Involvement of c-Myc/WWP1/TRIM65 Axis in Renal Fibrosis
Sonia Mazumder, Cody Gifford, Jiaqi Tang, Fortis Gaba, Varsha Mondal, Roel Goldschmeding, Rohan Samarakoon, Paul J. Higgins

TL;DR
This study identifies a new pathway involving c-Myc, WWP1, and TRIM65 that contributes to kidney fibrosis, suggesting WWP1 could be a target for treating chronic kidney disease.
Contribution
The paper discovers a novel c-Myc/WWP1/TRIM65 axis involved in renal fibrosis and identifies WWP1 as a potential therapeutic target.
Findings
WWP1 and TRIM65 are induced in human and experimental chronic kidney disease.
Silencing WWP1 or TRIM65 reduces fibrotic signaling in renal cells.
Restoring BMP-7 or SMAD5 signaling disrupts the fibrotic cascade and reduces fibrosis.
Abstract
Maladaptive tubular repair is a major contributor to fibrosis and chronic kidney disease (CKD), yet the molecular regulators of this process remain poorly understood. We report that the E3 ubiquitin ligases WWP1 and TRIM65 are novel regulators of tubular fibrosis. Both ligases were markedly induced in human and experimental CKD. WWP1 induction correlates with declining renal function in humans, highlighting the potential clinical relevance of WWP1. Profibrotic factor PAI-1 promotes a robust induction of WWP1 and TRIM65 in both primary human renal epithelial cells as well as cell line (HK-2). The silencing of WWP1 or TRIM65 significantly attenuated PAI-1-induced fibrotic signaling. Mechanistically, PAI-1 triggers a signaling cascade in which suppression of the regenerative BMP-7/SMAD5 pathway permits c-Myc induction, resulting in WWP1 and TRIM65 upregulation. The elevated expression of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
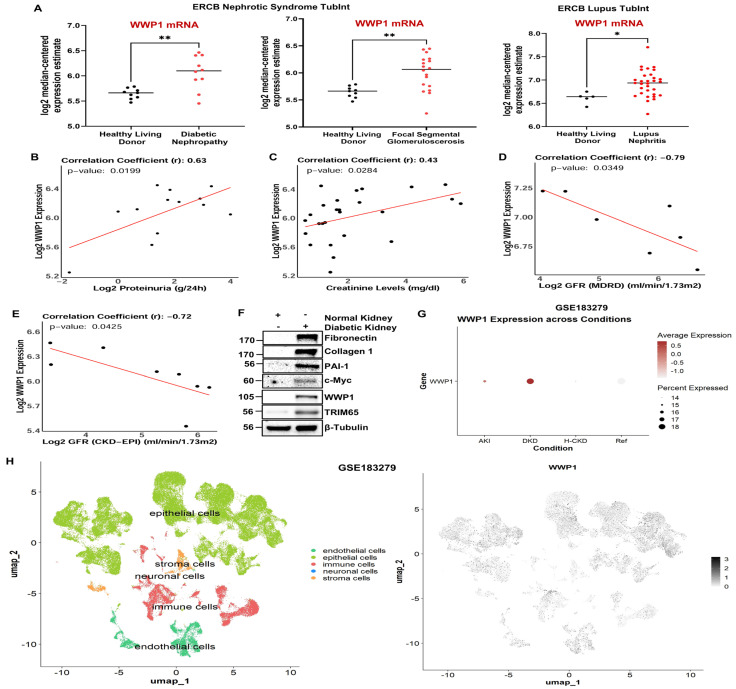 Figure 1
Figure 1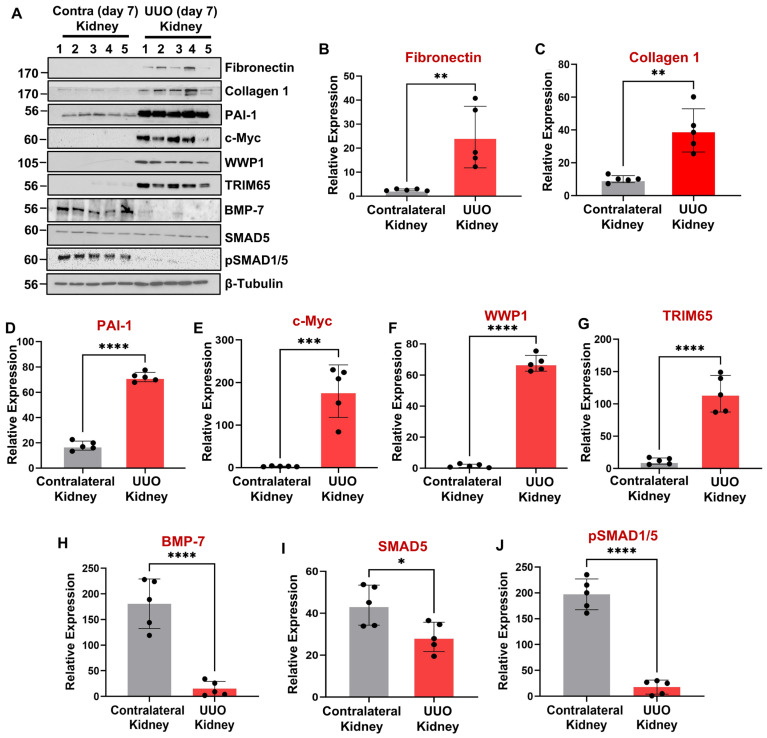 Figure 2
Figure 2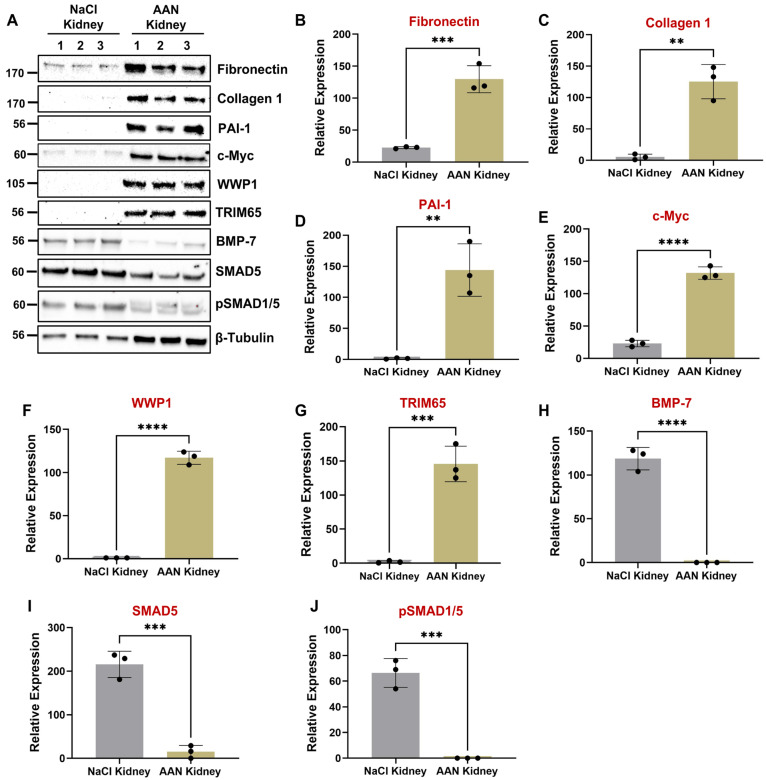 Figure 3
Figure 3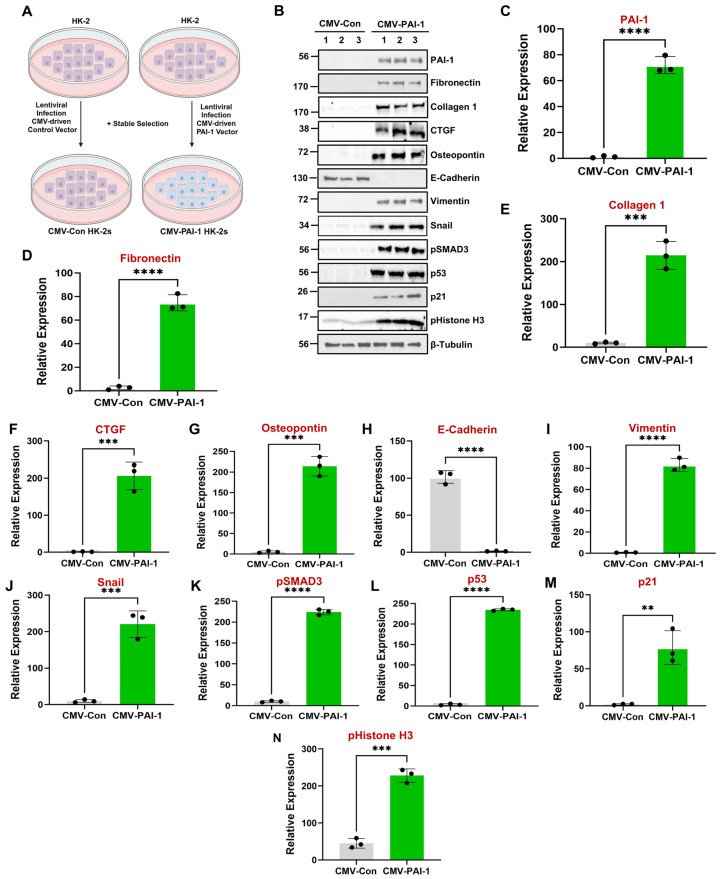 Figure 4
Figure 4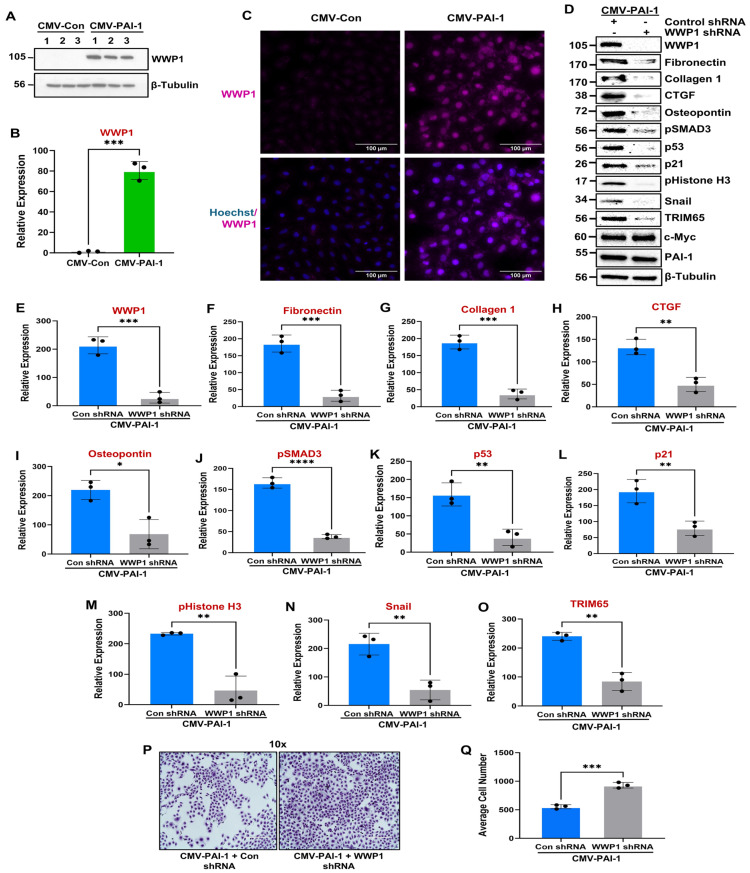 Figure 5
Figure 5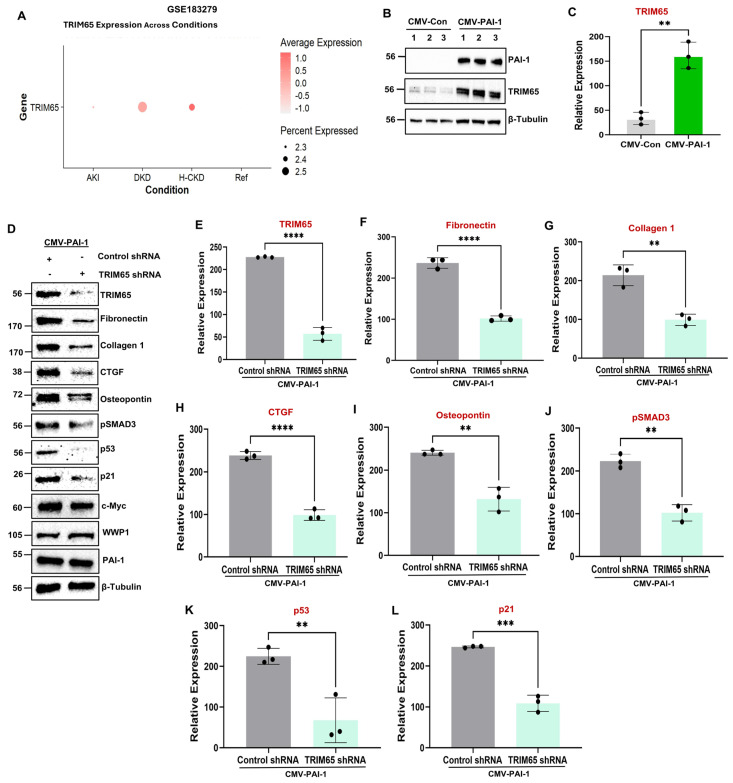 Figure 6
Figure 6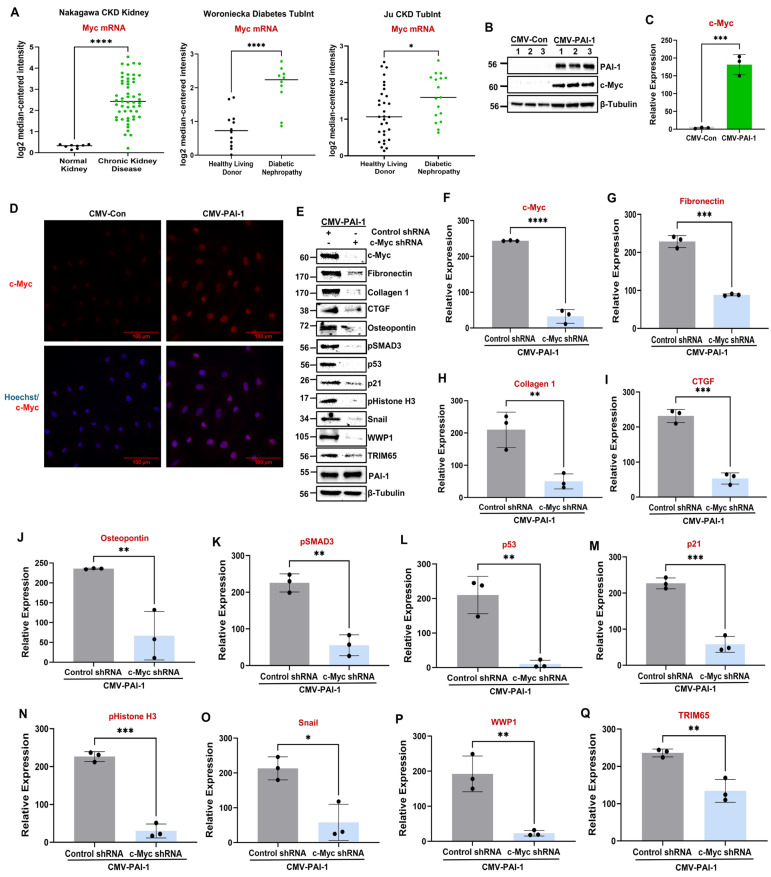 Figure 7
Figure 7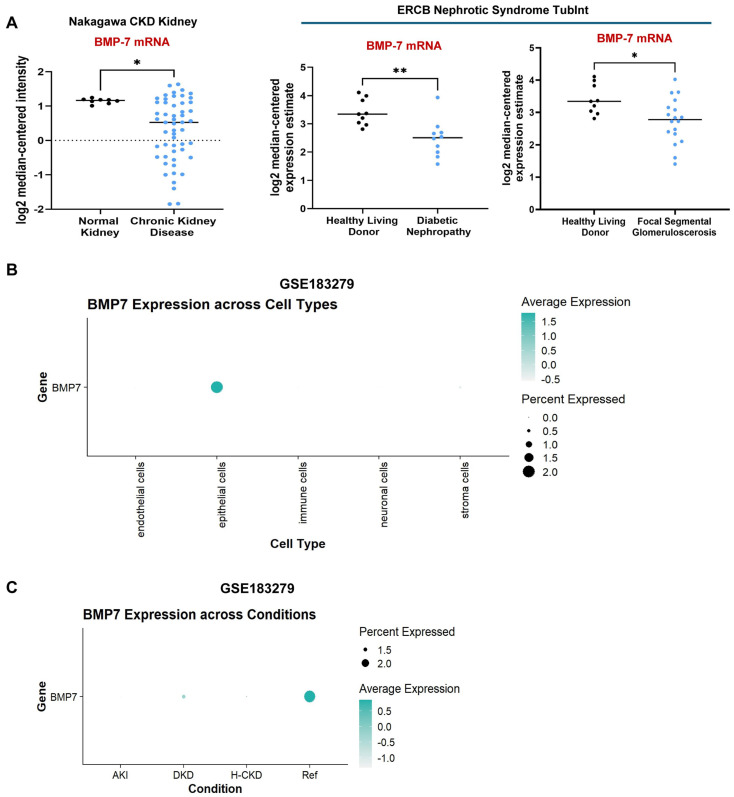 Figure 8
Figure 8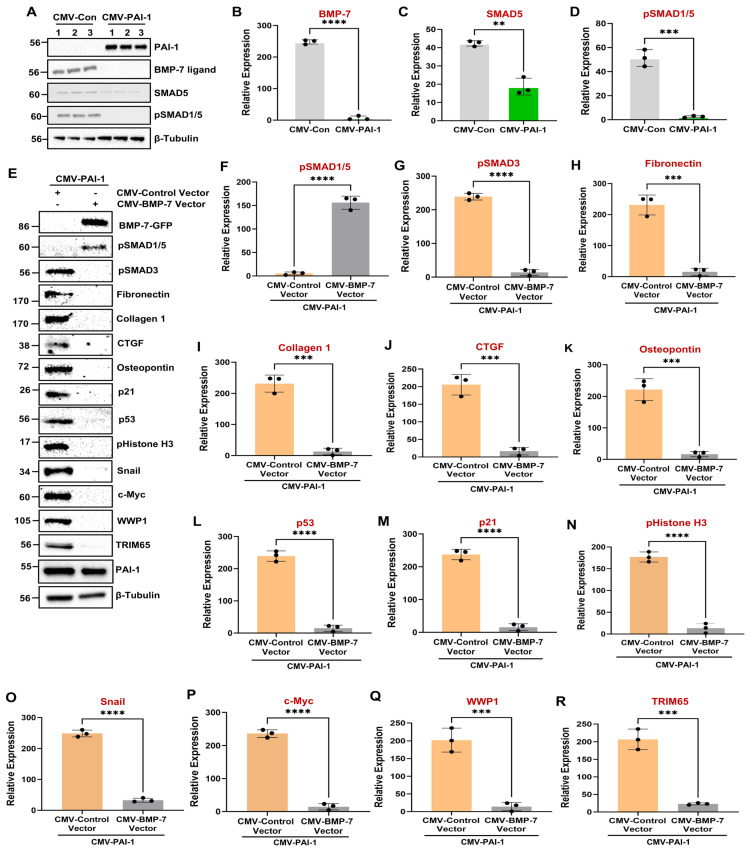 Figure 9
Figure 9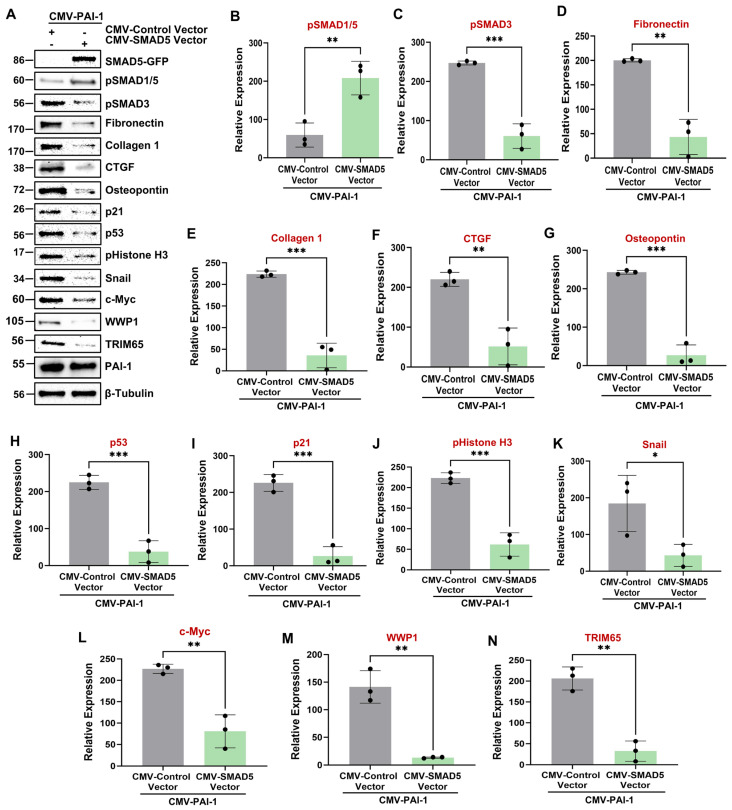 Figure 10
Figure 10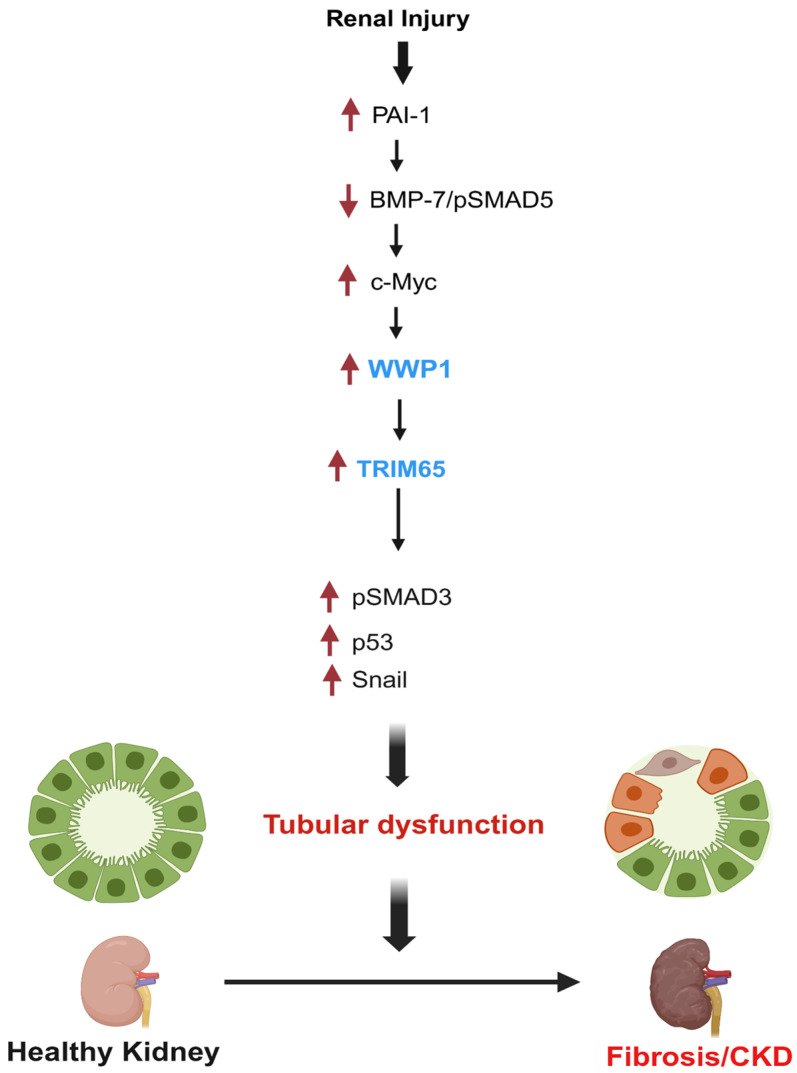 Figure 11
Figure 11- —NIH
- —Capital District Medical Research Institute
- —Friedman Family Research Fund
- —Charlotte Graver Foundation
- —John Faunce & Alicia Tracy Roach Fund
- —Edith Dickstein & Sylvan Kessler Estate Foundation
- —Butler Family Mesothelioma Research Fund
- —Mueller Family Cancer Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsUbiquitin and proteasome pathways · Chronic Kidney Disease and Diabetes · NF-κB Signaling Pathways
1. Introduction
Chronic kidney disease (CKD) is now regarded as a hidden epidemic affecting around 40 million individuals in the US and 850 million people worldwide [1,2,3,4,5]. Irrespective of etiology, recurring kidney injury triggers tubular maladaptive repair (tubular dysfunction), which culminates in interstitial fibrosis, CKD, and end-stage renal disease (ESRD) [6,7]. Due to the lack of effective antifibrotic drug regimens, inadequate renal replacement availability, and the high prevalence of risk factors, CKD is projected to become the fifth leading cause of death by 2040, further adding to the increasing global medical and financial burden [1,2,3,4,5,8]. Tubular maladaptive repair in response to repetitive renal injury triggers epithelial dedifferentiation, G2/M arrest, induction of a senescence-like secretory phenotype (SASP), inflammatory cytokine secretion and pathogenic epithelial-fibroblast crosstalk leading to myofibroblast accumulation, interstitial fibrosis, and progressive nephron loss [9,10,11,12].
The involvement of the ubiquitin–proteasome system (UPS) during CKD progression is far less understood, although aberrant expression and/or activity of UPS components such as E3 ligases are linked to the progression of neoplastic and fibrotic diseases [13,14]. Consistent with the causative role of certain E3 ligases in CKD [13,14,15,16,17], the clinical-grade proteasome inhibitor Bortezomib protects mice from progressive aristolochic acid nephropathy (AAN) [18]. E3 ligases are involved in protein ubiquitination, although their functions are not limited to protein degradation [19]. The WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) is highly upregulated in numerous cancers and drives tumor progression [20,21]. Although mice with WWP1 global ablation develop less cardiac hypertrophy and attenuated fibrotic ECM remodeling following pressure overload [22], WWP1 involvement in progressive nephropathies is not defined. The tripartite motif-containing (TRIM) family of E3 ubiquitin ligase proteins are involved in diverse cellular processes including cell cycle regulation and oncogenesis [23,24]. Among them, TRIM65 upregulation has been linked to carcinogenesis [25]. Although global silencing of TRIM65 in mice affords protection from renal fibrosis in general [26], its specific role as well as the mechanism of its induction during renal tubular injury is not well studied. A critical gap in the field is the identification of E3 ubiquitin ligases that integrate upstream fibrotic signals (such as transforming growth factor-β1/TGF-β1 and plasminogen activator inhibitor-1/PAI-1) and the loss of renal regeneration pathways (e.g., bone morphogenetic protein-7/BMP-7) that contribute to the establishment of a maladaptive renal epithelial state.
Among profibrotic networks, TGF-β1 is a well-known promoter of CKD via SMAD3 and non-SMAD3 transcription-factor-dependent mechanisms [15,27,28,29,30,31,32,33,34,35]. The expression of PAI-1, a major TGF-β1 target, is persistently upregulated in renal tubules regardless of renal insults [27,36]. Mice with PAI-1 ablation are, indeed, protected from renal fibrosis resulting from ureteral obstruction and diabetes, establishing PAI-1 as a CKD-promoting entity [37,38]. Although previous studies linked PAI-1 upregulation to extracellular matrix stabilization during fibrotic diseases, we were the first to implicate PAI-1 as a novel inducer of tubular maladaptive repair via p53 and pSMAD3 hyperactivation [39], two key transcription factors critical for renal maladaptive repair [35,40]. Activation of the transcription factor c-Myc in the kidney promotes fibrotic gene expression and CKD progression [41]. Whether c-Myc acts as a bridge between PAI-1 and downstream post-translational regulators such as E3 ligases, however, has not been addressed.
In contrast, bone morphogenetic protein-7 (BMP-7) and its downstream effectors SMAD1/5 comprise a well-established antifibrotic pathway in the kidney. BMP-7 promotes regenerative repair and counteracts TGF-β1–induced fibrosis by activating SMAD1/5 signaling [42,43,44,45]. During renal injury, loss of BMP-7/SMAD1/5 signaling shifts the balance toward fibrosis via hyperactivation of TGF-β1/SMAD3 pathway [46,47]. Whether BMP-7 suppression affects the activation of other profibrotic networks, including c-Myc and E3 ligases, remains unclear.
Collectively, these findings highlight a complex and poorly understood interplay among profibrotic (TGF-β1/PAI-1/c-Myc) and regenerative (BMP-7/SMAD1/5) networks that govern tubular cell fate and fibrosis progression during kidney injury. However, the mechanisms by which these opposing pathways converge on post-translational regulators such as E3 ubiquitin ligases remain unclear. To address this gap, we hypothesize that WWP1 and TRIM65 are induced by PAI-1- and c-Myc-dependent mechanisms during maladaptive tubular repair and that suppression of the BMP-7 signaling facilitates their activation. We investigated WWP1 and TRIM65 expression and regulation in experimental models of kidney fibrosis and human CKD transcriptomic datasets and examined their relationships to upstream fibrogenic and regenerative pathways to define novel mechanisms driving CKD progression.
2. Materials and Methods
2.1. Sex as a Biological Variable and Exclusion/Inclusion Criteria
All animal studies included both genders. All human samples from the Nephroseq (https://www.nephroseq.org (accessed on 5 June 2024)) database and single cell renal atlas (Accession No.: GSE183279) [48] include both males and females. In this study, sex was not considered as a biological variable. No exclusion and inclusion criteria were used for animal or cell culture studies.
2.2. Animals
C57Bl/6 mice were purchased from Charles River Laboratories (https://www.criver.com/ (Wilmington, MA, USA)). The mice were maintained in pathogen-free SPF conditions with a 12 h light–dark cycle and a constant environmental temperature. Mice were fed a standard pellet diet with free access to water. The sample sizes for our animal experiments were chosen based on our prior experience and previously published studies [30,49,50,51].
2.3. Unilateral Ureteral Obstruction (UUO) Nephropathy
Briefly, C57Bl/6 mice (6–8-weeks-old with a similar weight range) were anesthetized using isoflurane inhalation and under aseptic conditions, a small incision was made in the flank region to expose the kidneys. The ureter of the left kidney was ligated with two 5-0 silk sutures to generate a renal obstruction, and the unmanipulated contralateral right kidney served as a control. All animals were euthanized on day 7 post-ligation, and obstructed (UUO) and contralateral kidneys were harvested for biochemical analysis. The approximate mortality rate for the UUO procedure is 20%. The experiments were carried out in accordance with the European Community Guidelines and in compliance with the protocols approved (Approval no.: 2009.II.11.129) by the Animal Experiments Committee (DEC) of the University of Utrecht (Utrecht, The Netherlands).
2.4. Aristolochic Acid Nephropathy (AAN)
C57Bl/6 mice (6–8-weeks-old with a similar weight range) were intraperitoneally injected with aristolochic acid (AA) sodium salt (5 mg/kg body weight dissolved in distilled water; A9451; Sigma-Aldrich, St. Louis, MO, USA) once a day for 5 consecutive days (AAN group), while control mice received a NaCl vehicle (NaCl group). At 25 days, following the initial injections, all mice were euthanized, and the kidneys were harvested for biochemical analysis. The mortality rate for mice subjected to AAN is around 10%. All animals were randomly assigned to experimental (AAN group) and control groups (NaCl group). The experiments were conducted in compliance with the ethical guidelines and protocols approved (Approval no.: 2011.II.05.086) by the Animal Experiments Committee (DEC) of the University of Utrecht (Utrecht, The Netherlands).
2.5. Human Kidney Specimens
Diabetic renal tissue was obtained from a donor deemed unsuitable for transplantation due to advanced diabetic nephropathy, characterized by histological features such as Kimmelstiel–Wilson nodules. Normal human kidney tissue was collected from the non-neoplastic region of a nephrectomy specimen from a patient undergoing surgery for renal cell carcinoma. All patient samples were leftover body material from clinical biopsies (or resections) and were collected according to the ethical guidelines of the University of Utrecht (Utrecht, The Netherlands). Samples were anonymized, allowing us to use this residual tissue for research purposes without the consent of the patients.
2.6. Cell Culture and Generation of Stable Single and Double Transductants
Human renal proximal tubular epithelial cells (HK-2) (CRL-2190- ATCC, Manassas, VA, USA) were grown in DMEM media M (1X) + GlutaMAX-I (10567-014, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 5% FBS (fetal bovine serum) (16000-044, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and 5 units/mL penicillin + 5 µg/mL streptomycin (15140-122, Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Primary human tubular epithelial cells (RPTECs, #CC-2553) were purchased from Lonza Biosciences (Walkersville, MD, USA). In order to generate PAI-1 stable transductants, semiconfluent HK-2 cultures were infected with lentiviruses carrying a cytomegalovirus (CMV) promoter–driven PAI-1 cDNA construct (termed CMV-PAI-1) (LPP-F0606-Lv105) or an empty vector (termed CMV-Con) (LPP-NEG-Lv105) (GeneCopoeia, Rockville, MD, USA) in the presence of Polybrene at 5 μg/mL (sc-134220, Santa Cruz Biotechnology, Dallas, TX, USA) in 5% FBS/DMEM for 24 h. After a 24 h recovery, the cells were subjected to stable selection in complete medium (1X DMEM + GlutaMAX-I; 5% FBS; 5 units/mL penicillin + 5 µg/mL streptomycin) containing 5 µg/mL puromycin dihydrochloride (sc-108071, Santa Cruz Biotechnology, Dallas, TX, USA). Media were changed every 3 days to maintain the appropriate selection pressure. To generate RPTEC transgenic cells overexpressing PAI-1, cells were transiently infected with the LPP-F0606-Lv105 or control LPP-NEG-Lv105 lentiviral constructs (GeneCopoeia, Rockville, MD, USA) for 5 days prior to lysate collection. PAI-1 induction in transgenic cell types was confirmed by immunoblot analysis. Due to the proliferative defects following stable PAI-1 expression in HK-2 epithelial cells, CMV-PAI-1 cultures were initially seeded at a density three times higher than CMV-Con plates to achieve equivalent cell numbers at the time of lysate generation for Western blot analysis.
To generate stable double-transgenic epithelial cell lines for BMP-7 and SMAD5 expression rescue experiments, semiconfluent PAI-1 stable transductants were reinfected with CMV-BMP-7-GFP (LPP-A0309-Lv122) and CMV-SMAD5-GFP (LPP-I0510-Lv122) lentiviral particles, respectively, or CMV-Control vector lentiviral particles (LPP-NEG-Lv105) (GeneCopoeia, Rockville, MD, USA) for 1–2 days in complete medium with Polybrene at 5 μg/mL before stable selection. Successful restoration of BMP-7 and SMAD5 expression rescue was confirmed by immunoblotting for the GFP. To create stable double-transgenic epithelial cell lines with WWP1, c-Myc, and TRIM65 silencing in the context of PAI-1 upregulation, 40% confluent CMV-PAI-1 cultures were reinfected with either control (sc-108080) or WWP1 (sc-40366-V), c-Myc (sc-29226-V), and TRIM65 (sc-93883-V) short hairpin RNA (shRNA) lentiviral constructs (Santa Cruz Biotechnology, Dallas, TX, USA) for 1–2 days in complete medium containing Polybrene at 5 μg/mL followed by stable selection. WWP1, c-Myc, and TRIM65 depletion in PAI-1-overexpressing double-transgenic cultures were confirmed by Western blot analysis for respective proteins. WWP1-GFP expression lentiviral particles were purchased from Origene (RC206243L4, Rockville, MD, USA). All cells were routinely checked for contamination. Where appropriate, the blinding of the investigators was used as a strategy to reduce experimental bias. All cell culture experiments were repeated three times.
2.7. Western Blot Analysis and Antibodies
Cells were lysed in Laemmli sample buffer containing 5% β-mercaptoethanol and boiled for 5 min. Kidney tissues were extracted in 2% SDS/PBS. For SDS-PAGE electrophoresis, approximately 20–30 μg of protein from each sample was loaded into Bio-Rad Mini-PROTEAN TGX 10% pre-cast gels (4561034; Bio-Rad Laboratories, Hercules, CA, USA). Separated proteins were transferred to nitrocellulose membranes (1620112; Bio-Rad Laboratories, Hercules, CA, USA) and blocked in 5% non-fat dry milk in 0.05% Triton-X 100/PBS buffer. The membranes were then probed with the following primary antibodies overnight: rabbit anti-fibronectin (1:50,000; ab2413), rabbit anti-tubulin (1:20,000; ab6046), rabbit anti-phospho-SMAD3 (1:1000; ab52903), rabbit anti-WWP1 (1:1000; ab43791), rabbit anti-c-Myc (1:2000; ab32072), rabbit anti-BMP-7 (1:1000; ab84684), rabbit anti-osteopontin (1:2000; ab8448) from Abcam (Cambridge, UK), rabbit anti-vimentin (1:10,000; sc5565), mouse anti-p53 (1:500; sc126), mouse anti-GFP (1:500; sc-9996), goat anti-CTGF (1:1000; sc14939) from Santa Cruz Biotechnology (Dallas, TX, USA), rabbit anti-p21 (1:1000; 2947), rabbit anti-SMAD5 (1:1000; 12534), rabbit anti-phospho-SMAD1/5 (1:1000; 9516), rabbit anti-snail (1:1000; 3879), rabbit anti-phospho Histone H3 (1:1000; 9701) from Cell Signaling Technology (Danvers, MA, USA), mouse anti-E-cadherin (1:1000; 610181) from BD Biosciences, mouse anti-TRIM65 (1:250; H00201292-B01P) from Novus Biologicals, rabbit anti-collagen type 1 (1:5000; 234167) from Calbiochem, and rabbit anti-PAI-1 (1:3000) as described previously [39]. Membranes were washed three times and incubated with appropriate HRP-conjugated secondary antibodies (goat anti-rabbit, 31460; goat anti-mouse, 31430), both from Thermo Fisher Scientific (Waltham, MA, USA), and mouse anti-goat, sc-2354 from Santa Cruz Biotechnology (Dallas, TX, USA) at a dilution of 1:1000–5000 for 1 h at room temperature. Following three consecutive washes in 0.05% Triton-X 100/PBS, membranes were incubated in ECL (Bio-Rad Clarity Western ECL Substrate; 170-5061; Bio-Rad Laboratories, Hercules, CA, USA) and imaged with a ChemiDoc^TM^ Imaging system (Bio-Rad Laboratories, Hercules, CA, USA). Relative protein levels were quantified using the ImageJ software package (version 1.53t) (National Institute of Health, MD, USA). All the antibodies are commercially available and validated by the manufacturers and/or us.
2.8. Immunofluorescence
CMV-Con and CMV-PAI-1 cells were plated on coverslips in a 6-well culture chamber and allowed to reach 70–80% confluency. Cells were then washed 2 times with 1X PBST (1X PBS; Dulbecco’s phosphate buffer saline, 14190-144, Gibco, Thermo Fisher Scientific, Waltham, MA, USA + 0.05% tween-20; P7949, Sigma-Aldrich, MO, USA) followed by fixation with 4% paraformaldehyde (J61899.AP, Thermo Fisher Scientific, Waltham MA, USA) for 5 min and permeabilized with 0.5% Triton X-100 (T9284, Sigma-Aldrich, MO, USA) in 1X PBST for 10 min at room temperature. The cells were then blocked in 1% BSA in 1X PBST for 60 min in a humidity chamber (incubator) (Eppendorf, Hamburg, Germany) with occasional agitation followed by incubation with primary antibodies to rabbit anti-WWP1 (1:250; Abcam-ab43791) and rabbit anti-c-Myc (1:250; Abcam-ab32072) overnight at 4 °C. The cells were then washed three times (5 min each) before incubating with Alexafluor 647 (1:1000; A-21245; Invitrogen, CA, USA) and Alexafluor 594 (1:1000; A-11037; Invitrogen, CA, USA) secondary antibody, respectively, for 1 h in a humidity chamber in the dark. After washing, Hoechst stain in 1X PBST (1:15,000; H-3569, Molecular probes, OR, USA) was added to the cells for nuclear staining and incubated for 5 min with agitation at room temperature before coverslips were mounted using Prolong^TM^ Diamond anti-fade mounting media (P36961; Invitrogen, CA, USA). After 24 h post-curation, images were acquired at 40× magnification using a Nikon Eclipse Ti2-E inverted microscope operated by NIS elements software (version 5.41.2.17110) (Tokyo, Japan).
2.9. Analysis of Single Cell RNA-Seq Dataset on Human Renal Heathy and Diseased Specimens
One human renal atlas (Accession No.: GSE183279) [48], including specimens from several renal disease and healthy patients’ kidneys, was used in our study, and the original cell annotation provided by authors was used for downstream analysis. Dataset GSE183279 is one of the biggest renal cellular atlases, including 58 reference tissues and 52 diseased tissues. The raw count matrix and annotated metadata for single cell RNA sequencing dataset (Accession No.: GSE183279) were downloaded from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/ (accessed on 4 February 2025), followed by the creation of a Seurat object by merging both the count matrix and annotated data together. A series of downstream quality control steps were performed as listed by the authors [48], including filtering out low-quality cells or potential doublets (a cut-off of <50% mitochondrial reads per cell; >500 and <5000 genes per cell were applied), normalization, scaling, and identifying highly variable genes. All highly variable genes were utilized for linear dimensionality reduction (PCA, principal component analysis). Uniform manifold approximation and projection (UMAP) non-linear dimensionality reduction was performed using the top 50 principal components identified. Followed by dimensionality reductions, UMAP plots for annotated cell clusters and target gene expression were generated. All data processing, analysis, and visualization steps were performed using the R programming language (R software: version R 4.4.3) in the R studio environment (version 2024.12.1+563). Key packages utilized in this process included Seurat (version 5.2.1), ggplot2 (version 3.5.1), and dplyr (version 1.1.4). This study included all specimens described in the dataset (Accession No.: GSE183279) by the authors [48].
2.10. Analysis of Renal Disease Datasets from Nephroseq
Renal transcriptomics from the Nephroseq database (https://www.nephroseq.org (accessed on 5 June 2024)) were utilized for analyzing the mRNA expression of genes of interest. A minimum fold change of 1.5 and p-value of <0.05 were applied. The log2 median-centered intensity or expression estimates for both the healthy control and disease groups for the respective datasets (ERCB Nephrotic Syndrome TubInt, ERCB Lupus TubInt, Nakagawa CKD Kidney [52], Woroniecka Diabetes TubInt [53], and Ju CKD TubInt [54]) were downloaded from the Nephroseq database (https://www.nephroseq.org (accessed on 5 June 2024)). The expression estimates were then utilized to generate the histograms using GraphPad Prism (GraphPad Software, Inc., Boston, MA, USA) (version 10). The graphs and correlation analysis between WWP1 log2 expression values and GFR or proteinuria or serum creatinine levels were performed using the R programming language (version R 4.4.3) in the R studio environment (version 2024.12.1+563).
2.11. Statistical Analysis
Statistical differences between the biological groups were assessed using two-tailed Student’s T-tests or a one-way ANOVA followed by a Tukey’s post-hoc test, as appropriate. A p-value of less than 0.05 was considered statistically significant. All histograms and statistical plots were generated using GraphPad Prism (version 10; GraphPad Software, Inc., Boston, MA, USA). A Pearson correlation analysis was used to assess the relationship between WWP1 expression and GFR, serum creatinine levels, and proteinuria in human CKD patients using the R programming language (R software: version R 4.4.3) in the R studio environment (version 2024.12.1+563).
3. Results
3.1. WWP1 and TRIM65 E3 Ligases Are Highly Upregulated in Humans During Renal Injury, and Renal Upregulation of WWP1 Positively Correlates with Chronic Kidney Disease
Although recent studies suggest that proteosome inhibitors attenuate progression of certain chronic kidney diseases in mice [18], the identity of the ubiquitin ligases involved are not well understood. Therefore, we sought to identify novel E3 ligases important in renal tubular maladaptive repair and fibrosis and to characterize their regulation and associated pathogenic mechanisms. To investigate the role of WWP1 in renal fibrosis progression, we first analyzed its expression profile in healthy vs. renal disease specimens. An analysis of the available bulk RNA sequencing and human renal disease transcriptome datasets from Nephroseq (https://www.nephroseq.org (accessed on 5 June 2024)) revealed that WWP1 (WW domain-containing E3 ubiquitin protein ligase 1) mRNA expression is significantly upregulated in several progressive kidney disease types, including diabetic nephropathy (DN), focal segmental glomerulosclerosis (FSGS), and lupus nephritis (Figure 1A), compared to respective healthy human kidneys. A reduction in glomerular filtration rate (GFR) and elevated serum creatinine levels, as well as proteinuria (presence of protein in the urine), are widely used indicators of CKD progression. Our analysis revealed that WWP1 expression positively correlates with proteinuria (Figure 1B) and serum creatinine levels (Figure 1C), suggesting that WWP1 could contribute to the progression of renal injury. Moreover, WWP1 mRNA levels inversely correlate with glomerular filtration rate (GFR) (Figure 1D,E), indicating that WWP1 induction strongly associates with the decline in renal function. Immunoblot analysis of human diabetic kidneys, indeed, confirmed robust WWP1 protein induction in the fibrotic kidneys compared to respective controls (Figure 1F). Further analysis of a recent single cell RNA sequencing dataset (Accession No.: GSE183279) [48] from human renal specimens revealed that WWP1 mRNA expression is induced in the renal epithelial compartments of the diabetic kidneys (Figure 1G,H), which suggests potential involvement of WWP1 in tubular pathologies. Transcript levels of TRIM65 are also highly induced in human CKD patients compared to control groups as determined by analyzing the similar annotated single cell RNA sequencing human renal disease dataset (Accession No.: GSE183279) [48].
Western blot analysis of human diabetic kidneys further confirmed a dramatic increase in TRIM65 protein levels in fibrotic kidneys compared to healthy controls (Figure 1F). Although a recent study indicates that TRIM65 attenuates fibrotic lesions in obstructive and folic acid nephropathies in mice [26], whether this ligase impacts tubular pathologies during CKD progression is not defined.
3.2. WWP1 and TRIM65 Are Dramatically Upregulated in Mouse Fibrotic Kidneys Undergoing Ureteral Unilateral Obstruction (UUO) and Aristolochic Acid Nephropathy (AAN)
UUO and AAN are established models for investigating tubular-injury-initiated progression to interstitial fibrosis [55,56,57] as confirmed by an increased expression of fibronectin (Figure 2A,B; Figure 3A,B), collagen 1 (Figure 2A,C; Figure 3A,C), and PAI-1 (Figure 2A,D; Figure 3A,D). Immunoblot analysis of renal extracts confirmed that WWP1 (Figure 2A,F; Figure 3A,F) and TRIM65 (Figure 2A,G; Figure 3A,G) are robustly upregulated in the UUO (7 days post-ligation) and AAN (25 days post-AA administration) kidneys relative to contralateral or vehicle-treated controls. Furthermore, PAI-1 upregulation in fibrotic human (Figure 1F) and mouse kidneys (Figure 2A,D; Figure 3A,D) correlates with WWP1 induction (Figure 1F; Figure 2A,F; Figure 3A,F), suggesting a possible causal relationship between these entities.
3.3. PAI-1 Promotes WWP1 and TRIM65 Upregulation to Trigger Tubular Dysfunction
Our previous studies demonstrated that PAI-1 promotes tubular dysfunction via dedifferentiation, G2/M arrest, and the induction/secretion of ECM proteins, and the expression of profibrotic cytokines and growth factors, which is orchestrated by p53- and pSMAD3-dependent mechanisms [39]. Indeed, sustained PAI-1 upregulation (Figure 4B,C) results in a dramatic loss of E-cadherin (Figure 4B,H) and a significant upregulation of vimentin (Figure 4B,I) and snail (Figure 4B,J), indicative of dedifferentiation. Markers of cell cycle arrest p21 (Figure 4B,M) and pHistone H3 (Figure 4B,N) expression are also robustly induced following persistent PAI-1 induction. In addition, several fibrotic factors (Figure 4B, D-G) as well as pSMAD3 (Figure 4B,K) and p53 (Figure 4B,L) are significantly upregulated in the CMV-PAI-1 population relative to CMV-Con cultures. Notably, PAI-1 overexpression in primary human tubular epithelial cells (RPTECs) largely recapitulated the fibrotic tubular dysfunctional phenotype (Supplementary Figure S1).
Since we reported a strong correlation between PAI-1 (Figure 1F; Figure 2A,D; Figure 3A,D) and WWP1 (Figure 1F; Figure 2A,F; Figure 3A,F) upregulation during both UUO- and AAN-driven renal fibrogenesis as well as in human diabetic kidneys, we hypothesize that PAI-1 is a major regulator of WWP1 expression. Our screening of E3 ubiquitin ligases revealed that WWP1 is robustly induced in PAI-1-overexpressing HK-2 cells (Figure 5A–C) and primary human tubular epithelial cells (Supplementary Figure S1) relative to CMV-controls. To determine whether WWP1 is involved in PAI-1-driven tubular pathologies, we stably infected CMV-PAI-1 cultures with either control shRNA or WWP1 shRNA lentiviral constructs. WWP1 stable depletion (Figure 5D,E), indeed, mitigates the PAI-1-driven fibrotic phenotype, as marked by the downregulation of fibronectin, collagen 1, CTGF, osteopontin, and snail (Figure 5D,F–I and N). Crystal Violet-stained CMV-PAI-1 + Control shRNA and CMV-PAI-1 + WWP1 shRNA monolayers (initially seeded at similar densities and grown for 5 days) demonstrated that WWP1 depletion rescued PAI-1-induced growth defects (Figure 5P,Q), which is consistent with the reductions in p21 (Figure 5D,L) and pHistone H3 (Figure 5D,M) protein levels in our western analysis. Furthermore, WWP1 suppression in the context of sustained PAI-1 expression dramatically reduced p53 (Figure 5D,K) and pSMAD3 (Figure 5D,J) levels, both critical mediators of PAI-1-driven tubular maladaptive repair [39], compared to CMV-PAI-1 + Control shRNA cells. Indeed, activation of the transcription factors p53 and SMAD3 during renal injury is critical for fibrosis progression since proximal tubule-specific p53 knockout and global SMAD3 deficiency protects mice from renal tubular dysfunction and progressive CKD [35,40]. Therefore, tubular epithelial WWP1 upregulation is important for PAI-1 mediated tubular dysfunction, identifying WWP1 as a novel CKD-promoting entity. Moreover, stable expression of WWP1 (CMV-WWP1-GFP) in human renal tubular epithelial cells (HK-2), indeed, promotes a fibrotic response, as marked by an increase in fibronectin, collagen 1, p53, and snail expression compared to control cultures (CMV-Control) (Supplementary Figure S2), further highlighting its pro-fibrotic activity.
Extensive correlations between PAI-1 (Figure 1F; Figure 2A,D; Figure 3A,D) and TRIM65 upregulation (Figure 1F; Figure 2A,G; Figure 3A,G) in the human diabetic and mouse UUO and AAN kidneys necessitates testing whether PAI-1 promotes TRIM65 induction and contributes to tubular pathologies. Indeed, TRIM65 protein expression is prominently induced in PAI-1-overexpressing HK-2 (Figure 6B,C) and RPTEC cells (Supplementary Figure S1). Moreover, genetic depletion of TRIM65 (Figure 6D,E) also abrogates the PAI-1-driven fibrotic phenotype (Figure 6D,F–I), pSMAD3 (Figure 6D,J), p53 (Figure 6D,K), and p21 (Figure 6D,L) levels, identifying a novel pathogenic role for TRIM65 induction in PAI-1-mediated tubular maladaptive repair responses. Moreover, our mechanistic study reveals that WWP1 is an important regulator of PAI-1-driven TRIM65 expression in renal tubular epithelial cells (Figure 5D,O).
3.4. c-Myc Is a Major Upstream Regulator of WWP1 and TRIM65 Induction and PAI-1-Driven Tubular Dysfunction
The proto-oncogene c-Myc regulates multiple physiological processes including cell proliferation, differentiation, and apoptosis. Hyperactivation of this transcription factor, not surprisingly, is linked to cancer progression [58]. The involvement of c-Myc in CKD progression is not well understood, although a recent study implicates c-Myc upregulation in metabolic reprogramming during CKD [41]. Whether c-Myc induction is linked to renal tubular maladaptive repair, however, requires clarification.
Our analysis of human renal disease datasets from Nephroseq (https://www.nephroseq.org (accessed on 5 June 2024)) revealed a significant induction in c-Myc mRNA in diseased kidney (e.g., chronic kidney disease and diabetic nephropathies) relative to normal kidneys (Figure 7A). Our analysis of human fibrotic (diabetic) and mouse models (e.g., obstruction and toxin exposure) confirms not only a significant c-Myc renal induction but also a strong association between PAI-1 (Figure 1F; Figure 2A,D; Figure 3A,D) and c-Myc (Figure 1F; Figure 2A,E; Figure 3A,E) upregulation during renal injury, suggesting a potential relationship among these entities. PAI-1, in fact, promotes a dramatic increase in c-Myc protein levels in both HK-2 (Figure 7B–D) as well as RPTEC cells (Supplementary Figure S1), and c-Myc knockdown (Figure 7E,F) mitigates PAI-1-induced fibrotic reprogramming (Figure 7E,G–O). Therefore, PAI-1 is a novel upstream regulator of c-Myc induction and subsequent tubular dysfunction. Consistent with the previous studies demonstrating that c-Myc is a direct transcriptional activator of WWP1 during tumorigenesis [21], our western analysis revealed that c-Myc depletion in PAI-1 transductants also dramatically decreased WWP1 (Figure 7E,P) and TRIM65 protein levels (Figure 7E,Q). Collectively, these data indicate that PAI-1-driven WWP1 and TRIM65 induction requires c-Myc activation during tubular dysfunction.
3.5. PAI-1-Induced Repression of the Antifibrotic BMP-7/SMAD1/5 Pathway Is Causatively Linked to Tubular Dysfunction
BMP-7 is a member of the TGF-β superfamily of ligands that binds to its cognate receptors to initiate SMAD1/5 phosphorylation which, in turn, antagonizes profibrotic SMAD3 signaling triggered by TGF-β1 [42,47]. The expression of BMP-7 and SMAD5, as well as SMAD1/5 phosphorylation, is markedly downregulated in both UUO (Figure 2A,H–J) and AAN (Figure 3A,H–J) mouse models. While the mRNA expression of BMP-7 is particularly enriched in the renal epithelial compartment (Figure 8B), the levels are downregulated during human CKD progression (Figure 8A,C), consistent with previous observations [46,59]. Administration of recombinant BMP-7 protein during renal injury, on the other hand, mitigates fibrosis and improves kidney health, demonstrating its utility as a potent antifibrotic target [43,44,45,60]. Even though BMP-7 is an attractive antifibrotic target, upstream regulation of BMP-7 repression during the progression of renal disease, however, is not well understood. In this regard, whether PAI-1 represses BMP-7-SMAD1/5 pathways during CKD progression has not been tested. Immunoblotting of CMV-Con and CMV-PAI-1 cell extracts revealed that PAI-1 overexpression not only dramatically decreased BMP-7 ligand levels (Figure 9A,B) and SMAD1/5 phosphorylation (Figure 9A,D) but also repressed total SMAD5 protein levels in HK-2 cells (Figure 9A,C). Therefore, PAI-1 is a novel repressor of the renal tubular BMP-7/SMAD1/5 signaling axis, which is further confirmed in RPTEC cells (Supplementary Figure S1). To determine whether BMP-7 loss of expression is causatively linked to PAI-1-induced maladaptive repair, CMV-PAI-1 cells were infected with either CMV-Control Vector or CMV-BMP-7 lentiviral constructs prior to stable selection. Western blot analysis confirmed that restoration of BMP-7 expression in CMV-PAI-1 cells (Figure 9E) attenuates PAI-1-driven fibrotic response, as evident by major reductions in pSMAD3 (Figure 9E,G), fibronectin (Figure 9E, H), collagen 1 (Figure 9E,I), CTGF (Figure 9E,J), osteopontin (Figure 9E,K), p53 (Figure 9E, L), p21 (Figure 9E,M), pHistone H3 (Figure 9E,N), and snail (Figure 9E,O) levels compared to CMV-PAI-1 + CMV-Control Vector cells. As anticipated, BMP-7 restoration triggers pSMAD1/5 phosphorylation (Figure 9E,F). These findings indicate PAI-1-induced BMP-7 loss as a promoter of tubular dysfunction.
SMAD1/5 are direct downstream targets of the BMP-7 pathway by receptor-mediated phosphorylation [47]. Since PAI-1 is a novel repressor of SMAD5 protein levels, we tested whether SMAD5 repression is linked to tubular dysfunction. Ectopic restoration of SMAD5 expression (Figure 10A) in CMV-PAI-1 transgenic cells via lentiviral infection with CMV driven SMAD5 expression constructs significantly attenuates PAI-1 mediated tubular dysfunctional phenotype compared to vector-transduced double transgenic controls (CMV-PAI-1+CMV-vector) (Figure 10A–K). These data identify PAI-1 as a major repressor of BMP-7 signaling cascade and that disabling of BMP-7/SMAD5 signaling axis is linked to PAI-1-driven tubular maladaptive repair.
3.6. BMP-7 and SMAD5 Are Upstream Regulators of PAI-1-Driven c-Myc, WWP1, and TRIM65 Upregulation
The pathologic links among renal PAI-1 upregulation, BMP-7/SMAD1/5 repression, and c-Myc, WWP1, and TRIM65 induction are currently unclear. Intriguingly, BMP-7 repression downstream of PAI-1 promotes c-Myc, WWP1, and TRIM65 induction and renal tubular pathogenesis since rescue of BMP-7 expression in PAI-1 stable transductants abrogates PAI-1-driven c-Myc (Figure 9E,P), WWP1 (Figure 9E,Q), and TRIM65 (Figure 9E,R) expression. Similarly, restoration of SMAD5 levels (which is otherwise downregulated by PAI-1) also leads to a significant reduction in c-Myc (Figure 10A,L), WWP1 (Figure 10A,M), and TRIM65 (Figure 10A,N) expression downstream of PAI-1 overexpression, suggesting that SMAD5 downregulation is a critical upstream regulator of c-Myc, WWP1 and TRIM65 induction by PAI-1. Repression of BMP-7 and SMAD5 signaling in the UUO (Figure 2A,H-J) and AAN (Figure 3A,H-J) kidney injury models correlates with increased c-Myc (Figure 2A,E; Figure 3A,E), WWP1 (Figure 2A,F; Figure 3A,F) and TRIM65 (Figure 2A,G; Figure 3A,G) expression in mice, further highlighting the potential causative relationship among these entities during CKD progression. These data highlight a previously unknown pathogenic relationship between BMP-7/SMAD5 downregulation and induction of c-Myc, WWP1 and TRIM65 proteins during progressive renal injury.
4. Discussion
Our study uncovers several novel mechanistic insights into tubular maladaptive repair and renal fibrosis progression. We identify WWP1, an E3 ubiquitin ligase, as a previously unrecognized and highly upregulated factor in tubular epithelium during renal injury in both human CKD specimens (Figure 1) and murine UUO (Figure 2) and AAN (Figure 3) kidneys. Importantly, WWP1 expression correlates with CKD progression in humans (Figure 1), highlighting its potential clinical relevance. While proteasome inhibition is known to confer renoprotection [18], the specific contributions of E3 ligases in CKD pathogenesis remain underexplored. Our findings establish WWP1 as a novel mediator of tubular epithelial dysfunction and fibrogenesis. PAI-1 is robustly upregulated in renal tubules across injury models and promotes dysfunction via p53- and pSMAD3-dependent pathways [27,36,39]. We further identify PAI-1 as a novel upstream inducer of WWP1 (Figure 5; Supplementary Figure S1). Silencing WWP1 reverses PAI-1-driven maladaptive repair by reducing epithelial differentiation, relieving cell cycle inhibition, and decreasing extracellular matrix and fibrotic marker expression and pSMAD3/p53 signaling (Figure 5). To our knowledge, this is the first demonstration of PAI-1 regulating WWP1 in any pathological context. Moreover, WWP1 ectopic expression in renal tubular epithelial cells alone is sufficient to induce a fibrotic response (Supplementary Figure S2).
Furthermore, our study identifies TRIM65 as another novel E3 ligase regulated by PAI-1. Although global TRIM65 knockout mice are protected from UUO and folic acid-driven renal fibrosis [26], its precise role in tubular pathogenesis is unclear. PAI-1 robustly induces TRIM65 expression (Figure 6; Supplementary Figure S1), and stable silencing of TRIM65 inhibits PAI-1-driven fibrotic reprogramming (Figure 6), linking TRIM65 upregulation to tubular pathogenesis. While WWP1 depletion leads to the attenuation of PAI-1-driven TRIM65 expression (Figure 5), repression of TRIM65 in PAI-1 transductants does not impact WWP1 levels (Figure 6). Therefore, we also uncover WWP1 as an upstream regulator of TRIM65 during tubular dysfunction. Our current findings reveal that silencing either WWP1 or TRIM65 mitigates p53 levels and SMAD3 phosphorylation (Figure 5 and Figure 6), two key transcriptional regulators of renal tubular dysfunction and fibrosis [35,40]. WWP1 and TRIM65 upregulation is linked to PTEN and PPM1A ubiquitination, respectively [21,25]. Indeed, PPM1A and PTEN levels are dramatically decreased during kidney fibrosis originating from various etiologies [50,51], and tubular PPM1A and PTEN depletion alone triggers epithelial dedifferentiation, growth inhibition, and fibrosis via SMAD3 and p53 activation [50,51]. Therefore, WWP1- and TRIM65-mediated regulation of p53 and pSMAD3 could be linked to PTEN and PPM1A ubiquitination, leading to fibrotic maladaptive repair and fibrosis.
Previous studies demonstrated that BMP-7 is a potent CKD therapeutic target as the administration of recombinant BMP-7 mitigates UUO-driven fibrogenesis and improves renal health [43,44,45,60]. Here, we identify PAI-1 as a novel repressor of the BMP-7/SMAD1/5 axis. BMP-7 transcript levels are reduced in fibrotic human kidneys (Figure 8), consistent with the findings in CKD mouse models [46,59]. We demonstrate that PAI-1 represses BMP-7 protein levels, inhibits SMAD1/5 phosphorylation, and reduces total SMAD5 expression, highlighting a multilevel disruption of this signaling cascade (Figure 9; Supplementary Figure S1). Restoration of BMP-7 (Figure 9) or SMAD5 (Figure 10) reverses PAI-1-induced fibrotic reprogramming and suppresses c-Myc, WWP1, TRIM65, and downstream pSMAD3/p53 activation. Regardless of the genetic manipulation (either knockdown of c-Myc, WWP1, and TRIM65 or overexpression of BMP-7 and SMAD5), PAI-1 levels in the double transgenic population remain comparable (Figure 5, Figure 6, Figure 7, Figure 9 and Figure 10). Therefore, we define a previously unrecognized PAI-1–BMP-7/SMAD5–c-Myc–WWP1–TRIM65 regulatory hierarchy in tubular dysfunction. Future studies will determine the mechanism by which PAI-1 downregulates the BMP-7 ligand and how loss of BMP-7/SMAD5 signaling triggers c-Myc activation.
5. Conclusions
In summary, we identify WWP1 and TRIM65 as previously unrecognized mediators of tubular dysfunction. Mechanistically, PAI-1 suppresses the BMP-7/SMAD5 axis, thereby, triggering tubular fibrosis through activation of the c-Myc–WWP1–TRIM65 cascade (Figure 11). Our findings establish a novel paradigm in which PAI-1 acts as a dual-function mediator of renal fibrosis by suppressing regenerative pathways (BMP-7/SMAD5 axis) and activating profibrotic cascades (WWP1, c-Myc, p53, and SMAD3). Therefore, PAI-1 can be a compelling therapeutic target to restore the balance between pro- and anti-fibrotic signals during maladaptive repair. Inhibition of WWP1 could also offer a promising strategy to halt renal fibrosis progression.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kovesdy C.P. Epidemiology of chronic kidney disease: An update 2022 Kidney Int. Suppl.20221271110.1016/j.kisu.2021.11.00335529086 PMC 9073222 · doi ↗ · pubmed ↗
- 2Jha V. Garcia-Garcia G. Iseki K. Li Z. Naicker S. Plattner B. Saran R. Wang A.Y.-M. Yang C.-W. Chronic kidney disease: Global dimension and perspectives Lancet 201338226027210.1016/S 0140-6736(13)60687-X 23727169 · doi ↗ · pubmed ↗
- 3Perico N. Remuzzi G. Chronic kidney disease: A research and public health priority Nephrol. Dial. Transpl.201227 iii 19iii 2610.1093/ndt/gfs 28422764188 · doi ↗ · pubmed ↗
- 4Couser W.G. Remuzzi G. Mendis S. Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases Kidney Int.2011801258127010.1038/ki.2011.36821993585 · doi ↗ · pubmed ↗
- 5Centers for Disease Control and Prevention Chronic Kidney Disease Basics 2024 Available online: https://www.cdc.gov/kidney-disease/about/index.html(accessed on 7 May 2025)
- 6Boor P. Ostendorf T. Floege J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets Nat. Rev. Nephrol.2010664365610.1038/nrneph.2010.12020838416 · doi ↗ · pubmed ↗
- 7Humphreys B.D. Mechanisms of renal fibrosis Ann. Rev. Physiol.20188030932610.1146/annurev-physiol-022516-03422729068765 · doi ↗ · pubmed ↗
- 8Friedman S.L. Sheppard D. Duffield J.S. Violette S. Therapy for fibrotic diseases: Nearing the starting line Sci. Transl. Med.20135167 sr 110.1126/scitranslmed.300470023303606 · doi ↗ · pubmed ↗