Targeted Interference with USF2 Binding to the SERPINE1 Proximal Promoter E-Box in Dual Mutant p53R282Q,H179Y Human Keratinocytes Inhibits Serum-/TGF-β1-Induced SERPINE1 Expression and Stimulates Epithelial Cell Proliferation
Stephen P. Higgins, Ralf-Peter Czekay, Craig E. Higgins, Paul J. Higgins

TL;DR
Blocking USF2 binding to a specific DNA site in skin cells reduces SERPINE1 expression and boosts cell growth.
Contribution
Demonstrates that USF2 binding to the SERPINE1 promoter regulates cell proliferation in p53 mutant keratinocytes.
Findings
Interfering with USF2 at the PE2 E box reduces SERPINE1 expression in response to serum and TGF-β1.
Reduced USF2 occupancy increases the number of proliferating keratinocytes.
Overexpression of USF2 or PAI-1 inhibits HaCaT cell colony growth.
Abstract
The SERPINE1 gene encodes the serine protease inhibitor plasminogen activator inhibitor type-1 (PAI-1), a major negative regulator of the plasmin-dependent pericellular proteolytic cascade and a crucial determinant in the program of stromal remodeling. Recent omics approaches confirmed that high tumor SERPINE1 levels are prognostic for poor disease outcomes and shorter disease-free survival in various malignancies. Kinetic analysis of biomarkers of cell cycle transit in growth-synchronized p53 dual mutant human keratinocytes confirmed that PAI-1 transcription occurred early after growth activation of quiescent (G0) cells and prior to G1 entry. Previous evidence has confirmed that differential residence of USF family members (USF1→USF2 switch) at the PE2 region hexanucleotide E box motif (CACGTG) in the SERPINE1 proximal promoter characterizes the G0→G1 transition period and the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
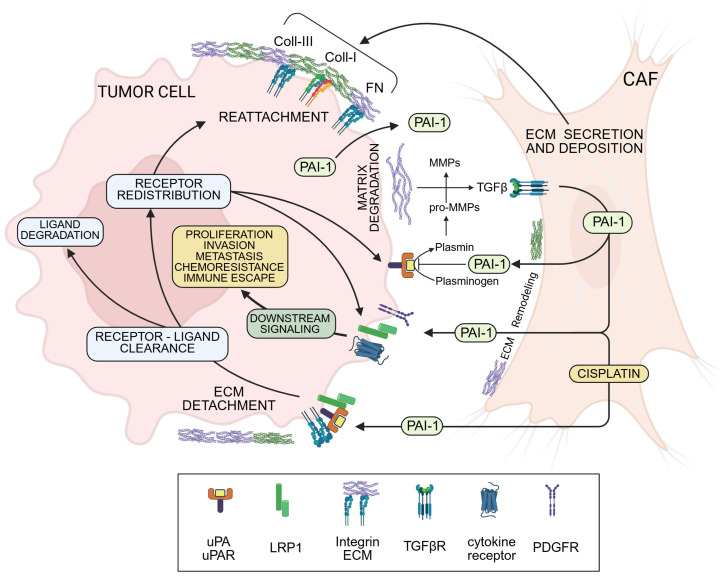 Figure 1
Figure 1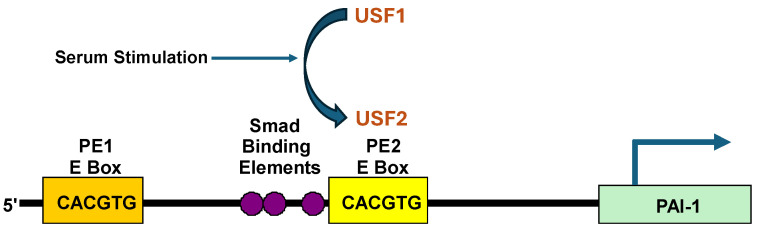 Figure 2
Figure 2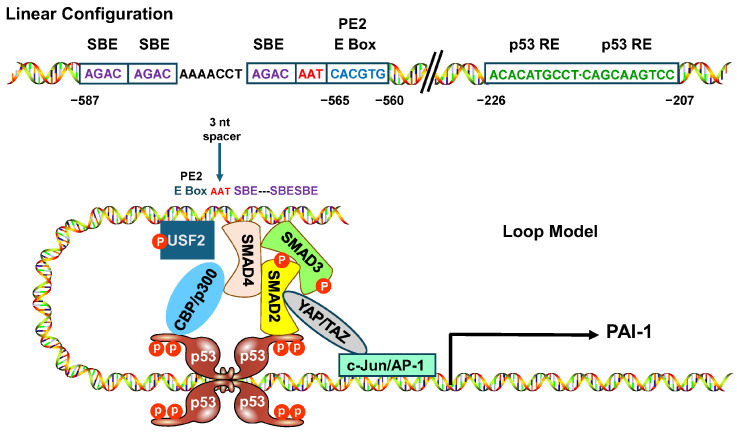 Figure 3
Figure 3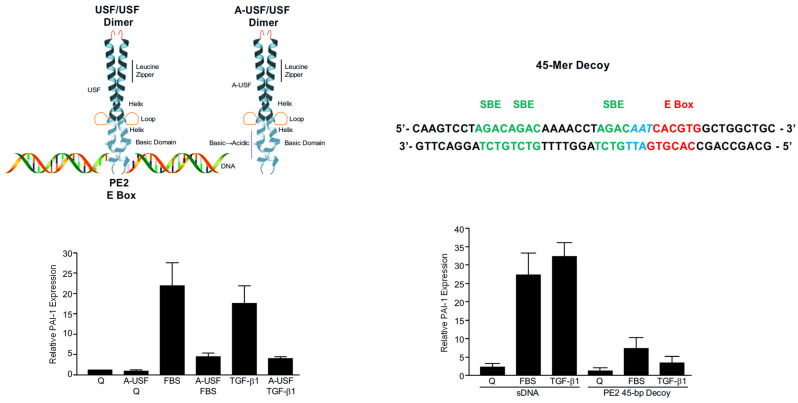 Figure 4
Figure 4- —NIH
- —AHA Fellowship
- —Friedman Family Cancer Fund
- —Graver Family Endowed Fund for Cancer Research
- —Butler Family Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtease and Inhibitor Mechanisms · TGF-β signaling in diseases · Bone and Dental Protein Studies
1. Introduction
Upstream stimulatory factor-1 and -2 (USF1/2) are members of the conserved basic helix-loop-helix/leucine zipper (bHLH-LZ) MYC family of transcription factors [1,2]. USF1/2 bind to core E box sequences (CANNTG) as tetramers in the promoter regions of target genes which encode proteins that regulate various basic processes, including development, cellular growth and proliferation, the immune response, metabolic pathways, and cancer progression [1,2,3]. Multiple signaling networks and their activated protein kinases influence USF1/2 function largely through site-specific phosphorylation (primarily on serine and threonine residues) while USF1/USF2 homo- and heterodimer composition and recruited co-factors dictate transcriptional controls on USF responsive genes [4,5,6,7]. Earlier work indicated that both USF1 and 2 inhibit myc oncogene-mediated cell transformation; however, USF2 has more widespread anti-proliferative properties and attenuates tumor cell focus-formation in response to E1A + ras and mutant p53 + ras oncogenes [8]. Perhaps not unexpectedly, therefore, Crispr-Cas9-directed elimination of USF2 in mouse embryonic fibroblasts promotes both cellular growth and migration [9]. Conversely, USF1/2 overexpression suppressed proliferation of normal and malignant thyrocytes, as well as other cell types, by delaying G_2_/M transit, likely as a consequence of down-regulation of cyclin B1 and CDK1, c-MYC and induction of p27 and p53 [7,10,11]. It is now apparent, however, that the cell cycle-related anti- vs. pro-proliferative functions of USF1/2 are context- and cell type-dependent and involve specific mitogen activated (p38, PKC, PKA) and cyclin-dependent (CDK1, CDK5) kinases as well as CK2, DNA-PK and GSK3β [6,7,8,9,12,13,14]. The generation of splice variants of USF1/2 also impacts transcriptional outcomes. The USF1 and USF2 genes, located on 1q22-q23 and 19q23, respectively, possess 10 protein encoding exons. Alternate transcript splicing involving exon 4 of USF2 generates the two isoforms USF2a (44 kDa) and USF2b (38 kDa), of which USF2b (lacking exon 4 sequences) functions as a dominant negative regulator of USF1 or USF2a target gene expression reviewed in [7].
Transcriptome analyses indicated that USF1/2 bind to E boxes in the promoter regions of over 2500 genes [15]. While loss of USF1/2 activity is evident in several tumor cell types, recent findings suggest that USFs may also be pro-tumorigenic [16,17] (reviewed in [18]). Omics approaches have illuminated the involvement of both USF1 and USF2 in several of the more lethal cancers, although the underlying mechanisms may vary or be tumor-type unique. USF1/2 abundance and, therefore, their transcriptional impact on individual responsive genes, appear critical in determining the tumor suppressive vs. promoting effects of USF family members [19,20,21]. In lung adenocarcinoma, for example, USF1 upregulated expression of the coiled-coil-helix-coiled-coil-helix domain containing 4 protein (CHCHD4) [22]. CHCHD4 is highly expressed in human malignancies where it is associated with disease progression, poor patient outcomes and increased recurrence. USF2 is also strongly expressed in the dysplastic pulmonary epithelium, in small cell lung cancer and squamous cell carcinoma [23]. The USF1 target genes lncRNA-NEAT1, TF-ETV5 and ZNF641 are similarly implicated in the progression of hepatocellular carcinoma [15,24], and USF1 stimulates growth and proliferation in breast cancer and glioblastoma by inducing expression of SEZ6L2 and the stemness protein CD90, respectively [25,26]. The LncRNA TUG1 recruits USF1 to increase transcription of ROMO1 and elevated ROMO1 expression induces an epithelial-to-mesenchymal transition (EMT) in hepatocellular carcinoma cells with increases in proliferation, migration and tissue invasion [27]. Elevated USF2 expression stimulates abnormal lipid metabolism in lung adenocarcinoma by transcriptionally activating PEX3 to upregulate and promote interactions with SLC25A17 [28]. USF2 knockdown inhibits abnormal lipid accumulation and attenuates tumor growth in nude mice while USF1 overexpression increases colony formation, survival and radio-resistance in prostate cancer cells [29]. USF2 induced transcription of the translational regulatory protein BZW2 facilitates colorectal cell proliferation, invasion, stemness and inhibition of apoptosis through a LAMP3-dependent mechanism [30].
At least some of these programmed changes appear due to a cooperative USF→TGF-β1 axis since both USF1 and USF2 stimulate TGF-β1 promoter activity and TGF-β1 protein secretion in specific cell types [10,31]. USF1 overexpression promotes both EMT and migration in melanoma cells by increasing TGF-β1 expression while USF1 knockdown attenuated both the morphologic transition and the motile response [32]. TGF-β1 may also function as an initiator, inducing USF2 to transcriptionally upregulate S100A8 in colorectal cells; S100A8, in turn, stimulates cell migration, invasion and EMT [33]. One interesting finding implicates USF2 as an inhibitor of Smurf1 and Smurf2 transcription [34]. Since Smurfs suppress TGF-β signaling, TGF-β reporter activity is significantly reduced by USF2 siRNA. USF2 promotes human breast cancer cell proliferation, invasion and migration [34]; it is tempting to speculate, therefore, that this may relate to USF2 activation of the TGF-β1 pathway leading to EMT and tumor aggressive behavior.
2. SERPINE1 (PAI-1) Is a Poor Prognosis Biomarker and Effector of Tumor Aggressiveness
The clade E member 1 serine protease inhibitor SERPINE1, also known as plasminogen activator inhibitor-1 (PAI-1), is a potent negative regulator of the pericellular proteolytic cascade and a key factor in the spatial/temporal program of stromal remodeling [35,36,37]. PAI-1 is also a prominent member of the serum-induced “wound-response” transcriptome [38,39]. In dual mutant p53^R282Q,H179Y^ immortalized keratinocytes (HaCaT cells), PAI-1 is required for TGF-β1-stimulated planar locomotion and invasion through Matrigel barriers, responses likely involving LRP1-dependent engagement of the Jak/Stat pathway [40,41,42]. Matricellular (i.e., extracellular matrix-anchored) PAI-1 also promotes acquisition of a mesenchymal-to-amoeboid phenotypic transition that correlates with activation of signaling networks and downstream target genes required for efficient 3-D “tissue” invasion [43]. The consistent association of PAI-1 expression with stromal restructuring [38,44,45] suggests that this SERPIN integrates cycles of cell-to-matrix adhesion/dis-adhesion with “scaffold” remodeling to initiate and maintain effective cellular migration [46]. The specific phenotypic impact of PAI-1, however, is significantly more complex and likely requires a balance between its intracellular and extracellular activities that may be both cell type- and stimulus-specific [47]. Indeed, several SERPINS (SERPINE1 [PAI-1], SERPINB1, SERPINB2) are prominent members of the stromal remodeling transcriptome and proteome [38,39]. PAI-1, most particularly, has several functions in the integrated control of focalized matrix restructuring, cell-to-substrate adhesion/detachment, migration and proliferation [44,45,46,48,49,50,51,52,53]. PAI-1 effectively limits plasmin generation within the pericellular space to maintain a supporting “scaffold” for cell movement [46] while also regulating urokinase plasminogen activator-dependent growth factor activation, thereby attenuating the associated proliferative response [54]. Concerning the latter, certain USF-target “growth arrest-associated” genes (i.e., p16^INK4a^, PAI-1) may synchronize the proliferative and motile phases of the adaptive and maladaptive tissue repair programs by inhibiting cell proliferation while promoting migration [54,55].
PAI-1 is also a major downstream TGF-β1 target gene; it is apparent that both PAI-1 and TGF-β1 promote tumor cell migration, amoeboid motility, and tissue invasion [56,57,58]. TGF-β1-induced cell movement is effectively attenuated by the small molecule PAI-1 inhibitor Tiplaxtinin, suggesting that the PAI-1 anti-proteolytic function is required for TGF-β1-stimulated locomotion. This is consistent with the recent finding that deficiencies in PAI-1 or TGF-β1 reduce cell motility [41]. Expression of a PAI-1-GFP fusion protein under the inducible control of +800 bp of the injury-activated PAI-1 promoter prominently “marks” keratinocyte migration trails. The use of PAI-1-null cells, knockdown approaches, PAI-1 add-back rescue, and neutralizing antibodies confirms the requirement for PAI-1 in cell movement [41]. Similarly, adipose mesenchymal stem cells stimulate cellular motility by modulating SERPINE1 expression in dermal fibroblasts and keratinocytes [59]. In glioblastoma cells, PAI-1 increased EMT characteristics, tumor cell invasion, and migration under hypoxic culture conditions, and these effects were mitigated by PAI-1 knockdown [60].
High tumor SERPINE1 levels predict poor outcomes and shorter disease-free survival in various human cancers [58,61,62,63,64,65]. SERPINE1 is a prominent member of the validated five-member EMT-related prognostic gene set in gastric carcinoma and the six-gene signature that is prognostic for reduced survival in head and neck squamous cell carcinoma patients, as well as indicative of a poor prognosis and higher risk score. SERPINE1 is a clinically important hub gene in a wide spectrum of human malignancies, and a strong indicator of reduced patient survival [66,67,68,69,70,71,72]. While many cell types in the tumor microenvironment (TME) synthesize and secrete PAI-1, cancer-associated fibroblasts (CAFs) are a particularly rich source of this SERPIN (Figure 1). PAI-1 co-localizes, in fact, to α-SMA-positive fibroblastoid cells at the tumor invasive margins and stroma-enriched regions [73,74,75,76,77,78,79]. The myofibroblastoid CAF phenotype (α-SMA^high^/PAI-1^high^), moreover, is involved in the development of tumor chemotherapeutic resistance and/or acquisition of a more aggressive, invasive phenotype [80,81]. Functional blockade of PAI-1 activity (e.g., with small molecule inhibitors) may be one approach to enhance chemotherapeutic efficacy [81].
3. Kinetics of Induced SERPINE1 Transcription in Dual Mutant p53R282Q,H179Y Human Keratinocytes
The SERPINE1 gene is a major TGF-β1 and p53 target and is robustly upregulated in response to serum-stimulation of growth-arrested G_0_ (quiescent) cells [5,40,54,82,83,84,85]. Kinetic modeling using established biomarkers of cell cycle transit (c-MYC; cyclin D_1_; cyclin A) in synchronized human (HaCaT) keratinocytes confirmed that PAI-1 transcription occurs early (prior to G_1_ entry) after serum-stimulation of quiescent (G_0_) keratinocytes [5,39,85]. Induced PAI-1 expression involves a USF1→USF2 subtype switch at the PE2 site E box motif (5′-CACGTG-3′) in the PF1 region (nucleotides −794 to −532) of the PAI-1 promoter [5] that resides three nucleotides downstream of a trio of TGF-β-responsive SMAD-binding elements (ACAG motif) at nucleotides −569 upstream of the transcription start site [85]. (Figure 2). Chromatin immunoprecipitation confirmed that the growth factor-responsive PE2 E box sites in the PAI-1 gene are, indeed, USF target sequences in vivo and that serum-stimulated PAI-1 expression reflected a dynamic USF1 → USF2 switch at this CACGTG motif [5]. A consensus PE2 E box motif (5′-CACGTG-3′) at nucleotides −565 to −560, moreover, is required for USF E box occupancy and serum-stimulated PAI-1 transcription. Indeed, a CG→AT substitution at the two central nucleotides ablated binding of USF to a mutant probe target and inhibits expression of a PAI-1 promoter-driven reporter [5], establishing the importance of an intact PE2 E box consensus sequence in serum-stimulated PAI-1 transcription.
PAI-1 mRNA levels rapidly decline several hours prior to the onset of S phase [5,55], suggesting that PAI-1 may negatively impact cell growth. PAI-1 knockdown in fact results in escape from senescence-associated proliferative arrest and TGF-β1-induced cytostasis in several cell types, including HaCaT keratinocytes [54,83]. The USF1 → USF2 transition at the PAI-1 PE2 E box, and subsequent PAI-1 transcription may regulate the time course of tissue remodeling progression through the cell cycle as part of the proliferative control program.
4. Interference with USF-DNA Binding Inhibits PAI-1 Expression and Stimulates Epithelial Cell Proliferation
Expression control by USF family members is distinct from simple site recognition, since positive or negative transcriptional outcomes likely depend on cell type, species, and adjoining motifs bound by non-USF factors [5,86,87]. The USF1→USF2 transition at the PE2 E box appears critical to the formation of the higher order transcriptional complex on the PE2 platform (Figure 3). Mobility shift assessments reinforced the finding (with regard to the PAI-1 promoter) that constructs with an intact consensus CACGTG sequence were effective competitors for USF binding while those lacking the CACGTG motif or containing the transcription-attenuating CG → AT mutation failed to inhibit complex formation with a wild-type probe [5,84]. Based on these results, it was important to determine if disruption of USF function would specifically affect PAI-1 transcription in response to serum or TGF-β1. Two approaches were selected to evaluate this possibility. Initially, keratinocytes were engineered to express a dominant-negative A-USF construct in which the wild-type DNA-binding domain was replaced with acidic residues; this modification greatly stabilizes heterodimers formed between A-USF and wild-type USFs and inhibits formation of endogenous USF/DNA complexes [5,6,13,85,88,89]. A-USF expression significantly attenuated both serum- and TGF-β1-induced PAI-1 levels. Secondly, a double-stranded 45 bp PE2 DNA construct was designed based on the previously identified requirements for an intact CACGTG motif for probe recognition by USF [5,84,85]. Similarly to the results obtained with A-USF, transfection of these double-stranded USF-binding “decoys” effectively reduced both serum- and TGF-β1-induced PAI-1 transcript levels in HaCaT keratinocytes [85] (Figure 4).
USF1/2 regulate the transcription of genes that impact cell growth and the proliferative program in various tumor cell types [3]. To address this relationship more specifically, a molecular genetic approach was taken involving transfection of a wild-type USF2 construct in dual p53 mutant HaCaT cells; USF2 overexpression significantly reduced colony formation [85]. To more specifically implicate USF in HaCaT cell proliferative control, a DOX-dependent A-USF expression system was developed to express an HA-tagged dominant-negative A-USF insert (DN-USF) in an inducible Tet-OFF system. Oxytetracycline removal and subsequent A-USF induction resulted in a marked increase in HaCaT cell growth and expression of the validated proliferative biomarker Ki-67 while suppressing both PAI-1 and PAI-2 mRNA levels [85]. Adenoviral-mediated PAI-1 overexpression, moreover, dramatically inhibited keratinocyte proliferation and colony expansion, mimicking the results of USF2 transfection. Collectively, these data demonstrate that the bHLH-LZ transcription factor USF2, and its target gene PAI-1, regulate keratinocyte proliferation.
5. Conclusions
Several members of the SERPIN gene family are implicated in tumor progression (SERPINE1, SERPINE2, SERPINE3, SERPINB9, SERPINI1), although SERPINE1 (PAI-1) is the SERPIN most prominently and causatively involved in the creation of a highly aggressive malignant phenotype [91,92,93,94]. Indeed, as previously detailed, PAI-1 regulates critical pathophysiological events in the TME, including angiogenesis and metastasis, immunosuppression (by recruiting M2-polarized macrophages and inducing PD-L1 expression), stromal remodeling, and resistance to chemotherapy (via and glycolytic reprogramming [36,37,46,48,73,76,77]. While high tumor levels are clearly associated with poor outcomes, in other cell types PAI-1 promotes a senescence-like growth arrest [54,83] which contributes to what is known as the PAI-1 “paradox” in cancer [95]. This review intended to provide a comprehensive overview of these developments, at least with regard to the tumor-promoting SERPINE1 gene, and the role of USF2 in SERPINE1 transcriptional control with a focus on various strategies that might have applicability as transcription-targetable agents.
Changes in transcriptional outputs is a hallmark of cancer and a convergence point of oncogenic signaling. Transformed cells often develop a dependency on such genomic reprogramming, highlighting the therapeutic potential of rectifying cancer-associated transcriptional abnormalities in malignant cells. Transcription therapy is an emerging strategy that intends to rectify aberrant gene expression in cancer cells through direct intervention in the transcription process. The transcriptional apparatus was previously considered undruggable due, in large measure, to the perception that (a) transcription is a nuclear event and therefore not readily accessible to therapeutic agents, and (b) many transcriptional components lack enzymatic activity and, thus, are unsuitable for chemically directed interventions [96]. In recent years, approaches and “therapeutic” agents, including transcription factor decoys and dominant-negative constructs, have been developed that target various levels of transcriptional regulation [96]. It has been estimated that at least 10% of FDA-approved anticancer drugs that were not initially developed to target transcription actually regulate this process in one way or another [96]. In recent years, however, great progress has been made in efforts to develop transcription-targeted therapeutic agents. Some of these agents are approved for clinical practice or have entered clinical trials for cancer treatments. Double-strand oligodeoxynucleotides (ODNs) or hairpin single strand ODNs are particularly effective decoys. These ODNs are designed to encompass recognition sequences for specific transcription factors or contain modified sequences that provide for significantly increased protein–DNA interactions [96]. Transfected decoys bind corresponding transcription factors, effectively sequestering them away from their target promoters, thereby regulating gene expression. Indeed, decoys targeting transcription factors including Sp1, AP-1, STAT3, and Ets-1 inhibit expression of cancer associated genes (e.g., VEGF, uPAR, and Bcl-XL) and attenuate growth and metastasis of various cancer cells [96]. Recent advances in design and fabrication have demonstrated the feasibility of developing decoys capable of discriminating among closely related transcription factors. However, as is true for other oligonucleotide-based “chemotherapeutic” agents, the delivery of transcription factor decoys to target cells in vivo and in vivo stability are two major challenges before clinical applications become a reality. As one example of a potential druggable target in a tumor progression gene, this review highlights the role of the USF1→USF2 transition in SERPINE1 transcription. Since PAI-1 is a central regulator of tumor progression and therapeutic resistance in cutaneous malignancies [94,95], this review focused on highlighting the consequences of disrupting the USF2/PAI-1 transcription network in p53 mutant human keratinocytes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Huang C. Xia M. Qiao H. Liu Z. Lin Y. Sun H. Yu B. Fang P. Wang J. Tetramerization of upstream stimulating factor USF 2 requires the elongated bent leucine zipper of the b HLH-LZ-domain J. Biol. Chem.202329910524010.1016/j.jbc.2023.10524037690682 PMC 10570711 · doi ↗ · pubmed ↗
- 2Littlewood T.D. Evan G.I. Transcription factors 2: Helix-loop-helix Protein Profile 199526217027553065 · pubmed ↗
- 3Corre S. Gailbert M.-D. Upstream stimulating factors: Highly versatile stress-responsive transcription factors Pigment Cell Res.20051833734810.1111/j.1600-0749.2005.00262.x 16162174 · doi ↗ · pubmed ↗
- 4Pawlus M.R. Wang L. Ware K. Hu C.J. Upstream stimulatory factor 2 and hypoxia-inducible factor 2α (HIF 2α) cooperatively activate HIF 2 target genes during hypoxia Mol. Cell. Biol.2012324595461010.1128/MCB.00724-1222966206 PMC 3486188 · doi ↗ · pubmed ↗
- 5Qi L. Allen R.R. Lu Q. Higgins C.E. Garone R. Staiano-Coico L. Higgins P.J. PAI-1 transcriptional regulation during the G 0→G 1 transition in human epidermal keratinocytes J. Cell. Biochem.20069949550710.1002/jcb.2088516622840 · doi ↗ · pubmed ↗
- 6Gailbert M.D. Carreira S. Goding C.R. The Usf-1 transcription factors is a novel target for the stress-responsive p 38 kinase and mediates UV-induced tyrosine expression EMBO J.2001205022503110.1093/emboj/20.17.502211532965 PMC 125271 · doi ↗ · pubmed ↗
- 7Horbach T. Gotz C. Kietzmann T. Dimova E.Y. Protein kinases as switches for the function of upstream stimulatory factors: Implication for tissue injury and cancer Front. Pharmacol.20156310.3389/fphar.2015.0000325741280 PMC 4332324 · doi ↗ · pubmed ↗
- 8Luo X. Sawadogo M. Antiproliferative properties of the USF family of helix-loop-helix transcription factors Proc. Natl. Acad. Sci. USA 1996931308131310.1073/pnas.93.3.13088577760 PMC 40076 · doi ↗ · pubmed ↗