Functional Characterization of the SCN5A p.D372H Variant Associated with Brugada Syndrome
Xianghuan Xie, Yunqi He, Yanghui Chen, Zhiqiang Li, Yang Sun, Guangzhi Chen

TL;DR
This study investigates how a specific genetic variant in the SCN5A gene affects heart function in Brugada syndrome, a condition that increases the risk of sudden cardiac death.
Contribution
The study provides new functional evidence on how the SCN5A p.D372H variant impairs sodium channel function in Brugada syndrome.
Findings
The D372H variant causes a near-complete loss of sodium currents in HEK293 cells.
Co-transfection with wild-type SCN5A and D372H reduces current density but does not alter activation or inactivation kinetics.
The mutant protein shows reduced fluorescence intensity, suggesting decreased expression levels confirmed by Western blot and RT-qPCR.
Abstract
Background: Brugada syndrome (BrS) is a genetic cardiac arrhythmia disorder inherited in an autosomal dominant manner, characterized by ST-segment elevation in the right precordial leads (V1–V3) on electrocardiograms (ECGs). This syndrome predominantly affects young individuals with structurally normal hearts and significantly increases the risk of ventricular arrhythmias and sudden cardiac death (SCD). The most common genotype found among BrS patients is caused by variants in the SCN5A gene, which lead to a loss of function of the cardiac sodium channel Nav1.5 by different mechanisms. Methods: Plasmids containing SCN5A were constructed using PCR and site-directed mutagenesis to create the D372H variant. HEK293 cells were cultured and transfected with the WT, D372H, or a combination of both plasmids. Patch-clamp recordings assessed sodium current characteristics. Confocal microscopy…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
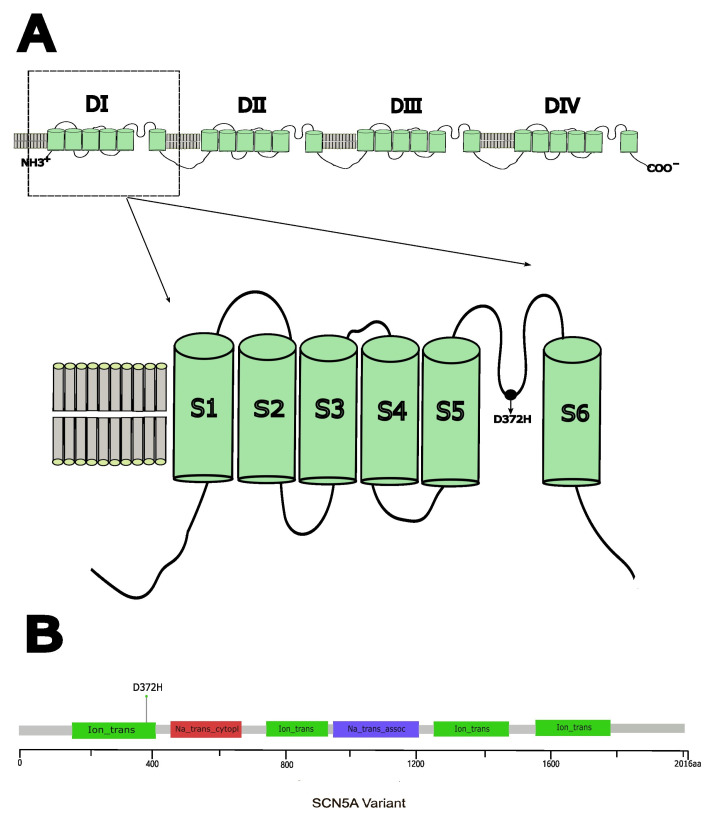 Figure 1
Figure 1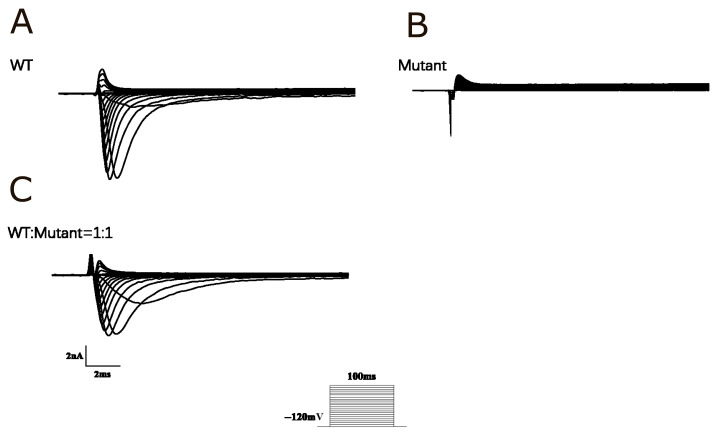 Figure 2
Figure 2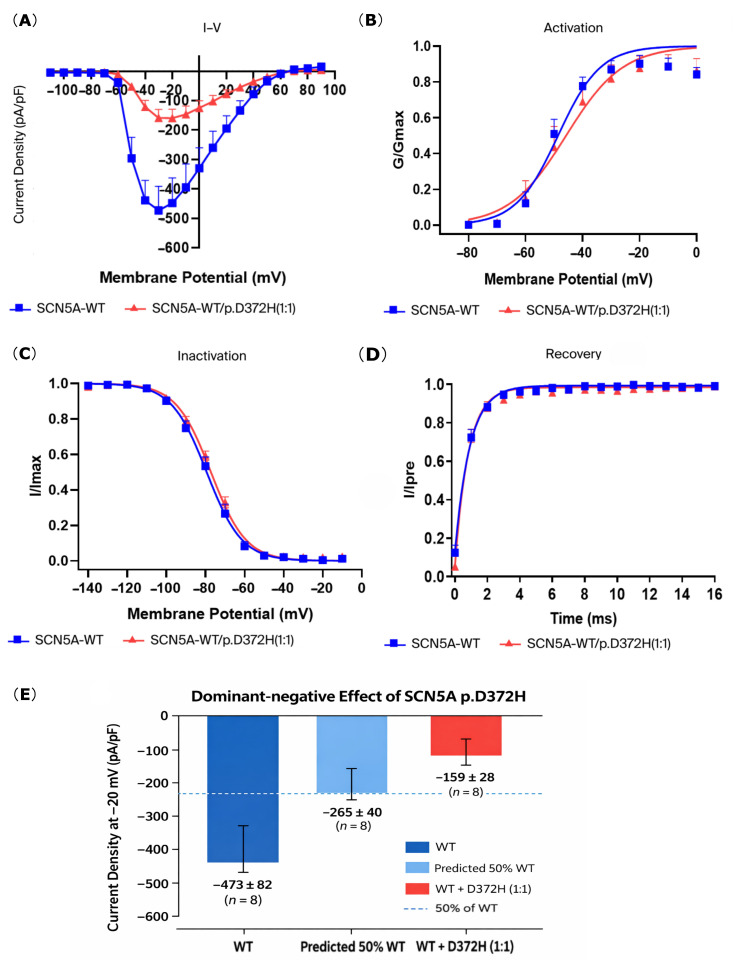 Figure 3
Figure 3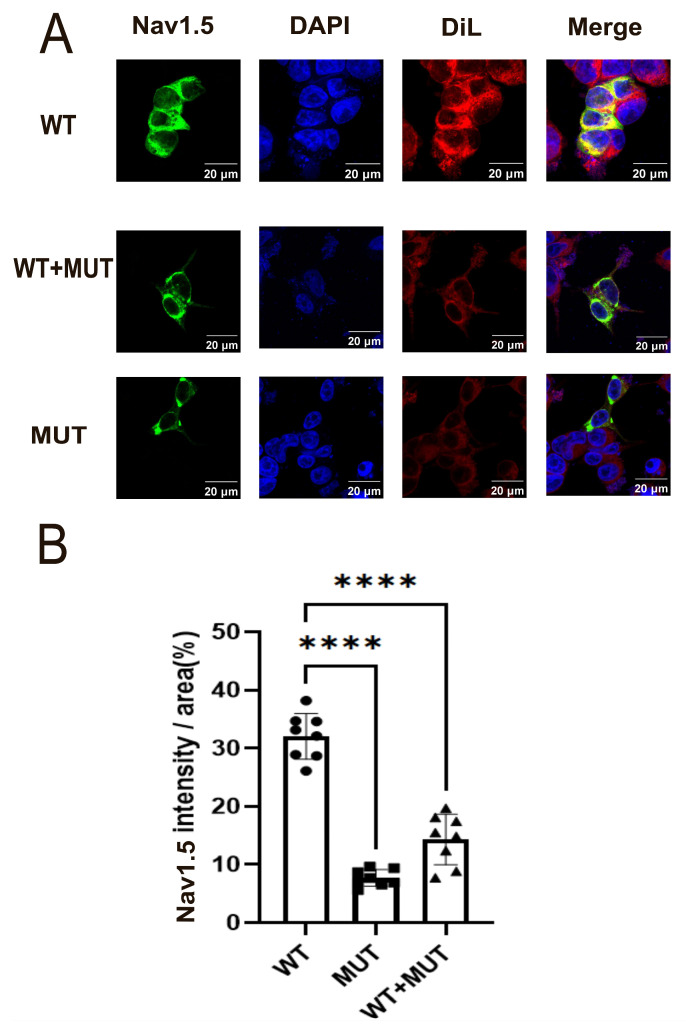 Figure 4
Figure 4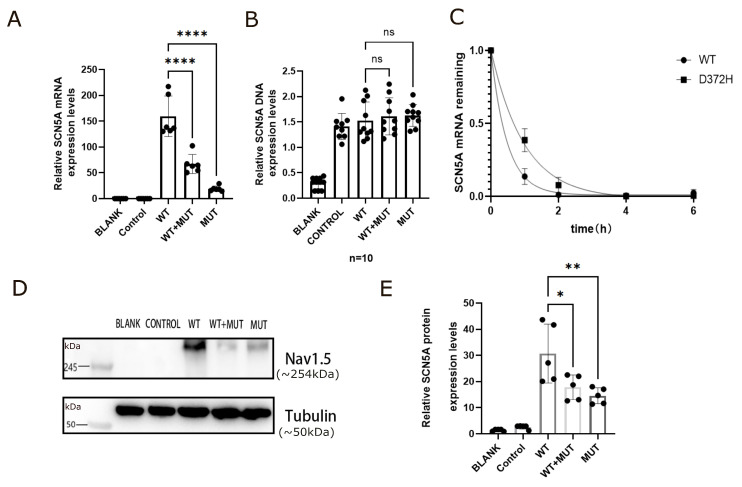 Figure 5
Figure 5- —International Science and Technology Cooperation Project of Hubei Province
- —Fund of Tongji Hospital, Huazhong University of Science and Technology
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac electrophysiology and arrhythmias · Ion channel regulation and function · ECG Monitoring and Analysis
1. Introduction
Brugada syndrome (BrS) is a genetic cardiac arrhythmia syndrome inherited in an autosomal dominant manner, characterized by distinctive electrocardiogram (ECG) changes, specifically ST-segment elevation in the right precordial leads (V1–V3) [1]. This condition primarily affects young individuals with structurally normal hearts and is associated with an increased risk of ventricular arrhythmias and sudden cardiac death (SCD) [2,3]. BrS is estimated to account for 4–12% of all sudden death cases and approximately 20% of sudden cardiac deaths in individuals under 50 years of age who have no identifiable structural heart disease [4,5]. The prevalence of BrS ranges from 1 in 2000 to 1 in 5000, with symptoms typically emerging in adulthood; the average age of SCD is reported to be 41 ± 15 years [6].
To date, more than 500 variants have been discovered in over 40 genes that may be associated with BrS [7]. These variants primarily involve genes that encode sodium [8], potassium [9], and calcium [10] channels, as well as related regulatory proteins. Nonetheless, most of these genetic variants remain functionally uncharacterized. At present, the SCN5A gene, encoding the cardiac sodium channel Nav1.5, is the only gene conclusively associated with BrS. Variants in SCN5A account for fewer than 30% of cases, leaving more than 60% of patients without an identified genetic cause [11,12].
The cardiac sodium channel α-subunit Nav1.5, encoded by the SCN5A gene, plays a critical role in generating action potentials in cardiac muscle. Nav1.5 is composed of 2016 amino acids arranged into four homologous domains (DI–DIV) [13,14]. Each of these domains contains six transmembrane α-helices (S1–S6), which are interconnected by intracellular loops. The voltage-sensing apparatus is primarily formed by the S1–S4 segments, which incorporate positively charged amino acids essential for channel activation. In contrast, the S5 and S6 segments, along with the S5–S6 loops, collectively create the channel’s pore and the selectivity filter for sodium ions (Na^+^). While SCN5A variants linked to Brugada syndrome (BrS) are dispersed throughout the entire channel structure, a combination of structural and functional analyses has identified that certain variants in specific regions are associated with distinct biophysical defects.
In our previous research, we discovered a novel variant, NM_198056.2: c.1114G > C (p.D372H) [15], situated in domain I of the Nav1.5 protein, specifically within the pore-forming intramembrane region between the S5 and S6 transmembrane segments (Figure 1) [16,17]. This variant was found in a 48-year-old male patient admitted with syncope, exhibiting a spontaneous type 1 Brugada syndrome ECG pattern characterized by concealed ST-segment elevation. The patient also suffered from sleep apnea syndrome accompanied by paroxysmal second-degree type I atrioventricular block, indicating arrhythmic events. Family history revealed the patient’s mother had a history of syncope and sudden cardiac death at age 50. Genetic testing of the patient’s siblings and daughter showed no variant and no symptoms, whereas his son carried the same variant, presenting with first-degree atrioventricular block and mild ST-segment elevation. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, the classification of a variant as pathogenic can be supported by well-established functional studies showing a deleterious effect (PS3 criterion). The p.D372H variant has been previously described in case reports as a potentially disease-associated variant; however, functional studies have not been performed to substantiate its pathogenicity and clinical significance. In 2020, Tadashi Nakajima and colleagues identified the SCN5A W374G variant, which is located in the pore loop of Domain I, in cases of severe Brugada syndrome (BrS) and is in close proximity to the D372H variant. Their functional studies indicated that the W374G variant led to a reduced current density, likely due to trafficking defects, and a depolarizing shift in steady-state activation (SSA), highlighting that variants in this region can be associated with altered channel function [18]. These findings suggest that variants within this domain significantly disrupt Na^+^ channel function; however, the impact of the D372H variant on channel activity has not been thoroughly investigated. The aim of this study was to investigate the functional consequences of the SCN5A p.D372H variant identified in a patient with Brugada syndrome. Using heterologous expression in HEK293 cells combined with electrophysiological recordings, confocal imaging, and expression analyses, we evaluated the effects of this variant on Nav1.5 channel function and protein expression. These experiments were designed to determine whether the p.D372H variant alters sodium channel properties in a manner consistent with previously reported Brugada syndrome-associated SCN5A variants, thereby providing functional evidence to aid interpretation of its potential clinical relevance.
2. Materials and Methods
This study employed an in vitro expression system to examine the functional effects of the SCN5A p.D372H variant on Nav1.5 channel properties. Electrophysiological, imaging, and molecular approaches were used to assess alterations in sodium current characteristics and channel expression.
2.1. Plasmid Construction
According to the gene sequence of SCN5A provided by PubMed (reference sequence NM_198056), we designed primers for the flanks of introns to amplify the entire SCN5A gene by polymerase chain reaction (PCR) and incorporated GFP to create a fusion gene. Plasmid encoding SCN5A (Nav1.5) was generated using a Vazyme site-directed mutagenesis kit (Vazyme Biotech Co., Ltd., Nanjing, China). according to the manufacturer’s instructions. All plasmid constructs were verified by Sanger sequencing across the entire coding region to confirm the presence of the intended variant and absence of unwanted sequence changes. The cloning of the relevant fragment of SCN5A variant (D372H) was generated using overlap extension PCR and inserted as an EcoR1/HindIII fragment into the EcoR1/HindIII site of SCN5A cDNA of pcDNA3.1-EGFP-3×FLAG, which AuGCT Biotechnology provided. The head and tail primers used are as follows:
5′-CCAAGCTGGCTAGCGTTTAAACTTAAGCTTGCCACCATGATTCCTGGTAACCGAA-3′
5′-GATGGTAGTAGAGGGATGTGGGTGCCGCTGAGAATTCTGCAGATATCCAGCACAGTGGTGCG-3′
All constructs were purified using Qiagen columns (QIAGEN Inc., Hilden, Germany). The cDNA was sequenced to confirm the presence of the correct fragment, containing the variant.
2.2. Cell Culture and Transient Expression in HEK293 Cells
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; BOSTER, Pasching, Wuhan, China) supplemented with 10% fetal bovine serum (FBS; BOSTER, Pasching, Wuhan, China) and 1% penicillin–streptomycin, and maintained at 37 °C in a humidified atmosphere containing 95% air and 5% CO_2_.
For transfection, cells were seeded in 6-well plates at approximately 60–70% confluence and transfected using HighGene transfection reagent (5 μL per well) with plasmids encoding pcDNA3.1-EGFP-3×FLAG wild-type SCN5A (WT, 2 μg), pcDNA3.1-EGFP-3×FLAG-D372H mutant SCN5A (MUT, 2 μg), or a 1:1 combination of WT and MUT plasmids (WT+MUT, 1 μg each; total DNA 2 μg).
Following transfection, cells were incubated for 48 h at 37 °C before being subjected to patch-clamp electrophysiological recordings, confocal imaging, and biochemical analyses.
2.3. Electrophysiology
Whole-cell patch-clamp recordings were performed on HEK293 cells transiently transfected with SCN5A-WT or SCN5A-D372H. Sodium currents were recorded using a patch-clamp amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) and analyzed with PatchMaster software, version 2x73 (HEKA Elektronik, Lambrecht/Pfalz, Germany). Patch pipettes were pulled from borosilicate glass capillaries with a resistance of 2–5 MΩ when filled with internal solution. Series resistance was compensated by 70–80%, and cells with leak currents > 10% of peak current were excluded from analysis. The extracellular solution contained the following components (in mM): 140 NaCl, 3.5 KCl, 1 MgCl_2_, 2 CaCl_2_, 10 glucose, 10 HEPES, and 1.25 NaH_2_PO_4_ (pH 7.4). The pipette (intracellular) solution contained the following components (in mM): 50 CsCl, 10 NaCl, 10 HEPES, 60 CsF, and 20 EGTA (pH 7.2).
2.4. Sodium Current Characteristics
The membrane was held at −120 mV, and test voltages ranging from −80 mV to +60 mV were applied for 100 ms. Representative schematic traces and voltage-clamp protocols illustrating sodium channel activation, steady-state inactivation, and recovery from inactivation under normal conditions are shown in Supplementary Figure S1. Sodium current amplitudes were converted to conductance and normalized to the maximal conductance to generate the steady-state activation curve. The data were fitted with the Boltzmann equation y = 1/[1 + exp (−(x − V1/2)/κ)] to determine the half-activation voltage (V_1/2_) and the slope factor (κ).
A double-pulse protocol was employed, with the membrane held at −120 mV and conditioning voltages ranging from −140 mV to +20 mV applied for 100 ms, followed by a 50 ms test pulse at −20 mV. Steady-state inactivation curves were obtained by normalizing the peak current during the test pulse to the maximal current and fitting the data with the Boltzmann equation to derive V1/2 and κ.
Recovery from inactivation was assessed using a triple-pulse protocol. Cells were held at −120 mV, depolarized to −20 mV for 100 ms to induce channel inactivation, and then allowed to recover at −120 mV for intervals ranging from 1 to 1000 ms before a second 50 ms pulse at −20 mV. Recovery curves were generated by normalizing the peak currents of the second pulse to the maximal current. Data were fitted using either a double-exponential function y = 1 − A1 exp(−x/τ1) − A_2_ exp(−x/τ2) or a single-exponential function y = A1 exp(−x/τ1) + B, where A1 and A2 represent the relative contributions of the fast and slow recovery components, and τ1 and τ2 denote their respective time constants. All voltage-clamp protocols and analyses were performed using the same acquisition settings across experimental groups to ensure comparability.
2.5. Confocal Microscopy
To determine the subcellular localization of NaV1.5 channels, including the wild-type (WT) and the D372H mutant, in HEK293 cells, confocal microscopy was performed on cells transfected with GFP-tagged NaV1.5 constructs. HEK293 cells were transfected with plasmids encoding GFP-tagged NaV1.5 and incubated for 48 h. After incubation, cells were washed three times with phosphate-buffered saline (PBS) to remove residual medium and unbound plasmid. The cells were then fixed with 4% formaldehyde for 30 min at room temperature, followed by three additional washes with PBS. To visualize the plasma membrane and assess membrane localization of NaV1.5, cells were incubated with the lipophilic membrane dye DiI (BOSTER, Pasching, Wuhan, China) according to the manufacturer’s instructions. Subsequently, nuclear staining was performed using DAPI (BOSTER, Pasching, Wuhan, China) for 15 min. After staining, cells were washed with PBS and mounted on glass coverslips. A microscope (Leica, Microsystems, Wetzlar, Germany) was used to visualize the localization of the fluorescent channels. Images were acquired using identical laser power, detector gain, and exposure settings across groups. Fluorescence intensity was quantified using ImageJ software, version 1.54g (National Institutes of Health, Bethesda, MD, USA). with background subtraction performed before analysis. At least three independent transfections were analyzed.
2.6. Quantitative Real-Time RT-PCR Analysis
To evaluate the effect of the SCN5A p.D372H variant on SCN5A mRNA expression, total RNA was extracted from transfected cells using TRIzol reagent (TaKaRa, Dalian, China) according to the manufacturer’s instructions. RNA concentration and purity were assessed spectrophotometrically. Complementary DNA (cDNA) was synthesized using a reverse transcription kit (Vazyme Biotech Co., Ltd., Nanjing, China). Quantitative real-time PCR (qPCR) was performed using a qPCR 900 system and ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China). Each reaction contained 2 μL cDNA template, 10 μL SYBR Green master mix (including ROX), and 200 nM of each forward and reverse primer in a final volume of 20 μL. The thermal cycling conditions were as follows: initial denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 10 s and annealing/extension at 60 °C for 30 s. Relative mRNA expression levels were calculated using the 2^−ΔΔCt^ method, with tubulin serving as the internal reference gene. All samples were analyzed in technical triplicate, and at least three independent biological replicates were performed. Each sample was analyzed in technical triplicate, and at least three independent biological replicates were performed. The primer sequences used are listed below:
SCN5A, 5′-GTCTCAGCCTTACGCACCTT-3′ (forward);
5′-GGCAGAAGACTGTGAGGACC-3′ (reverse);
TUBULIN, 5′-GACAAGACCATTGGGGGAGG-3′ (forward);
5′-ACAGGCAGCAAGCCATGTAT-3′ (reverse).
To evaluate whether the D372H variant affects SCN5A transcript stability, mRNA decay was assessed following transcriptional inhibition. Forty-eight hours after transfection (WT or D372H), cells were treated with actinomycin D (5 μg/mL) to block new RNA synthesis. Total RNA was collected at 0, 1, 2, 4, and 6 h after treatment. SCN5A mRNA levels were quantified by RT-qPCR and normalized to tubulin. Relative mRNA remaining was calculated by setting the 0-h time point as 1.0. Decay curves were fitted using nonlinear regression (one-phase exponential decay model) in GraphPad Prism, version 9.1.0 (GraphPad Software, San Diego, CA, USA).
2.7. Western Blot
To assess the expression levels of Nav1.5 channels, Western blot analysis was performed on transfected HEK293 cells. The cells were seeded in six-well plates and incubated for 48 h. Following this incubation period, IP lysate buffer was added to each well, and the plates were placed on a shaker and mixed at 4 °C for 15 min to extract total proteins. Protein concentrations were determined using the BCA quantification method. Subsequently, the proteins were separated using SDS-PAGE with a gradient of 4% to 12% (Boster, Wuhan, China). Equal amounts of total protein (20–30 µg) were loaded per lane. After electrophoresis, the proteins were transferred to a nitrocellulose membrane. The membrane was then incubated with primary antibodies against Nav1.5 (1:1000, Cat# 23016-1-AP, Proteintech, Wuhan, China) and Tubulin (1:000, Cat# A03989-1, Boster, Wuhan, China) to detect the target protein and serve as a loading control, respectively. Following incubation with the primary antibodies, the membrane was washed three times with TBST (Tris-buffered saline with Tween 20) to remove unbound antibodies. The membrane was then incubated with a goat anti-rabbit secondary antibody ((1:20,000, Cat# 926-32111, LI-COR Biosciences, Lincoln, NE, USA) to visualize the protein bands. Band intensities were quantified using ImageJ and normalized to tubulin. All experiments were repeated independently at least three times.
2.8. Statistical Analysis
All data are presented as mean ± SD (standard deviation). Statistical analyses were performed using SPSS version 17.0 (SPSS, Chicago, IL, USA) or GraphPad Prism (version 9). Data distribution was assessed for normality before applying parametric tests. Comparisons between two groups were performed using unpaired two-tailed Student’s t-tests when data were normally distributed. For comparisons involving more than two groups, one-way analysis of variance (ANOVA) followed by appropriate post hoc tests was used. A p value < 0.05 was considered statistically significant. At least three independent biological replicates were performed for each experiment.
This study was conducted in a heterologous HEK293 expression system, which allows controlled assessment of channel properties but does not fully replicate the cellular environment of human cardiomyocytes. Therefore, the observed functional effects reflect channel behavior under in vitro conditions and may not capture the full complexity of cardiac electrophysiology in vivo.
3. Results
3.1. D372H Variant Significantly Impairs Nav1.5 Sodium Current Expression
To investigate the pathophysiological mechanisms underlying the phenotype of Brugada syndrome, Nav1.5 channels were transiently expressed in HEK 293 cells, and the functional impact of the D372H variant was assessed using whole-cell patch-clamp recordings. In cells transfected with wild-type (WT) Nav1.5 channels, robust sodium currents were observed. In contrast, sodium currents were almost completely abolished in cells transfected with the D372H mutant. To further explore the effects of this variant, we designed a subsequent patch-clamp experiment where equal amounts of wild-type (WT) and D372H plasmids were co-transfected into HEK293 cells at a 1:1 ratio. Specifically, 1 μg of WT plasmid (approximately 751.7 ng/μL) and 1 μg of D372H plasmid (approximately 791 ng/μL) were used per well, keeping the total DNA amount consistent with single-transfection experiments. Whole-cell patch-clamp recordings were conducted after transfection to assess the resulting sodium currents. Analysis of the currents recorded from WT-expressing cells revealed a significant reduction in current density after co-expression with the D372H variant, as demonstrated by the I-V curve (Figure 2). This finding indicates that SCN5A-WT/p.D372H (1:1) co-expression leads to the down-regulation of channel activity, with statistical significance observed between the groups (p < 0.05).
As summarized in Table 1, co-expression of SCN5A-WT and p.D372H (1:1) resulted in a marked reduction in peak sodium current density at −20 mV compared with WT alone. The I–V relationship (Figure 3A) further demonstrated attenuated inward sodium currents across the tested voltage range in the WT+D372H group. In contrast, the voltage dependence of activation and inactivation was largely preserved, with no significant differences in V1/2, κ, steady-state inactivation, or recovery from inactivation kinetics between WT and WT+D372H channels (Table 1 and Figure 3B–D). These findings suggest that the D372H variant predominantly reduces current amplitude rather than altering channel gating. To test whether the reduction in current density in the WT+D372H (1:1) group exceeded that expected from simple haploinsufficiency, we compared the observed current density with the theoretical 50% of WT current. The predicted haploinsufficiency level was −236.55 ± 41.13 pA/pF. The observed current density in WT+D372H cells (−158.54 ± 28.37 pA/pF) was significantly lower than the predicted 50% WT level (one-sample t-test vs. predicted value, p < 0.01), supporting a dominant-negative effect of the D372H variant (Figure 3E).
3.2. Reduced Nav1.5 Expression in Cells Expressing the SCN5A-D372H Variant
To further investigate the effect of the SCN5A-D372H variant on the functional expression of the NaV1.5 sodium channel protein, confocal microscopy was performed in transfected HEK293 cells. As shown in Figure 4A, Nav1.5-WT displayed strong and concentrated green immunofluorescence, whereas the Nav1.5-D372H mutant and WT+MUT groups exhibited markedly reduced fluorescence intensity. Quantitative immunofluorescence analysis revealed that Nav1.5 fluorescence intensity was highest in the WT group, significantly reduced in the WT+MUT co-transfection group compared with the WT group (p < 0.05), and lowest in the MUT group (p < 0.05). Notably, the fluorescence intensity in the WT+MUT group was markedly decreased and closer to that observed in the MUT group than in the WT group (Figure 4B).
3.3. D372H Reduces SCN5A mRNA Abundance and Stability Independent of Transfection Efficiency
The impact of the SCN5A-D372H variant on Nav1.5 expression was first evaluated at the transcript level. RT-qPCR analysis demonstrated that SCN5A mRNA expression was highest in the WT group and significantly reduced in cells expressing D372H (**** p < 0.0001, Figure 5A). Importantly, the WT+MUT co-transfection group also exhibited a marked decrease in transcript levels compared with WT (**** p < 0.0001 Figure 5A), with expression levels substantially lower than the theoretical 50% of WT, suggesting that the mutant allele affects overall transcript abundance rather than acting through simple dilution. To exclude unequal transfection efficiency as a potential confounding factor, intracellular plasmid DNA levels were first quantified across experimental groups. No significant differences were detected among WT, WT+MUT, and MUT conditions (ns, Figure 5B), demonstrating comparable plasmid uptake. These findings indicate that the observed reduction in SCN5A mRNA levels is not attributable to differences in plasmid abundance. Having excluded unequal transfection efficiency, we next investigated whether reduced transcript levels were due to impaired mRNA stability. Transcription was inhibited using actinomycin D, and time-course analysis revealed a more rapid decay of SCN5A mRNA in D372H-expressing cells compared with WT (Figure 5C). Nonlinear regression fitting confirmed accelerated transcript degradation in the mutant group, indicating that the D372H variant compromises mRNA stability through a post-transcriptional mechanism.
Consistent with the mRNA findings, Western blot analysis demonstrated that total cellular Nav1.5 protein expression was highest in the WT group and markedly reduced in cells expressing D372H (Figure 5D,E). The uncropped full-length Western blot images with molecular weight markers are presented in Supplementary Figures S2–S5. Molecular weight markers are indicated in kilodaltons (kDa). The predicted molecular weight of native Nav1.5 is approximately 227 kDa (Figure 5D). In the present study, SCN5A constructs were expressed as GFP-tagged fusion proteins, which increased the apparent molecular weight and contributed to band migration above the 245 kDa marker. Quantitative analysis further showed that protein levels in the WT+MUT group were significantly lower than those in WT (** p < 0.01, Figure 5E) and were more comparable to the MUT group than to the expected 50% WT level.
4. Discussion
A key strength of this study lies in the comprehensive evaluation of the SCN5A p.D372H variant across molecular and electrophysiological levels, providing mechanistic insight beyond simple current reduction. Importantly, SCN5A plasmid DNA quantification demonstrated comparable plasmid abundance among WT, WT+MUT, and MUT groups, excluding unequal transfection efficiency as a potential confounder. Despite equivalent plasmid levels, SCN5A mRNA expression was significantly decreased in cells expressing D372H, and actinomycin D chase experiments revealed accelerated transcript decay, indicating that the variant impairs mRNA stability and reduces overall transcript abundance. This reduction at the RNA level was paralleled by markedly decreased Nav1.5 protein expression, as confirmed by Western blot and immunofluorescence analyses. Notably, in the WT+MUT co-expression condition, both mRNA and protein levels were substantially lower than the theoretical 50% WT level, suggesting that the mutant allele affects overall channel expression beyond simple haploinsufficiency. Functionally, whole-cell patch-clamp recordings showed near-complete abolition of sodium current in D372H-expressing cells, and co-expression with WT resulted in a reduction in peak current density that fell significantly below the predicted 50% WT value. This observation supports a dominant-negative-like effect under the present experimental conditions. Importantly, voltage-dependent activation, inactivation, and recovery kinetics were not significantly altered, indicating that the predominant defect arises from reduced channel abundance rather than altered gating behavior. Collectively, these findings demonstrate that the p.D372H variant produces a severe loss-of-function phenotype characterized by impaired transcript stability, decreased Nav1.5 expression, and markedly diminished sodium current in vitro. However, it should be noted that these functional and expression defects were characterized in a heterologous HEK293 expression system, which lacks cardiac-specific sodium channel auxiliary subunits and regulatory proteins. Consequently, the trafficking, gating, and regulatory properties of NaV1.5 observed in this model may not fully recapitulate those in native cardiomyocytes in vivo, and further validation in cardiomyocyte-based systems will be necessary to confirm the physiological relevance of these findings
Missense variants in SCN5A reduce sodium current through diverse mechanisms, including altered gating kinetics, defective trafficking, protein misfolding, and reduced transcript abundance [18]. Some variants disrupt activation or inactivation kinetics [19], whereas others impair channel maturation and surface delivery, as exemplified by ER-retained variants such as R1432G [20] and D1690N [21]. The pore-forming S5–S6 linker (pore loop) is a structurally sensitive region essential for channel folding and selectivity filter integrity, and variants within this microdomain frequently produce severe loss-of-function phenotypes, supporting its designation as a functional hotspot. The p.D372H variant identified in this study resides within the Domain I pore loop. Comparison with neighboring variants further emphasizes the pathogenic relevance of this region. For instance, the adjacent p.W374G variant causes markedly reduced INa, depolarizing shifts in activation, and impaired trafficking with partial rescue by mexiletine, while truncating variants such as p.E375X (Table 2) similarly result in profound loss-of-function phenotypes due to structural disruption. Despite close spatial proximity, p.D372H exhibits a distinct mechanistic profile. It did not significantly alter voltage-dependent gating but instead caused a marked reduction in Nav1.5 expression driven by decreased mRNA stability and reduced protein abundance. Co-expression experiments demonstrated sodium current reduction exceeding that predicted by simple haploinsufficiency, consistent with a dominant-negative-like effect under experimental conditions. Although impaired transcript stability appears to represent the primary mechanism, whether D372H additionally affects protein folding or intracellular trafficking remains to be determined. Given the structural sensitivity of the pore-loop region, subtle conformational perturbations impacting channel maturation cannot be excluded. Collectively, these findings indicate that the DI pore loop constitutes both a structural vulnerability site and a zone of mechanistic heterogeneity, where closely positioned substitutions may produce loss-of-function phenotypes through distinct molecular pathways.
The functional data presented here also provide evidence relevant to variant interpretation under the ACMG/AMP framework. The marked reduction in sodium current and decreased protein expression are consistent with the PS3 functional criterion. Its location within a functionally important pore-loop region supports PM1, and its rarity in population databases is consistent with PM2. In silico tools further predict a deleterious effect (PP3). Together, these criteria support classification of D372H as likely pathogenic; however, such classification should be interpreted in conjunction with clinical and genetic evidence. Importantly, the present findings should be viewed as mechanistic functional evidence that may assist variant interpretation rather than as confirmatory clinical proof.
From a clinical perspective, severe SCN5A loss-of-function variants have been associated with arrhythmic risk in Brugada syndrome and have informed risk stratification strategies. Variants that markedly reduce inward sodium current in vitro, such as D372H, may help inform future studies exploring how severe SCN5A loss-of-function variants relate to arrhythmic risk in Brugada syndrome, although this relationship requires confirmation in clinical and in vivo studies. As such, functional characterization of variants like D372H may ultimately help refine genotype–phenotype correlations and guide future studies of substrate-directed therapies.
5. Limitations
The results of this study indicate that the SCN5A-D372H variant, located in a critical domain of the NaV1.5 sodium channel, leads to functional impairment characterized by reduced sodium current density and decreased channel expression levels, potentially contributing to the pathogenesis of Brugada syndrome. However, several limitations should be noted. Firstly, we utilized HEK293 cells for our experiments, which, although useful for studying ion channel properties through plasmid-mediated overexpression, do not fully replicate the physiological environment of cardiac myocytes. As a result, factors such as polygenic modifiers relevant to Brugada syndrome may not be adequately represented, limiting the applicability of our findings to native cardiac tissues [22]. Secondly, earlier research has demonstrated that the W374G variant results in a decrease in sodium current density, which can be partially recovered with mexiletine (MEX) [18]. This suggests that trafficking defects associated with SCN5A variants in the pore-S6 regions of Domains I, III, and IV can be rescued by MEX, whereas variants in Domain II do not exhibit this property. Nevertheless, further investigation is necessary to clarify the potential therapeutic role of MEX in rescuing the sodium current loss associated with the D372H variant in Brugada syndrome.
6. Conclusions
In conclusion, our study demonstrates that the SCN5A p.D372H variant markedly impairs Nav1.5 channel function and expression in vitro. These findings provide mechanistic insight and support a potential role of this variant in altering sodium channel function, which may be relevant to Brugada syndrome, though further clinical and in vivo studies are required to confirm its pathogenic role. These findings underscore the importance of further research to explore therapeutic strategies that could mitigate the functional deficits associated with this and similar variants.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mizusawa Y. Wilde A.A.M. Brugada syndrome Circ. Arrhythmia Electrophysiol.2012560661610.1161/CIRCEP.111.96457722715240 · doi ↗ · pubmed ↗
- 2Marsman E.M.J. Postema P.G. Remme C.A. Brugada syndrome: Update and future perspectives Heart Br. Card. Soc.202210866867510.1136/heartjnl-2020-31825834649929 · doi ↗ · pubmed ↗
- 3Brugada J. Campuzano O. Arbelo E. Sarquella-Brugada G. Brugada R. Present status of brugada syndrome: JACC state-of-the-art review J. Am. Coll. Cardiol.2018721046105910.1016/j.jacc.2018.06.03730139433 · doi ↗ · pubmed ↗
- 4Saffitz J.E. Structural Heart Disease, SCN 5A Gene Mutations, and Brugada Syndrome: A Complex Ménage à Trois Circulation 20051123672367410.1161/CIRCULATIONAHA.105.58714716344397 · doi ↗ · pubmed ↗
- 5Antzelevitch C. Brugada P. Borggrefe M. Brugada J. Brugada R. Corrado D. Gussak I. Le Marec H. Nademanee K. Perez Riera A.R. Brugada syndrome: Report of the second consensus conference: Endorsed by the heart rhythm society and the european heart rhythm association Circulation 200511165967010.1161/01.CIR.0000152479.54298.5115655131 · doi ↗ · pubmed ↗
- 6Rattanawong P. Mead-Harvey C. Fatunde O.A. Van Der Walt C. Ko N.K. Hooke P. Yinadsawaphan T. Kulthamrongsri N. Shen W.-K. Sorajja D. Prevalence and incidence of type 1 brugada pattern: A 30-year experience at mayo clinic Mayo Clin. Proc.2025100809310.1016/j.mayocp.2024.05.02839641715 · doi ↗ · pubmed ↗
- 7Frosio A. Micaglio E. Polsinelli I. Calamaio S. Melgari D. Prevostini R. Ghiroldi A. Binda A. Carrera P. Villa M. Unravelling Novel SCN 5A Mutations Linked to Brugada Syndrome: Functional, Structural, and Genetic Insights Int. J. Mol. Sci.2023241508910.3390/ijms 24201508937894777 PMC 10606416 · doi ↗ · pubmed ↗
- 8Brunklaus A. Feng T. Brünger T. Perez-Palma E. Heyne H. Matthews E. Semsarian C. Symonds J.D. Zuberi S.M. Lal D. Gene variant effects across sodium channelopathies predict function and guide precision therapy Brain J. Neurol.20221454275428610.1093/brain/awac 006PMC 989719635037686 · doi ↗ · pubmed ↗