEGF bispecific antibodies and multiple molecules are in advanced stages (phase 2 or above) of clinical development (Table 4). Anti-PD-1 and anti-TGF-β bispecific antibody is another combination of targets that has shown promising activity in preclinical studies, and multiple bispecific molecules targeting PD-1 along with TGF-β are in early clinical studies [63].
Though bispecific antibodies as a class demonstrated clinical success with multiple regulatory approvals, they are limited by emergence of on-target and off-target toxicities, development of anti-drug antibodies that can increase the elimination or inhibit the activity of the drugs and by the complexities associated with the tumor microenvironment that can impact the bioavailability of the antibodies at the target site and also inhibit the anti-tumor activity of the effector T-cells [63]. Further research on optimization of antibody components, binding affinity and avidity of the antibody, immunogenicity and pharmacokinetic properties may be needed to improve the application of bispecific antibodies in the clinical practice.
3. T-Cell-Redirecting Therapies
BiTEs
The concept of using bispecific antibodies to direct the T-cells to target tumor cells and induce tumor cell killing by forming immune synapses was introduced nearly four decades ago in the 1980s [8]. The idea of linking effector T-cells and tumor cells was optimized over time to develop the currently available bispecific antibodies. Research on monoclonal antibody structures revealed the significance of the neonatal Fc Receptor (FcRn) in the recycling of antibodies and extending the lifecycle [81], and the significance of IgG binding to Fcγ receptors on myeloid cells and natural killer (NK) cells in the activation of antibody-dependent cellular cytotoxicity (ADCC) [82,83]. As the technology of monoclonal antibodies advanced, various formats of bispecific antibodies were developed, which mainly differed based on the core structure of the molecule (IgG vs. non-IgG) and subsequent Fc-mediated functions (Figure 3) [84,85]. Bispecifics based on the IgG format retain Fc-mediated functions, have a long half-life due to FcRn-mediated recycling and are able to induce ADCC through Fcγ binding depending on the type of IgG subtype, whereas BiTEs based on the non-IgG format have higher tissue penetration but have a short half-life and lack the ability to induce antibody-induced cellular cytotoxicity [63]. Preclinical head-to-head experiments comparing the activity of IgG versus non-IgG documented the significance of the structure on the half-life and activity, and showed that the Fc portion of the molecule can determine the overall target binding property, potency and stability of the BiTE, and thereby influence the mechanism of action and cytotoxicity [86].
The approval of blinatumomab, an anti-CD19 and anti-CD3 BiTE, in 2014 for the treatment of acute lymphoblastic leukemia is considered a breakthrough in immunotherapy after ICBs, and has introduced a unique method of modulating the anti-tumor activity of effector T-cells. Blinatumomab approval triggered a wave of research on bispecific antibodies and T-cell engagers, resulting in the development and approval of several BiTE molecules for the treatment of cancer. To date, nine T-cell-redirecting bispecifics and BiTE molecules, including blinatumomab, have been approved by the US FDA for the treatment of different types of hematological cancers (Table 3). It is to be noted that almost all of the recently introduced BiTEs have been approved by the US FDA through an accelerated pathway and have received conditional approval pending confirmatory randomized controlled trials.
The characteristic feature of BiTEs is their ability to bypass TCR specificity, antigen presentation through MHC molecules and costimulatory pathways, and to induce T-cell-mediated cytotoxicity in the target cells [87]. The main advantage with the BiTE approach is that the T-cells can overcome MHC downregulation on tumor cells and effectively eliminate the target cells. Indeed, BiTEs demonstrated promising potential in improving the overall survival in certain hematological cancers [88]. A recent phase 3 study with multiple myeloma patients also demonstrated the potential of combination therapy including a BiTE in improving the survival of patients [89]. However, there has been limited success for BiTEs outside of hematological cancers and the application of BiTEs in solid tumors has been challenging due to the complex tumor microenvironment [87,90,91]. To date, two BiTEs, including tebentafusp-tebn, a bispecific fusion protein designed to connect gp100 expressing cells with CD3 of T-cells, and tarlatamab-dlle, a bispecific antibody targeting delta-like ligand (DLL) 3 on cancer cells and CD3 on T-cells, have been conditionally approved for solid tumors, including uveal melanoma and small cell lung cancer respectively (Table 3) [92,93].
T-cell engagers: mechanism of action (A) and structure (B). Reproduced from an article by Spinazzola et al. [91]. Copyright © Spinazzola et al. Published by Frontiers in Immunology [91] under Creative Commons License CC.BY 4.0.
The main hurdles in the clinical utilization of BiTEs include limited efficacy in solid tumors due to factors associated with the tumor microenvironment (TME) and the emergence of adverse events, such as cytokine release syndrome and infections [94]. Despite the strong rationale in preclinical studies, several BiTE molecules fail in clinical studies, and the failure is commonly attributed to pharmacokinetic properties and on-target but off-tumor toxicity [95,96]. The TME is known to be immunosuppressive and devoid of T-cells or unable to infiltrate the tumor in ‘cold’ tumors, or may consist of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T-cells (Tregs), that can inhibit the activity of effector T-cells [17,18]. Tumor stroma can also be a physical barrier for the distribution of drugs and may result in the reduced availability of BiTE molecules at the target site [19,20]. Tumor-associated antigen (TAA) heterogeneity and the presence/emergence of tumor cell clones that do not express the TAA targeted by BiTEs can also result in reduced efficacy. The use of step-up dosing strategies and increased monitoring were proposed to address the severity of CRS and infections associated with BiTEs, and has been successfully adopted into clinical practice [97]. The selection of TAAs that are abundantly expressed on tumor cells as targets for the development of BiTE molecules is considered as a promising strategy to address tumor heterogeneity. Indeed, DLL3, a transmembrane protein known to suppress the Notch signaling pathway and regulate the neural tube closure during pregnancy [98], is shown to be overexpressed in neuroendocrine and non-neuroendocrine tumors [99], and has been successfully targeted by a BiTE molecule (tarlatamab-dlle) for the treatment of small cell lung cancer (Table 3). A list of selected ongoing advanced (phase 2 or above) studies evaluating BiTE molecules is presented in Table 4. The findings from the studies can provide further guidance on strategies for the optimization of the efficacy and safety of BiTEs.
4. Adoptive Cell Therapies
4.1. CAR T-Cells
CAR T-cells are T-cells designed to recognize specific antigens on tumor cells through their antigen-specific chimeric receptors and trigger cytotoxicity against tumor cells. The idea of chimeric T-cell receptors was first proposed in the late 1980s and involved combining variable regions (VH/VL) from the antibody with the constant regions from the T-cell receptor (TCR) [11]. The study demonstrated the feasibility of expressing chimeric receptors on T-cells and the activation of T-cells expressing chimeric receptors in response to antigens. Two years later, another team of researchers demonstrated the target antigen-dependent binding and subsequent IL-2 production and cytotoxicity of target cells using T-cells expressing chimeric receptors [13]. However, the initial design of CARs, called the ‘first generation of CAR’, which included scFv fused to the CD3ζ signaling endodomain, showed little to no anti-tumor activity in humans when evaluated in clinical studies using respective autologous CAR T-cells in patients with ovarian cancer, metastatic renal cell cancer, neuroblastoma and mantle cell lymphoma [100,101,102]. To address the concerns on clinical efficacy, the second generation of CAR T-cells designed to incorporate both stimulatory signals of T-cell activation, including TCR as well as CD28, or other costimulatory receptors, such as 4-1-BB, were evaluated [103]. The second generation of CAR thus included the CD3ζ signaling endodomain and CD28 or 4-1-BB endodomain [15]. The design of CAR further evolved to include two costimulatory endodomains, such as CD28 and 4-1-BB, along with the CD3ζ signaling endodomain, and to T-cells redirected for universal cytokine-mediated killing (TRUCKs) that secrete a cytokine, such as IL-12, upon activation. It further evolved to CARs that contain three stimulatory signals, including the CD3ζ signaling endodomain, CD28 or 4-1-BB endodomain, and a cytokine signaling endodomain (Figure 4) [23,104,105,106]. Research on the CAR structure and the resultant improvements aimed at enhancing the cytotoxic activity of the CAR T-cells and to reduce T-cell exhaustion [107,108]. Preclinical studies showed that the latest generation of CAR T-cells, including the TCR signaling domain, costimulatory domain or domains and a cytokine signaling domain or expressed proteins that blocked T-cell exhaustion ligands, improved the target specificity, increased the proliferative capacity and enhanced the cytotoxicity [109,110]. The second generation of CAR T-cells have seen clinical success, especially in patients with hematological cancers, whereas the recent iterations are still in clinical trials.
The T-cells used for the generation of CAR T-cells are usually collected from the patient (autologous) through apheresis, and genetically modified using various gene modification techniques and expanded further ex vivo before administration. Retroviral transduction is the commonly used technique for the transfer of the CAR gene into T-cells in the CAR T-cell therapies that are approved by the US FDA, whereas CAR T-cells generated using other gene modification techniques, such as electroporation, CRISPR-CAS and mRNA, are currently under evaluation [111].
The potential of CAR T-cells in providing durable responses has been documented mainly in hematological cancers, such as mantle cell lymphoma, multiple myeloma, B-cell lymphoma, including diffuse large B-cell lymphoma and follicular lymphoma, and acute lymphoblastic leukemia [112,113,114,115]. To date seven CAR T-cell therapies (all autologous T-cells) have been approved by the US FDA for hematological malignancies (Table 5). Tisagenlecleucel (Tisa-cel) was the first CAR T-cell therapy to be approved by the US FDA, and obecabtagene autoleucel (obe-cel) was the most recently approved CAR T-cell therapy (Table 5). Autologous CAR T-cell therapies have shown encouraging potential in clinical trials as well as real-world studies, with documented responses in the majority of patients and improvements in progression-free and overall survival in responding patients after a single dose of CAR T-cell therapy [116,117,118,119,120,121]. CAR T-cell therapies are also associated with serious adverse effects, such as cytokine release syndrome (CRS), neurotoxicity, prolonged cytopenia and recurrent infections [115]. Recently, rare cases of second primary malignancies of T-cell origin were reported in patients treated with CAR T-cell therapies [122,123]. However, the benefits of CAR T-cell therapy outweigh the risks associated with the treatment and better algorithms are now available for the management of serious adverse events in patients treated with CAR T-cell therapy.
While autologous CAR T-cell therapies have shown promising clinical efficacy, their utilization has been limited by manufacturing and supply chain challenges [23]. To address the expected time delays associated with the manufacturing and shipping of autologous CAR T-cells, allogeneic/donor-T-cell-derived CAR T-cell therapies are proposed. Though the concept of allogeneic CAR T-cell therapy is interesting, the approach has the risks of ‘graft versus host disease’, which occurs when the T-cell receptors present on the allogeneic CAR T-cell surface recognize the antigens on the surface of patient’s healthy tissues and trigger immune response, and ‘host versus graft disease’, which occurs when the patient’s immune system rejects the allogeneic CAR T-cells. The former reaction can be serious and life-threatening, whereas the latter reaction can lead to treatment failure [24,25,26]. To address the concerns of GvHD and HvG, multiple approaches such as selecting T-cells with low TCR signaling during manufacture, knocking out of TCR and CD52 from T-cells, and expressing CD47 on T-cells have been proposed [24]. The optimized allogeneic CAR T-cells with reduced GvHD and HvG potential are currently under advanced stages of clinical evaluation. The list of key allogeneic CAR T-cell therapies under clinical evaluation is presented in Table 6. Interestingly, despite the promising potential to address manufacturing delays associated with autologous CAR T-cells, progress in allogeneic CAR T-cells is relatively slow and most studies are in early-phase trials. Results from clinical studies are needed to support the continued interest in allogeneic cell therapies [124].
Schematic representation of CAR T-cell structure: intracellular, transmembrane, and extracellular domain (upper panel). Evolution of the five generation of CARs: from the first generation, containing only one activation domain, to the last next generation CARs, aiming to improve their safety and efficacy (lower panel). Reproduced from article by Pinto et al. [125]. Copyright © Pinto et al. Published by Journal of Translational Medicine [125] under Creative Commons License CC.BY 4.0.
Ongoing research is also focused on developing CAR T-cell therapies for solid tumors, which has been elusive despite the success in hematological cancers. Utilization of CAR T-cell therapies for solid tumors has been challenging mainly due to difficulties in identifying the tumor-specific target antigen, immunosuppressive tumor microenvironment, restricted T-cell chemotaxis and also due to the occurrence of severe toxicity associated with CAR T-cells in solid tumors [21]. Recently multiple antigens, such as claudin (CLDN) 18.2, EGFR, CD70, and mesothelin, have been shown to be viable targets for the utilization of CAR T-cell therapies, and multiple approaches to overcome the extreme conditions in tumor microenvironment, such as armoring CAR T-cells to express stimulatory cytokines including IL-2, IL-12 and IL-15, and expressing proteins to block T-cell exhaustion pathways have been proposed [126,127]. The list of key phase 1/phase 2 studies evaluating CAR T-cells in solid tumors is summarized in Table 7. Among the studies that reported clinical results, satricabtagene autoleucel (satri-cel)/CT041, an autologous CAR T-cell therapy targeting CLDN18.2, has shown manageable safety and promising efficacy in early clinical trials in patients with gastrointestinal cancers and has received Regenerative Medicine and Advanced Therapeutics (RMAT) designation from the US FDA [128,129,130]. However, it is to be noted that the clinical evidence generated to date is from early-phase trials and randomized phase 3 studies are needed to validate the findings from the studies. Secondly, while the outcomes are promising, the efficacy seen with solid tumor indications is still not comparable to the efficacy in hematological malignancies. Further research is needed to improve the efficacy of CAR T-cells in solid tumor indications.
Currently approved ex vivo CAR T-cell therapies are limited by manufacturing process and supply chain complexities and high costs. To address the concerns associated with manufacturing process, an in vivo CAR T-cell approach was proposed [131]. The in vivo CAR T-cell strategy involves the generation of CAR T-cells from T-cells within the patient by delivering the CAR gene to the patient through gene editing tools, such as viral vectors, mRNA and CRISPR technologies. In vivo CAR T-cell therapies are in the early stages of development compared to allogeneic and next-gen CAR T-cell therapies, and the interest in the clinical development of CAR T-cell therapies has increased recently [125,131]. Major challenges that need to be addressed in the development of in vivo CAR T-cell therapies include the development of delivery methods (e.g., viral vectors and nanocarriers) for CAR transgenes that are stable and have adequate bioavailability at the target site, and which also selectively and effectively transfect T-cells in the body [131]. Insertional mutagenesis risk is another concern with viral vectors that would need long-term monitoring of the patients for the development of secondary malignancies [132]. If successful in addressing the challenges, in vivo CAR T-cell therapies have the potential to have a paradigm-shifting impact on the treatment of cancer. The list of selected biotech companies developing in vivo CAR T-cell therapies is summarized in Table 8.
4.2. TILs
TIL therapy involves the isolation of physiologically infiltrating lymphocytes from the tumor tissues, expanding the isolated lymphocytes in vitro and infusing them back into the patients along with cytokines, such as IL-2, typically following lymphodepletion therapy similar to CAR T-cell therapy (Figure 5) [133,134]. The concept of using lymphocytes infiltrating the tumors to treat cancer was first proposed in 1986 [10]. Using a murine MC-38 colon adenocarcinoma model, researchers showed that the treatment of tumor-bearing mice with TILs and cyclophosphamide combination resulted in the elimination of liver and lung metastatic deposits, and that the addition of IL-2 to the combination of TILs and cyclophosphamide resulted in the complete elimination of liver metastatic deposits in 100% of mice and lung metastatic deposits in 50% of mice [10]. The team continued the research and extended their experience from murine models to clinical trials in melanoma patients, showing the feasibility of extracting lymphocytes from freshly resected tumors, expanding them in vitro and reinfusing them into patients [135]. The study reported objective responses in 9 out of 15 IL-2 treatment-naïve patients, and in 2 out 5 patients who previously failed IL-2 therapy and regression of tumors lungs, liver, bone, skin and subcutaneous sites. The response reportedly lasted for 2–13 months in responding patients [135].
TIL therapy is thought to be advantageous over CAR T-cell therapy, especially in solid tumors where tumor heterogeneity is a challenge. TILs have the ability to target multiple antigens, increasing the likelihood of tumor killing in solid tumors. TILs are also primed specifically for the neoantigens of the tumors, reducing the possibility of ‘off-tumor’ cytotoxicity, and are superior to CAR T-cells in their ability to infiltrate the tumor microenvironment due to the expression of chemokine receptors on their surface. The risk of severe and life-threatening cytokine release syndrome seen with CAR T-cell therapy is also low with TIL therapy [136,137,138]. Clinical research on the utilization of TILs for the treatment of cancers continued for nearly two decades and led to the approval of lifileucel in 2024 for the treatment of metastatic melanoma, which was previously treated with a PD-1 blocking antibody and, if BRAF V600-positive, a BRAF inhibitor with or without a MEK inhibitor [139].
While TIL therapy has advantages over CAR T-cell therapy, it is limited by similar manufacturing and supply chain-related challenges as seen with CAR T-cell therapy. In addition, the quality of TILs extracted is dependent on the tumor tissue used for extraction and can result in an inconsistent quality of the product. Secondly the TIL product is heterogenous and may include lymphocytes that are not primed for tumor tissues and include exhausted T-cells [139]. Furthermore, TIL therapy is not recommended for all patients, and the patients are carefully selected based on the expected quality of TILs in the tumors, the overall condition and immune status of the patient and the heterogeneity of the tumors [139]. The latest research is aimed at addressing the concerns associated with TIL therapy, and novel approaches, such as using genetically modified TILs that can secrete IL-15 to stimulate T-cell proliferation and sorting TILs based on specific T-cell markers to select the most effective T-cells, have been proposed [139]. Multiple clinical studies have been initiated to evaluate the next generation of TIL therapies and are in the early stages of clinical research (Table 9). Clinical evidence from randomized controlled trials is needed to conclude the benefits of TIL therapies in other cancer subtypes. Findings from ongoing early-stage studies could guide the design of advanced phase 3 clinical studies and eventually the approval of additional TIL therapies.
5. Conclusions
Targeting T-cell-mediated immunity has fundamentally transformed cancer therapy and represents one of the most significant advancements in contemporary oncology. Among T-cell-based strategies, the approach of targeting the T-cell activation and T-cell exhaustion, especially through the inhibition of the PD-1/PD-L1 axis, has attained the most extensive clinical success and is now firmly recognized as a standard element of treatment for various malignancies (Table 10). Anti-PD-1/PD-L1 blockers as monotherapy or as combination therapy demonstrated efficacy in several solid tumors and heme malignancies and improved patient survival (Table 10). However, the approach of targeting T-cell activation and exhaustion has been limited by the incidence of treatment-limiting adverse events and lack of response in a significant proportion of patients. The latest iterations of bispecific antibodies aiming to improve response rates demonstrated success in NSCLC and need to be evaluated in other cancer types (Table 10). T-cell-redirecting therapies such as BiTEs have the benefits of showing cytotoxic activity independent of MHC restriction and avoiding concerns of manufacturing and supply chain issues (Table 10). While most of the BiTEs have been successful in hematological malignancies, two BiTEs have shown activity in solid tumor indications, such as metastatic melanoma and SCLC. However, it is to be noted that the majority of BiTE molecules have only received conditional approval and randomized clinical trials are needed to confirm their efficacy and safety. Also, while the incidence of CRS and ICANS is comparatively low, patients need to be carefully monitored for adverse events such as infections, and labels for all BiTE molecules carry a ‘black box’ warning for severe and life-threatening CRS and ICANS (Table 11). Cell therapies, including CAR T-cell therapy and TIL therapy, aim to personalize the treatment and improve the response rates in patients. CAR T-cell therapy was successful in improving response rates, and a majority (and in some cases almost all) of the treated patients showed objective responses, which deepened over time and were durable. However, the efficacy of CAR T-cells has been limited to hematological malignancies, and clinical success in solid tumors is still awaited. Secondly, CAR T-cell therapy is associated with incidences of CRS and ICANS, which can be severe and life-threatening, prolonged cytopenias and secondary T-cell malignancies, which are highlighted through a ‘black box’ warning on the labels (Table 11). TIL therapy, on the other hand, demonstrated efficacy in metastatic melanoma patients refractory to ICB treatment and improved patient survival, but carries risk profile and ‘black box’ warning similar to CAR T-cell therapy (Table 11).
It is to be noted that all therapeutic classes of immunotherapies discussed in this review have been studied in carefully designed clinical trials. The inclusion and exclusion criteria for the clinical trials vary widely across the trials and the patients are screened based on specific biomarker criteria. For that reason, it is not possible to make cross-trial comparisons or draw conclusions on the comparative efficacy of the therapeutic classes. Though propensity-matched scores are introduced to account for the differences in selection criteria, accurate comparative claims can only be made when appropriately powered head-to-head RCTs are conducted. This review does not intend to compare the efficacy and safety across therapeutic classes, but to describe the current clinical status of the T-cell-targeting classes and discuss the latest advances within.
Despite the achievements and improvement in anti-tumor activity, the clinical benefit of T-cell-targeting therapies remains uneven across patient populations and cancer types. Further research is needed to develop the next iterations of the therapies. Among the latest advancements within the therapeutic classes, bispecific and multispecific antibody platforms, including dual immune checkpoint and immune checkpoint–angiogenesis combinations, represent an evolving class of therapies designed to simplify combination regimens and improve anti-tumor efficacy. Bispecific antibodies and BiTEs are in phase 3 clinical trials designed to confirm the clinical efficacy and safety and provide rationale for regulatory approval. Next-gen CAR T-cell therapies developed to enhance efficacy in solid tumors are in early stages of development, where the safety of the product is evaluated along with preliminary evidence of efficacy. Additional time is needed to optimize the next-gen CAR T-cell therapy and move to the confirmatory phase 3 trials. Similarly, allogeneic CAR T-cell products are still in phase 1 and early phase 2 trials, and evidence is awaited to demonstrate that the depth and durability of responses from allogeneic CAR T-cell therapies are on par with autologous CAR T-cell therapies. In vivo CAR T-cell therapies are promising and generating excitement across the research community for their potential to address several manufacturing and supply chain concerns seen with CAR T-cell therapy. However almost all of the in vivo CAR T-cell products are either in the preclinical/investigative new drug (IND) application enabling stage or early phase 1 dose finding/first-in-human (FIH) stage, and the results from the clinical trials may provide evidence for the feasibility of in vivo therapy. In conclusion, new T-cell-targeting therapies may be introduced into the clinic in the coming years and further improve the prognosis of cancer patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yang Y. Cancer Immunotherapy: Harnessing the Immune System to Battle Cancer J. Clin. Investig.20151253335333710.1172/JCI 8387126325031 PMC 4588312 · doi ↗ · pubmed ↗
- 2Rosenberg S.A. IL-2: The First Effective Immunotherapy for Human Cancer J. Immunol.20141925451545810.4049/jimmunol.149001924907378 PMC 6293462 · doi ↗ · pubmed ↗
- 3Atkins M.B. Kunkel L. Sznol M. Rosenberg S.A. High-Dose Recombinant Interleukin-2 Therapy in Patients with Metastatic Melanoma: Long-Term Survival Update Cancer J. Sci. Am.20006 S 11S 1410685652 · pubmed ↗
- 4Dutcher J.P. Schwartzentruber D.J. Kaufman H.L. Agarwala S.S. Tarhini A.A. Lowder J.N. Atkins M.B. High Dose Interleukin-2 (Aldesleukin)—Expert Consensus on Best Management Practices-2014 J. Immunother. Cancer 201422610.1186/s 40425-014-0026-031546315 PMC 6889624 · doi ↗ · pubmed ↗
- 5Mansh M. Ipilimumab and Cancer Immunotherapy: A New Hope for Advanced Stage Melanoma Yale J. Biol. Med.20118438138922180676 PMC 3238313 · pubmed ↗
- 6Shu C.A. Rizvi N.A. Into the Clinic with Nivolumab and Pembrolizumab Oncologist 20162152752810.1634/theoncologist.2016-009927026678 PMC 4861376 · doi ↗ · pubmed ↗
- 7Rotte A. D’Orazi G. Bhandaru M. Nobel Committee Honors Tumor Immunologists J. Exp. Clin. Cancer Res.20183726210.1186/s 13046-018-0937-630376854 PMC 6206712 · doi ↗ · pubmed ↗
- 8Huehls A.M. Coupet T.A. Sentman C.L. Bispecific T-Cell Engagers for Cancer Immunotherapy Immunol. Cell Biol.20159329029610.1038/icb.2014.9325367186 PMC 4445461 · doi ↗ · pubmed ↗