Expanded Clinical Spectrum of Autosomal-Dominant STT3A-CDG
Hamdan Al-Shahrani, Evelin Szabó, Caroline Staccone, Georgia MacDonald, Yutaka Furuta, Daniel Schecter, Andrew C. Edmondson, Anne McRae, Josh Baker, Eva Morava, Rory J. Tinker

TL;DR
This study expands the known clinical features of autosomal-dominant STT3A-CDG, a rare genetic disorder affecting glycosylation, and highlights new symptoms and biochemical patterns.
Contribution
The paper reports new clinical features and biochemical variability in autosomal-dominant STT3A-CDG, including previously unreported symptoms like anorectal malformation and bleeding diathesis.
Findings
Abnormal transferrin glycosylation was present in nearly all individuals (20/21).
Newly reported features include anorectal malformation, morbid obesity, and bleeding diathesis.
Variants clustered in conserved catalytic regions, with p.Arg405 as a recurrent hotspot.
Abstract
STT3A encodes the catalytic subunit of the oligosaccharyltransferase A (OST-A) complex and is classically linked to severe autosomal-recessive congenital disorder of glycosylation (CDG). To define the distinct autosomal-dominant disorder, we reviewed all published cases and integrated three previously unpublished individuals from the CDG natural history study. Across 21 individuals, abnormal transferrin glycosylation was present in nearly all individuals (20/21), and subtle facial dysmorphism was common (18/21). Neurodevelopmental involvement was frequent, including motor delay (13/21), learning difficulties (13/21), speech delay (12/21), and intellectual disability (10/21). Musculoskeletal manifestations were also common, including skeletal abnormalities (12/21), short stature (11/21), muscle cramps (8/21), and early-onset osteoarthritis in adults (6/21). Less frequent features…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
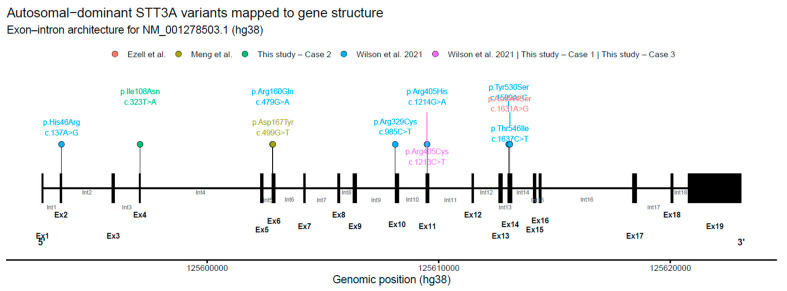 Figure 1
Figure 1- —National Institute of General Medical Sciences
- —National Institute of Neurological Disorders and Stroke (NINDS)
- —National Center for Advancing Translational Sciences (NCATS)
- —Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlycosylation and Glycoproteins Research · Carbohydrate Chemistry and Synthesis · Genomics and Rare Diseases
1. Introduction
Congenital disorders of glycosylation (CDGs) are a heterogeneous group of inherited metabolic diseases caused by defects in the synthesis, processing, or attachment of glycans to proteins and lipids, leading to multisystem disease most often characterized by neurodevelopmental impairment, growth abnormalities, and variable hepatic, endocrine, hematologic, and immune involvement; however, their clinical heterogeneity and limited disease-specific biomarkers frequently result in substantial diagnostic delay [1,2,3,4,5,6]. In this context, STT3A gene (MIM 601134), located on 11q24.2, encodes the catalytic subunit of the OST-A complex responsible for co-translational N-glycan transfer in the endoplasmic reticulum [7,8,9]. STT3A was long believed to cause disease only through autosomal recessive inheritance (MIM 615596), based on early reports of consanguineous families with biallelic missense variants that reduced STT3A abundance, impaired N-glycosylation, and produced a severe neurodevelopmental phenotype [10,11]. Recent genomic and functional studies, however, have revealed a second mechanism: heterozygous, catalytic-site missense variants that exert a dominant-negative effect on OST-A, establishing autosomal dominant STT3A-CDG (MIM 619714) [12]. This shift demonstrates that STT3A defects span both recessive and dominant inheritance and raises new questions regarding penetrance, pathogenic mechanisms, and clinical variability [13].
Dominant STT3A-CDG is characterized by missense variants that alter residues essential for substrate binding or catalytic function, impairing oligosaccharide transfer despite near-normal protein levels. Functional studies (including S. cerevisiae complementation, OST substrate assays, and patient fibroblast analyses) confirm reduced N-glycosylation and a characteristic type I transferrin pattern [12]. Clinically, individuals present with developmental delay, craniofacial differences, short stature, musculoskeletal anomalies, and EEG abnormalities, though severity varies widely [12,14,15]. The coexistence of both dominant and recessive pathogenic mechanisms within STT3A underscores the sensitivity of OST-A to even partial catalytic disruption. Yet the literature remains limited, and the full phenotypic spectrum, natural history, and biochemical consequences of heterozygous STT3A variants are not well defined. More broadly, even as diagnostic yield improves with next-generation sequencing, large rare-disease cohorts continue to show that a substantial fraction of inherited metabolic and rare disorders are missed by traditional screening approaches and are increasingly identified through integrated genomic and multidisciplinary evaluation [16,17,18].
The aim of this study is to synthesize current knowledge of dominant STT3A-CDG, integrate newly described cases and patients from the CDG natural history study with existing mechanistic and clinical data, and clarify how heterozygous catalytic-site variants disrupt OST-A function [19]. We hypothesize that dominant STT3A-CDG is an under-recognized glycosylation disorder with broader phenotypic variability than currently appreciated. Our objectives are to (1) summarize the molecular spectrum underlying the dominant-negative disruption of STT3A, (2) delineate the emerging clinical phenotype and, and (3) identify key gaps that should guide future diagnostic, functional, and natural history studies.
2. Materials and Methods
2.1. Data Sources
We conducted a targeted literature review to identify all published STT3A-CDG cases using terms including “STT3A,” “congenital disorder of glycosylation,” “CDG type Iw,” “dominant-negative glycosylation,” and “oligosaccharyltransferase.” PubMed, Embase, and OMIM were searched without date or language limits, and reference lists were screened to ensure complete ascertainment.
2.2. Natural History Study
We also identified three previously unpublished individuals with heterozygous STT3A variants from the Frontiers of Congenital Disorders of Glycosylation Consortium (FCDGC) natural history study (IRB: 19-016991; ClinicalTrials.gov NCT04199000), drawn from 410 participants in the NIH RDCRN RedCAP database [19]. All three were enrolled at Mount Sinai (New York, NY, USA) within the past year. Data were collected prospectively during study visits and supplemented by retrospective chart review under the IRB-approved protocol.
2.3. Phenotypic Extraction and Transferrin Glycoform Reference Intervals
For published and FCDGC cases, phenotypes were abstracted using a structured framework and mapped to standardized domains (neurodevelopment, growth, craniofacial, musculoskeletal, EEG/neuroimaging, ophthalmologic/systemic findings, and biochemical glycosylation markers). Molecular details (inheritance, variant position, catalytic-site proximity, and predicted functional impact) were recorded where available. Terminology was harmonized across reports to enable comparisons and qualitative synthesis of recurrent features, variability, and genotype–phenotype patterns [20,21]. Transferrin glycoform analysis and ApoC-III isoform profiling were abstracted from clinical laboratory reports and prior publications; testing was performed in CLIA-certified laboratories using standard methods (e.g., isoelectric focusing/capillary electrophoresis with immunofixation and/or mass spectrometry). Because assay platforms and cutoffs varied across sources, we report results as issued by the performing laboratories. Where available, transferrin results are summarized using laboratory-reported glycoform ratios (e.g., mono-oligo/di-oligo, a-oligo/di-oligo, tri-sialo/di-oligo), which reflect enrichment of under-glycosylated transferrin species relative to the normally glycosylated di-oligo fraction; elevations above each laboratory’s upper reference limit were interpreted as consistent with a type I CDG pattern.
2.4. Variant Curation and Gene-Structure Visualization
All reported autosomal-dominant STT3A variants (literature + this study) were curated (cDNA/protein change, inheritance, clinical/biochemical annotations) and standardized to NM_001278503.1 (hg38) using HGVS nomenclature and HPO terms [22,23]. Recurrent variants were retained with provenance but collapsed to a single genomic position for visualization. STT3A exon–intron structure was derived from UCSC hg38 RefSeq (TxDb.Hsapiens.UCSC.hg38.refGene), transcript coordinates were reconstructed from exon intervals, and variants were projected onto the gene model. Schematics were generated in R (v4.5.2) using ggplot2 (exons as boxes, introns as segments, variants as lollipops) and exported at publication resolution.
3. Results
3.1. Initially Reported Autosomal-Dominant STT3A-CDG
The initial report described 16 individuals from 9 families with heterozygous STT3A missense variants, establishing an autosomal-dominant CDG with recurrent features including short stature, macrocephaly, craniofacial dysmorphism, skeletal anomalies, hypertonia, and muscle cramps, with intellectual disability in 50% [12]. Pathogenic variants clustered at the catalytic site, and functional studies supported a dominant-negative mechanism (abnormal glycosylation despite normal STT3A expression; supportive yeast data). Abnormal transferrin glycoforms were notable and helped distinguish dominant STT3A-CDG from recessive STT3A-CDG.
3.2. Recently Reported Individuals Expanding the Phenotypic and Mechanistic Spectrum
Subsequent reports have described two additional individuals with de novo heterozygous STT3A variants, further expanding the recognized clinical and mechanistic spectrum of autosomal-dominant STT3A-CDG (Table 1, main text). These individuals shared core features with the originally reported cohort, including developmental delay, short stature, craniofacial dysmorphism, and epilepsy or abnormal EEG findings, while also exhibiting broader neurobehavioral phenotypes such as autism spectrum disorder and attention-deficit/hyperactivity disorder in some cases. Molecular and functional data from these studies further strengthened the dominant disease model, demonstrating variant-specific disruption of glycosylation sites, reduced STT3A protein levels in vitro, and in vivo validation using heterozygous zebrafish knockdown models that recapitulated key patient phenotypes, including craniofacial and skeletal anomalies, developmental delay, behavioral alterations, and electrophysiological abnormalities.
3.3. Unpublished Cases Expanding the Phenotypic Spectrum of Dominant STT3A-CDG from the Natural History Study
Patient 1. An 18-month-old female with a de novo heterozygous pathogenic STT3A variant (c.1213C>T; p.Arg405Cys) was diagnosed at birth and followed for growth and developmental concerns. She had an anorectal malformation and a sacral dimple. Family history was noncontributory. Her course included short stature, feeding intolerance, chronic otitis media and developmental delay (early speech delay requiring therapy). Cardiac findings included a resolved PFO and a bicuspid aortic valve. By 18 months she had achieved major motor milestones without regression. Growth remained <10th percentile. Laboratory testing showed mild anemia, transient hypoglycemia, and low factor VIII activity. CDG biomarker testing demonstrated elevated mono-oligo/di-oligo and asialo/di-oligo ratios, consistent with a type-I CDG pattern. Examination showed good social engagement, preserved tone, and a stable gait with mild truncal sway, along with dysmorphic features (hypertelorism, flat nasal bridge, prominent glabella, smooth philtrum). Overall, her presentation is consistent with the expanding phenotype of dominant STT3A-CDG; the anorectal malformation may represent a rare or previously underrecognized feature. Her Nijmegen Progression CDG Rating Scale score was 9, indicating mild severity.
Patient 2. A 10-year-old male twin B born premature at 30 weeks of gestation who had a multisystem disease including spontaneously resolved ASD/VSD, repaired laryngeal cleft, early-life seizures (with normal brain MRI), recurrent respiratory/skin infections, easy bruising, livedo reticularis, scoliosis, and severe recurrent muscle cramps. The spine MRI showed a small thoracic syrinx. He had early growth delay followed by rapid pubertal weight gain (weight 99th percentile; height 54th), persistent learning difficulties and behavioral abnormalities requiring medication. Family history was noncontributory. Chromosomal microarray was normal. Whole-exome sequencing identified a heterozygous STT3A likely pathogenic variant (c.323T>A; p.Ile108Asn). Biochemical testing showed mild ApoC-III hypo-glycosylation with normal transferrin glycosylation (mono/di-oligo 0.03; asialo/di-oligo 0.000; trisialo/di-oligo 0.02), consistent with a atypical biochemical profile despite a clear molecular diagnosis of autosomal-dominant STT3A-related CDG. The Nijmegen Progression CDG Rating Scale score was 20 (moderate severity).
Patient 3. An 8-year-old female was diagnosed with autosomal-dominant STT3A-CDG during evaluation for lifelong growth delay. Born at 27 weeks, she had early feeding intolerance (resolved) but ongoing cyclic vomiting and global developmental delay. Additional features include short stature on growth hormone, hypotonia with mild coordination difficulties, speech/cognitive delays, ADHD, and anxiety. Dysmorphology included hypertelorism with vertically narrow palpebral fissures, bushy eyebrows, and a bulbous nasal tip with a midline nasal dimple, as well as a prominent forehead and mild facial asymmetry. Ophthalmology showed mild hyperopia; cardiac workup was unremarkable aside from a resolved PFO. She has a significant bleeding phenotype with very low vWF activity (<20%) and factor VIII (10%) with prolonged aPTT, consistent with reported STT3A-CDG-associated coagulopathy. She has had no regression and no confirmed seizures. Biochemical testing supported a CDG type I transferrin pattern. Serum transferrin glycoform analysis was interpreted as abnormal and suggestive of a CDG type I profile, with elevated mono-oligo/di-oligo ratio (0.24) and a-oligo/di-oligo ratio (0.017), while tri-sialo/di-oligo ratio was 0.02. Apolipoprotein CIII ratios were also reported (Apo CIII-1/CIII-2 2.55; Apo CIII-0/Apo CIII-2 0.48). Exam showed short stature with preserved mobility. She was diagnosed with a heterozygous likely pathogenic STT3A variant (c.1213C>T; p.Arg405Cys), inherited from her mother with evidence of maternal mosaicism on Genome sequencing. The Nijmegen Progression CDG Rating Scale: 14 (mild severity).
3.4. Genotype Summary of Autosomal-Dominant STT3A-CDG
Across published reports and the present study, we identified multiple heterozygous missense variants in STT3A reported in individuals with an autosomal-dominant congenital disorder of glycosylation phenotype (Table 1; Supplementary Table S2; Figure 1). The variants affect highly conserved residues and, based on prior structural and functional work, are consistent with involvement of sequence elements important for STT3A catalytic function within the oligosaccharyltransferase complex. Most variants were reported as de novo, although several were inherited, including variants associated with milder or atypical biochemical phenotypes (Supplementary Table S2). A recurrent p.Arg405 variant was observed in multiple unrelated individuals, including de novo, inherited, and parental mosaic cases, suggesting a mutational hotspot and variable inheritance mechanisms (Supplementary Table S2; Figure 1). While variants are distributed across multiple exons, they appear to cluster in regions likely to be functionally important, compatible with a shared pathogenic mechanism. When mapped onto the exon–intron structure of STT3A, disease-associated variants were not evenly distributed and were enriched in coding exons corresponding to the central portion of the transcript (Figure 1). Collectively, these observations support STT3A as a gene in which specific missense variants can be associated with dominant disease and variable expressivity.
3.5. Phenotypic Summary of Dominant STT3A-CDG from All Published and Unpublished Cases
Across a combined cohort of 21 unrelated families, including all published cases and the new cases reported here, with autosomal dominant STT3A associated congenital disorder of glycosylation, a variable but recognizable multisystem phenotype was observed (Table 2). Biochemically, abnormal serum transferrin glycosylation consistent with a type I CDG was identified in nearly all individuals (20 of 21), and fibroblast studies uniformly demonstrated glycoprotein hypoglycosylation when tested (Table 2). Phenotypically, first, subtle facial dysmorphism was present in most individuals (18 of 21), with recurrent features including high anterior hairline, short palpebral fissures, and thin upper lip vermilion. Second, neurodevelopmental involvement was common, with motor delay (13 of 21), learning difficulties (13 of 21), and speech delay (12 of 21) frequently reported; intellectual disability was present in approximately half of the cohort (10 of 21). Third, musculoskeletal manifestations were prominent, including skeletal abnormalities (12 of 21), muscle cramps (8 of 21), and early-onset osteoarthritis in several adults (6 of 21). Short stature (11 of 21) and macrocephaly (7 of 21) were also observed. Seizures and behavioral diagnoses were infrequent. Stratification by inheritance demonstrated substantial phenotypic overlap between de novo and inherited autosomal-dominant cases (Supplementary Table S3). Neurodevelopmental features were more frequently reported among individuals with de novo variants, whereas musculoskeletal and biochemical findings were shared across both groups. Given the limited cohort size and potential ascertainment bias, these differences should be interpreted cautiously.
Compared with autosomal-recessive STT3A-CDG, the autosomal-dominant form is distinguished by heterozygous missense variants clustered in highly conserved catalytic/active-site residues, consistent with a dominant-negative, qualitative disruption of OST-A enzymatic activity despite near-normal STT3A mRNA/protein levels (Table 3). In contrast, recessive STT3A-CDG reflects loss-of-function with reduced STT3A abundance/function and a quantitative reduction in overall OST-A activity, and typically presents as a more severe, early-onset, relatively homogeneous neurodevelopmental disorder with broader multisystem involvement (including prominent feeding difficulties and reported factor VIII/von Willebrand factor deficiency). Overall, dominant STT3A-CDG shows marked inter-individual variability with prominent musculoskeletal features and variable neurodevelopmental impact, whereas the recessive phenotype is generally more consistently severe neurologically.
4. Discussion
In this study, we integrate all published evidence on autosomal-dominant STT3A-related congenital disorder of glycosylation with three newly described individuals from the CDG natural history study, expanding both the genotypic and phenotypic spectrum of dominant STT3A-CDG. Our novel cases support that heterozygous missense variant in the catalytic core act through a dominant-negative mechanism, disrupting OST-A-mediated N-glycosylation [24]. Importantly, we identify novel phenotypic features in autosomal-dominant STT3A-CDG that broaden the recognized organ system involvement beyond prior reports. In addition to cardiac malformations and morbid obesity, which were not emphasized in earlier descriptions, we further extend the phenotype to include anorectal malformation, clinically significant bleeding diathesis characterized by marked reductions in von Willebrand factor activity and factor VIII levels, and behavioral abnormalities including attention-deficit/hyperactivity disorder and anxiety. These findings expand the clinical boundaries of dominant STT3A-CDG to encompass congenital structural anomalies, more severe hematologic involvement than previously appreciated, and neurobehavioral manifestations, thereby refining both diagnostic recognition and anticipatory management of affected individuals (Table 2) [9]. We also observed some variability in biochemical signatures: predominantly classic Type I patterns, with infrequent subtle or atypical deviations [25,26]. Recurrent involvement of p.Arg405, including de novo occurrence, inheritance, and parental mosaicism, highlights a mutational hotspot and underscores variable expressivity and penetrance [27,28].
In the context of prior reports, the original description of dominant STT3A-CDG emphasized short stature, craniofacial differences, macrocephaly, skeletal anomalies, and variable neurodevelopmental involvement, with variants localizing to the catalytic site [12,14,15]. Subsequent publications added neurobehavioral phenotypes and provided in vivo validation in zebrafish models [15]. Our expanded cohort is broadly concordant with these observations, but suggests a wider clinical range, including congenital malformations and clinically meaningful coagulation abnormalities. We also confirm that transferrin testing can be insensitive in most dominant cases; but in addition to very mild, a normal transferrin profile does not exclude the diagnosis when clinical and genetic findings are supportive.
Compared with autosomal-recessive STT3A-CDG, the dominant disorder differs substantially, as recessive STT3A-CDG typically reflects biallelic variants that reduce STT3A abundance, produce more global impairment of N-glycosylation, and cause a more severe, early-onset neurodevelopmental phenotype. While both conditions reflect OST-A dysfunction, the clinical consequences diverge in severity and consistency: recessive disease is generally more severe and homogeneous, whereas dominant disease shows greater inter-individual variability and survival into adulthood with relatively preserved function in some individuals [10]. Together, these patterns support a model in which quantitative loss of STT3A activity drives severe multisystem disease, while qualitative catalytic disruption in the presence of wild-type protein produces a dominant-negative, context-dependent glycosylation defect.
Several limitations should be acknowledged. Case numbers remain modest, limiting genotype–phenotype inference. Phenotypes were derived partially from heterogeneous sources (literature review), increasing reporting and ascertainment bias; however, the newly described individuals were ascertained through and systematically phenotyped as part of a CDG natural history study, providing more standardized clinical characterization. Biochemical evaluation was not standardized across individuals, and mild abnormalities may be missed by transferrin-only assays. Adult natural history data remain sparse, limiting conclusions about long-term morbidity and late-onset complications.
Despite these constraints, the work has diagnostic and research implications. Dominant STT3A-CDG should be considered in individuals with unexplained short stature, skeletal anomalies, neurodevelopmental differences, bleeding diathesis, or multisystem involvement. Even when transferrin studies are normal. Mechanistically, STT3A defect provides a clear example of dominant-negative enzymatic dysfunction within a multiprotein complex, with broader relevance to glycosylation biology. Future priorities include standardized biochemical phenotyping, functional validation of additional variants, and prospective natural history studies to clarify disease trajectory, penetrance, and late complications. The potential role of modifier genes and environmental factors in shaping expressivity also warrants investigation.
5. Conclusions
Autosomal-dominant STT3A-CDG is an under-recognized, variably expressive multisystem glycosylation disorder driven by dominant-negative catalytic-site missense variants, and our integrated cohort broadens its diagnostic spectrum, highlighting that normal or subtle transferrin profiles do not exclude the diagnosis and that congenital malformations and clinically significant bleeding can be key clues.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Francisco R. Brasil S. Poejo J. Jaeken J. Pascoal C. Videira P.A. dos Reis Ferreira V. Congenital disorders of glycosylation (CDG): State of the art in 2022 Orphanet J. Rare Dis.20231832910.1186/s 13023-023-02879-z 37858231 PMC 10585812 · doi ↗ · pubmed ↗
- 2Tinker R.J. Fisher M. Gimeno A.F. Gill K. Ivey C. Peterson J.F. Bastarache L. Diagnostic delay in monogenic disease: A scoping review Genet. Med. Off. J. Am. Coll. Med. Genet.20242610107410.1016/j.gim.2024.101074 PMC 1114058838243783 · doi ↗ · pubmed ↗
- 3Lipiński P. Tylki-Szymańska A. Congenital Disorders of Glycosylation: What Clinicians Need to Know?Front. Pediatr.2021971515110.3389/fped.2021.71515134540767 PMC 8446601 · doi ↗ · pubmed ↗
- 4Sparks S.E. Krasnewich D.M. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview Gene Reviews® Adam M.P. Bick S. Mirzaa G.M. Pagon R.A. Wallace S.E. Amemiya A. University of Washington Seattle, WA, USA 199320301507 · pubmed ↗
- 5Ferreira C.R. Rahman S. Keller M. Zschocke J. ICIMD Advisory Group An international classification of inherited metabolic disorders (ICIMD)J. Inherit. Metab. Dis.20214416417710.1002/jimd.1234833340416 PMC 9021760 · doi ↗ · pubmed ↗
- 6Shah R. Eklund E.A. Radenkovic S. Sadek M. Shammas I. Verberkmoes S. Ng B.G. Freeze H.H. Edmondson A.C. He M. ALG 13-Congenital Disorder of Glycosylation (ALG 13-CDG): Updated clinical and molecular review and clinical management guidelines Mol. Genet. Metab.202414210847210.1016/j.ymgme.2024.10847238703411 PMC 11402470 · doi ↗ · pubmed ↗
- 7Shrimal S. Cherepanova N.A. Gilmore R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum Semin. Cell Dev. Biol.201541717810.1016/j.semcdb.2014.11.00525460543 PMC 4442082 · doi ↗ · pubmed ↗
- 8Kelleher D.J. Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase Glycobiology 20061647 R 62R 10.1093/glycob/cwj 06616317064 · doi ↗ · pubmed ↗