BRAF Mutations in Myeloid Neoplasms: Prevalence, Co-Mutation Landscape, and Clinical Outcomes—A Comprehensive Review
Shehab F. Mohamed, Ali Mohamed, Mohamed Fawzi Mudarres, Azza E. A. Abdalla, Abdulrahman F. Al-Mashdali, Mohammed Abdulgayoom, Rowan Mesilhy, Tareq Abuasab, Honar Cherif, Gautam Borthakur

TL;DR
This paper reviews the role of BRAF mutations in myeloid cancers, showing they are rare, often linked to poor outcomes, and rarely respond well to targeted therapies.
Contribution
The paper provides a comprehensive synthesis of BRAF mutation prevalence, co-mutations, and clinical outcomes in myeloid neoplasms.
Findings
BRAF mutations are rare in myeloid neoplasms, occurring in less than 1% of cases.
V600E and non-V600E BRAF mutations are often found alongside other mutations like ASXL1 and TET2.
Targeted MAPK inhibition shows limited long-term effectiveness in treating BRAF-mutated myeloid cancers.
Abstract
Background: BRAF is a core component of the RAS–MAPK signaling pathway and an established oncogenic driver in several solid tumors and selected hematologic malignancies. In myeloid neoplasms, BRAF mutations are rare, and their prevalence, molecular context, and clinical significance remain incompletely defined. Available evidence is scattered across heterogeneous reports involving acute myeloid leukemia, myelodysplastic syndromes, myeloproliferative neoplasms, and overlap myelodysplastic/myeloproliferative neoplasms, with variable descriptions of mutation subtypes, co-mutational profiles, cytogenetic associations, therapeutic approaches, and clinical outcomes. To address these gaps, this review synthesizes data from the published literature up to 2025, summarizing the distribution, genetic landscape, and clinical impact of molecularly confirmed BRAF mutations across the spectrum of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
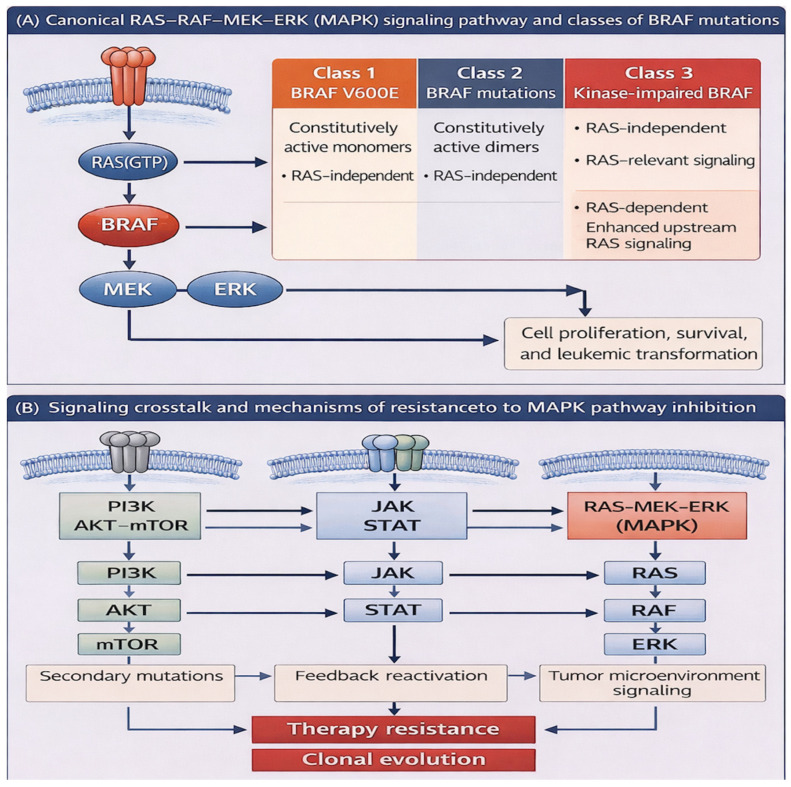 Figure 1
Figure 1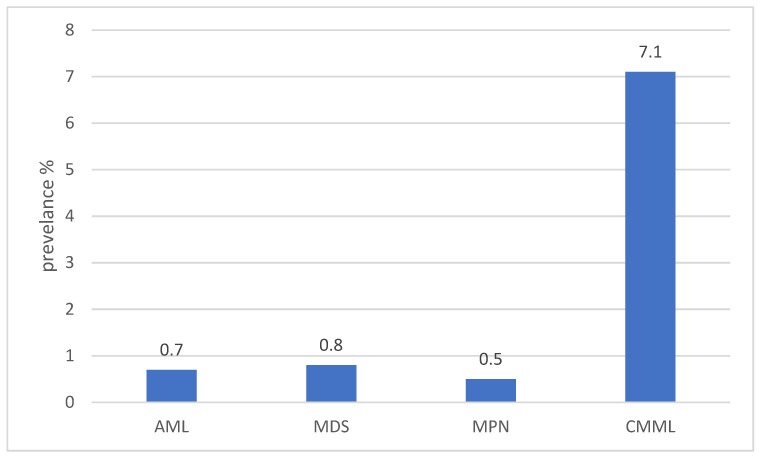 Figure 2
Figure 2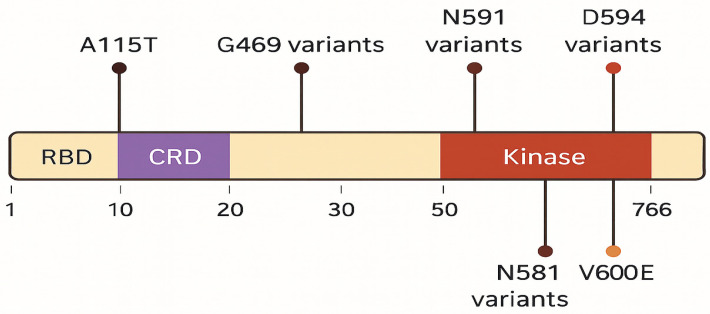 Figure 3
Figure 3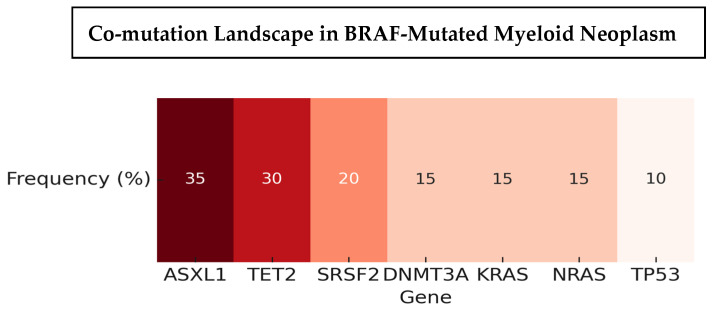 Figure 4
Figure 4| ID | Study | Total BRAF-Mutated Cases | Underlying Disease Category (n) | Reported Subtype/Clinical Classification (n) |
|---|---|---|---|---|
| 1 | Abuasab et al., 2024 [ | 48 | AML (18), CMML (12), MDS (10), MF (5), MDS/MPN (1), APL (1), Myeloid sarcoma (1) | s-AML (12), t-MDS (4) |
| 2 | Zhang et al., 2014 [ | 5 | CMML | CMML-1 (2), CMML-2 (3) |
| 3 | Christiansen et al. (2005) [ | 3 | Therapy-related AML | AML-M5 |
| 4 | Fei et al. (2024) [ | 14 | AML (7), MPN (3), MDS (2), MDS/MPN (1), unclear (1) | Acute monocytic AML; AML with monocytic differentiation; ET |
| 5 | George et al. (2024) [ | 2 | s-AML | AML with monocytic features |
| 6 | Abu-Shihab et al. (2023) [ | 32 | AML | De novo AML (19), relapsed/refractory AML (8), secondary AML (15) |
| 7 | Kandarpa et al. (2017) [ | 2 | MPN | Post-ET myelofibrosis |
| 8 | Santos et al. (2014) [ | 1 | MDS/MPN overlap | Ph-negative MDS/MPN-U |
| 9 | Papaemmanuil et al. (2016) [ | 9 | AML | De novo AML; therapy-related AML; secondary AML |
| 10 | Xu et al. (2017) [ | 4 | AML | Monoblastic AML |
| 11 | Lee et al. (2004) [ | 2 | AML | Biphenotypic AML (1); AML with maturation (1) |
| 12 | Lee et al. (2025) [ | 50 | AML | De novo AML (21), secondary AML (20), relapsed/refractory AML (9), AML-MR (34) |
| ID | Study | BRAF Mutation/VAF | Co-Mutations | Karyotype/Cytogenetics |
|---|---|---|---|---|
| 1 | Abuasab et al., 2024 [ | G469A, V600E, others; median VAF 8.6% (1.3–86.6) | KRAS, NRAS, ASXL1, TET2, SRSF2, TP53, CBL, DNMT3A | 44% diploid; 19% high-risk; poor risk in AML |
| 2 | Zhang et al., 2014 [ | D594E, N581S, L597Q, G466E (exon 11/15) | RAS WT | Low risk |
| 3 | Christiansen et al. (2005) [ | V600E | AML1, CBFb, MLL, RARa, KRAS | Recurrent balanced translocations; +8; MLL-rearrangements |
| 4 | Fei et al. (2024) [ | V600E, D594G, N581S, others (exons 6, 11, 15, 17) | NRAS, KRAS, DNMT3A, TET2, ASXL1, IDH1, JAK2, TP53, etc. | Normal: 6; Abnormal: 7 (del5q, +8, del9q, del20q, etc.); KMT2A fusions |
| 5 | George et al. (2024) [ | V600E, N581S | TET2, KRAS, ZRSR2, EZH2, RUNX1T1, RAF1 | Complex karyotype; KMT2A rearrangements |
| 6 | Abu-Shihab et al. (2023) [ | G469, D594, others; VAF 1–83% | TET2, ASXL1, NRAS, KRAS, RUNX1, DNMT3A, FLT3, NPM1, SRSF2 | NA |
| 7 | Kandarpa et al. (2017) [ | D594E, V600E, G469V | JAK2, ASXL1, ASXL2, TP53, NF1, PIK3R3, KMT2C | NA |
| 8 | Santos et al. (2014) [ | D594G | JAK2 | del(5q) |
| 9 | Papaemmanuil et al. (2016) [ | V600E, D594N, L597Q, A115T | ASXL1, DNMT3A, EZH2, FLT3, IDH1/2, NRAS, RUNX1, TP53, etc. | Favorable: 205; Intermediate: 960; Adverse: 253 |
| 10 | Xu et al. (2017) [ | V600E, D594G, K601E (exons 11,15) | NPM1, ASXL | Normal: 2; Abnormal: +8, der (1;12), t(10;11) |
| 11 | Lee et al. (2004) [ | Exon 11 | NA | NA |
| 12 | Lee et al. (2025) [ | V600, G469, D594, others | TET2, NPM1, NRAS, KRAS, BRAF, TP53, and SRSF2. | MECOM-rearrangements, RUNX1:RUNX1T1 fusion |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Retinoids in leukemia and cellular processes · Chronic Myeloid Leukemia Treatments
1. Introduction
The B-Raf proto-oncogene, serine/threonine kinase (BRAF) is a key component of the RAS–MAPK/ERK signaling pathway, which regulates cellular proliferation, differentiation, and survival [1]. Located on chromosome 7q34, BRAF encodes a 766–amino acid protein with three conserved regions: CR1, which contains the RAS-binding domain (RBD); CR2, a serine-rich hinge region; and CR3, the kinase domain responsible for MEK phosphorylation [2]. Under normal conditions, BRAF remains inactive through intramolecular interactions, with activation occurring when GTP-bound RAS engages the RBD. This conformational shift halts autoinhibition, promotes membrane localization and dimerization, and enables phosphorylation of downstream MEK–ERK targets [1,3,4]. The canonical RAS–RAF–MEK–ERK signaling cascade and its interaction with other major signaling pathways implicated in leukemogenesis are illustrated in Figure 1. Oncogenic mutations disrupt this regulation, driving constitutive pathway activation and uncontrolled proliferation [2].
BRAF mutations are established oncogenic drivers in multiple solid tumors, including melanoma (approximately 60%), papillary thyroid carcinoma (40–60%), colorectal cancer (10–15%), and non-small-cell lung cancer (1–4%), as well as subsets of glioblastoma and lymphoma [2,5,6]. More than 200 variants have been described, with nearly 30 functionally characterized. These mutations fall into three classes: class 1 (e.g., V600E), which act as constitutively active monomers; class 2, which signal as active dimers independent of RAS; and class 3, which are kinase-impaired but amplify upstream RAS activity [1,7]. This classification has therapeutic relevance, as class 1 and 2 tumors often respond to direct BRAF inhibition, whereas class 3 mutations are typically dependent on upstream RAS or receptor tyrosine kinase signaling [7].
In hematologic malignancies, BRAF mutations are rare. They are clinically significant in hairy cell leukemia, where V600E serves as both a diagnostic marker and therapeutic target [8,9], but have been reported in fewer than 1–2% of myeloid neoplasms. Notably, the spectrum of BRAF mutations in myeloid disease appears to include a higher proportion of non-V600E variants, whose functional and clinical significance remain unclear [5]. Given the scarcity of cases, available data is limited to small series and case reports with heterogeneous methods and outcomes. This review synthesizes published evidence up to 2025 on molecularly confirmed BRAF mutations across the major categories of myeloid neoplasms, including acute myeloid leukemia, myelodysplastic syndromes, myeloproliferative neoplasms, and myelodysplastic/myeloproliferative overlap disorders. We integrate available data on mutation prevalence, variant subtype, variant allele frequency, co-occurring genetic alterations, cytogenetic features, treatment strategies, and clinical outcomes, with particular attention to patterns of mutation acquisition, therapeutic response, and prognostic implications. By consolidating these dispersed observations, this review aims to clarify the emerging biological and clinical landscape of BRAF-mutated myeloid neoplasms and to highlight areas requiring further investigation.
2. Results
2.1. Study Characteristics
Across 12 published studies, research designs included retrospective and prospective cohorts, cross-sectional analyses, and case series. Sample sizes ranged from 2 patients to more than 6000. Mutation detection methods also varied over time.
Early studies used PCR-based assays, such as restriction fragment length polymorphism (RFLP) analysis [10], single-stranded conformational polymorphism (SSCP) [11,12], and allele-specific PCR for BRAF V600E [13].
In contrast, more recent studies relied on next-generation sequencing (NGS). These included targeted myeloid panels of different sizes, such as 28-gene, 53-gene, or 81-gene assays [14], as well as broader clinical panels like the HopeSeq Heme panel [15]. Other approaches included capture-based and amplicon-based sequencing [16,17,18,19,20].
2.2. Prevalence, Clinical Features, and Genetic Landscape
The overall prevalence of BRAF mutations in myeloid neoplasms was consistently low, generally below 1%. Reported frequencies were 0.5–1% in acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), and chronic myelomonocytic leukemia (CMML), though CMML-specific cohorts demonstrated higher rates, reaching up to 7.1% (Figure 2). Sporadic cases were also described in acute promyelocytic leukemia (APL), myeloid sarcoma, and biphenotypic acute leukemias (Table 1). biomedicines-14-00672-t001_Table 1Table 1Summary of published studies on BRAF mutations in myeloid neoplasms.IDStudyDOIStudy DesignTotal Sample SizeBRAF PrevalenceAgeGender (M/F)1Abuasab T et al. (2024) [14]10.1080/10428194.2024.2347539Retrospective cohort study666748/6667 (0.7%)- AML: 18/2438 (0.7%)- MDS/CMML: 22/2538 (0.8%)- MPN: 5/939 (0.5%)Median: 70 yo (24–89)28/202Zhang et al. (2014) [16]10.1002/ajh.23652Retrospective cohort study705/70 (7.1%)Median: 67.8 yo (28–86)37/333Christiansen et al. (2005) [10]10.1038/sj.leu.2404009Retrospective cohort study140t-AML: 3/51 (5.8%)t-MDS: 047, 65, 69 yo1/24Fei et al. (2024) [15]10.3390/ijms25105183Retrospective cohort study263214/2632 (0.53%)Mean: 63.9 yo (23–89)10/45George et al. (2024) [17] 10.3390/genes15111383Case series216/1600 (1%)2 cases detailed2, 75 yo2/06Abu-Shihab et al. (2023) [18] 10.1093/heqpro/daad094Cross-sectional study42NAMedian: 67 yo (19–84)24/187Kandarpa et al. (2017) [19] 10.1002/ajh.24728Retrospective cohort study82/765, 83 yo1/18Santos et al. (2014) [21]10.1182/blood.V124.2.1.3172.3172Case series871/87 (1.2%)NANA9Papaemmanuil et al. (2016) [20] 10.1056/NEJMoa1516192Prospective cohort study15409/1540 (0.6%)18–84 yo823/71910Xu et al. (2017) [22]10.1080/10428194.2016.1213830Cross-sectional study3394/339 (1%)0.5, 49, 59, 60 yo2/211Lee et al. (2004) [11]10.1038/sj.leu.2403201Retrospective cohort study902/90 (2.2%)20–80 yoNA12Lee et al. (2025) [23]10.1101/2025.10.14.682328Retrospective cohort study577950/5779 (1%)Median: 67 yo (19–84)27/23Abbreviations: yo = years old; M = male; F = female; NA = not available; AML = acute myeloid leukemia; MDS = myelodysplastic syndrome; MPN = myeloproliferative neoplasm; CMML = chronic myelomonocytic leukemia; t-AML = therapy-related AML. Note: For studies reporting multiple myeloid neoplasm (MN) subtypes, the number of cases in each subtype is indicated within the corresponding table cell. Across larger cohorts, BRAF-mutated patients were typically older adults, with median ages in the sixth to seventh decade; sex distribution was generally balanced when reported [10,11,14,15,16,17,18,19,20,21,22]. Mutations occurred across a range of myeloid neoplasms (Table 2). However, they were relatively enriched in chronic myelomonocytic leukemia and in therapy-related or secondary AML, where frequencies exceeded those seen in de novo AML [11,14,18].
The spectrum of BRAF variants was heterogeneous, including V600E, G469A/V, D594E/G, L597Q/R, N581S/I/K, and K601E, with variant allele frequencies ranging from 1% to over 80% (Figure 3). Co-mutations were frequent and recurrent across studies. The most common partners included ASXL1, TET2, DNMT3A, and splicing factor mutations such as SRSF2, along with recurrent RAS-pathway lesions (KRAS/NRAS) (Figure 4). Other reported partners included IDH1, EZH2, FLT3-ITD, RUNX1, JAK2, TP53, and NPM1. Only a minority of cases carried BRAF mutations as solitary events, suggesting that BRAF usually occurs within a multi-hit genomic context. Cytogenetic profiles ranged from normal karyotype to high-risk abnormalities, such as monosomy 7, complex karyotypes, and KMT2A rearrangements (Table 3).
2.3. Survival Outcomes
Survival outcomes varied by disease subtype and genetic background. In AML, prognosis was generally poor, with median overall survival (OS) ranging from 126 days to 7 months [14,15,17,18]. Clearance of the BRAF mutation, when documented, correlated with improved outcomes (OS 34.8 vs. 10.4 months, p = 0.047) [11,14]. In MDS and CMML, survival was somewhat longer, typically 16–22 months [11,14,17], particularly in patients with diploid cytogenetics and fewer co-mutations (Supplementary Table S2) In many cases, BRAF mutations occurred within complex genomic backgrounds that included established adverse-risk mutations (e.g., ASXL1, TP53, DNMT3A, and RAS-pathway genes), making it difficult to attribute clinical outcomes specifically to BRAF alterations.
2.4. Treatment Approaches and Outcomes
Differences in treatment strategies and clinical outcomes across disease subtypes are summarized in Table 4. In AML, most patients received intensive induction chemotherapy, with generally poor outcomes; occasional prolonged survival was observed in those who achieved BRAF clearance. MAPK inhibitors (dabrafenib, trametinib, vemurafenib) were used mainly in relapsed or refractory settings, producing transient hematologic or morphologic responses without durable molecular remissions [24,25,26,27,28,29]. In MDS and CMML, hypomethylating agents were the most common therapy, with modest clinical improvements but rare molecular clearance; hydroxyurea was also frequently used in CMML. In MPN, patients generally received conventional agents such as hydroxyurea or ruxolitinib, with no consistent evidence for BRAF-targeted therapy.
Outcomes differed substantially by disease subtype. AML harboring BRAF mutations carried the poorest prognosis, with median OS typically ranging from 4 to 7 months [14,17,19]. Clearance of the BRAF mutation, when achieved, was associated with longer survival (34.8 vs. 10.4 months) and may be enhanced by HSCT [11]. Secondary and therapy-related AML were enriched for high-risk cytogenetic abnormalities, including KMT2A rearrangements and monosomy 7, and were associated with inferior outcomes, often measured in weeks [10,15,21]. In contrast, de novo AML cases could achieve initial remissions with intensive chemotherapy, although relapse was common and durable molecular clearance of BRAF was rare [17,18]. Patients with MDS and CMML demonstrated relatively more favorable outcomes, with median OS of approximately 16–22 months in larger cohorts [11,14,17]. Notably, Zhang et al. (2014) reported improved survival among CMML-1 patients with low-risk cytogenetics, a finding that was not consistently reproduced in subsequent datasets [16]. Data on MPN were limited; however, co-mutation with RAS-pathway genes was associated with aggressive disease biology and progression to AML despite therapy [20,22].
2.5. Published Case Reports of BRAF-Altered Myeloid Neoplasms
Six detailed case reports published between 2015 and 2025 were also identified, originating from Australia, the United States, France, and India. Patient ages ranged from early childhood to 78 years, with equal sex distribution (Table S1). Underlying diagnoses included chronic myeloid leukemia (CML), therapy-related AML, CMML, therapy-related MDS, and post–acute lymphoblastic leukemia AML. There was also one case of CMML with Langerhans cell histiocytoma, the mutation was harbored in both myeloid neoplasms. BRAF p.V600E was present in five of six cases, while the remaining report described a BRAF-wild-type patient with MAPK pathway activation. Co-mutations included KRAS, TET2, and SRSF2, often accompanied by high-risk cytogenetics such as t(9;11) KMT2A–MLLT3, t(9;22) BCR–ABL1, +8, del(7q), and +5. Treatments ranged from conventional chemotherapy and hypomethylating agents to MAPK inhibitors, with responses varying from transient cytoreduction to complete remission. Durable responses were rare; relapses often coincided with KRAS-mutated subclones. Death occurred in four cases, typically within a short time frame due either to disease progression or treatment-related complications. One patient was reported to be in preparation for haploidentical bone marrow transplantation at last follow-up.
3. Discussion
BRAF mutations are rare in myeloid neoplasms, occurring in <1% of unselected cohorts and more commonly in CMML and therapy-related AML [10,11,14,15,16,17,18,19,20,21,22]. Most reported BRAF mutations were clustered within the kinase domain, specifically involving the activation segment (exon 15, including V600). Despite the overall poor outcomes, achieving clearance of BRAF mutations was associated with longer survival, and this clearance may be augmented by HSCT. These findings highlight the clinical relevance of identifying BRAF mutations despite their low prevalence.
When compared with other hematologic malignancies, the role of BRAF in myeloid disease is far less definitive. In hairy cell leukemia, BRAF V600E is disease-defining and therapeutically targetable [8,9]. In Langerhans cell histiocytosis and Erdheim–Chester disease, BRAF or MAPK pathway mutations occur in 50–70% of cases and underpin the dramatic success of BRAF/MEK inhibitors, now approved in these settings [30,31,32,33,34]. In multiple myeloma, BRAF V600E is rare but associated with more aggressive disease and inferior outcomes [35,36]. By contrast, in myeloid neoplasms, BRAF mutations arise infrequently and within a heterogeneous genomic background, limiting their utility as stand-alone biomarkers or therapeutic targets [10,11,14,15,16,17,18,19,20,21,22]. Indeed, some studies such as Shin et al. (2016) have reported no BRAF mutations at all, underscoring the variability across cohorts [37].
The biological implications of these findings are important. Canonical class 1 mutations, such as V600E, act as constitutively active monomers, while class 2 and class 3 variants rely on dimerization or upstream RAS activation. Their frequent co-occurrence with epigenetic regulators (ASXL1, TET2, DNMT3A), splicing factors (SRSF2), and RAS pathway mutations (KRAS/NRAS) suggest that BRAF is rarely an initiating lesion in myeloid disease but rather contributes to clonal evolution and progression. These co-occurrences likely reflect shared clonal architecture and disease biology rather than a direct functional interaction between BRAF and specific co-mutated genes. Given the reported adverse prognostic impact of many of these co-mutations, the available data do not allow reliable assessment of an independent biological or prognostic contribution attributable specifically to BRAF mutations. However, we also acknowledge that some cases describe BRAF as the sole detectable abnormality (within the limits of the assays used), raising the possibility that BRAF could act as a more central driver in select patients. Because the included studies span different testing eras and platforms (PCR vs broader NGS), improvements in sequencing breadth and sensitivity may increasingly clarify whether there exists a subset with more “isolated” BRAF lesions or more interpretable clonal architecture.
Reports of clonal dynamics support this view: in some patients, BRAF persists or expands at relapse, while in others it is replaced by alternative drivers, underscoring its instability as a therapeutic target [11,14].
From a clinical standpoint, BRAF testing may be most relevant in contexts where it is enriched, such as CMML and therapy-related AML [11,14,19]. The relative enrichment of BRAF mutations in myeloid neoplasms with prominent monocytic differentiation, including CMML and acute monocytic leukemia, suggests a potential lineage-specific biological context. Although MAPK signaling is ubiquitous across cell types, monocytic lineage cells exhibit heightened dependence on tightly regulated MAPK activity during differentiation and inflammatory activation [38]. Parallels can be drawn with histiocytic disorders such as Langerhans cell histiocytosis and Erdheim–Chester disease, which arise from monocyte-derived cells and are characterized by frequent MAPK pathway mutations. These observations raise the possibility that monocytic-lineage cells may be particularly permissive to MAPK-driven clonal expansion; however, this association remains speculative and requires dedicated mechanistic and functional studies for validation. Standard therapies remain ineffective, with short survival in AML and modest benefit in MDS and CMML. Responses to BRAF/MEK inhibitors have been observed but are typically transient, without durable molecular clearance [11,14,30,31,32,33,34]. Achieving molecular mutation clearance has been reported to be prognostically meaningful, and HSCT may offer a survival benefit when clearance is attained. However, this observation is currently supported primarily by a single retrospective study (Tariq et al.) and therefore remains hypothesis-generating and in need of validation in larger, independent cohorts and prospective studies [11,14]. These observations suggest that BRAF clearance could be explored as a potential biomarker of treatment response.
Several limitations affect interpretation. Most data come from retrospective cohorts or case reports with a small number of BRAF-positive cases. Testing strategies varied; while earlier studies used PCR-based hotspot assays, more recent cohorts used NGS, complicating prevalence estimates [10,11,12,13,14,15,16,17,18,19,20,21,22,39]. PCR-based approaches are inherently limited in their ability to detect non-hot-spot substitutions, indels, and low–allele frequency variants, potentially leading to underestimation of both the true prevalence and the full mutational spectrum of BRAF alterations in earlier cohorts. Treatment approaches were heterogeneous and often anecdotal, and publication bias may favor reports of positive targeted therapy outcomes. Although publicly available genomic databases may provide additional insights into mutation frequency and co-mutation patterns, the rarity of BRAF alterations in myeloid neoplasms, coupled with variable diagnostic annotation and clinical granularity, limits their utility for definitive genotype–phenotype correlations in this setting.
Future work should prioritize prospective multicenter studies with standardized sequencing to define prevalence and outcomes more precisely. Functional studies are needed to clarify the significance of non-V600E variants, particularly class 2 and 3 mutations. Clinical trials of BRAF and MEK inhibitors, ideally in rational combinations with chemotherapy, HMAs, or HSCT, will be required to establish whether targeting MAPK signaling can provide a durable benefit. Finally, systematic evaluation of molecular clearance as a biomarker should be incorporated into trial design.
4. Conclusions
BRAF mutations in myeloid neoplasms are rare but clinically relevant. They are enriched in CMML and therapy-related AML, usually occur in a complex genomic background, and portend poor outcomes. Unlike HCL or histiocytic disorders, where BRAF is a defining driver, in myeloid neoplasms, these mutations appear secondary and unstable. While targeted therapy responses have been modest and short-lived, clearance of BRAF mutations and subsequent HSCT may provide a path to improved outcomes. Collaborative efforts will be essential to clarify the prognostic and therapeutic role of this rare subset.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wellbrock C. Karasarides M. Marais R. The RAF proteins take centre stage Nat. Rev. Mol. Cell Biol.2004587588510.1038/nrm 149815520807 · doi ↗ · pubmed ↗
- 2Davies H. Bignell G.R. Cox C. Stephens P. Edkins S. Clegg S. Teague J. Woffendin H. Garnett M.J. Bottomley W. Mutations of the BRAF gene in human cancer Nature 200241794995410.1038/nature 0076612068308 · doi ↗ · pubmed ↗
- 3Chambard J.-C. Lefloch R. Pouyssegur J. Lenormand P. ERK implication in cell cycle regulation Biochim. Biophys. Acta 200717731299131010.1016/j.bbamcr.2006.11.01017188374 · doi ↗ · pubmed ↗
- 4Avruch J. Khokhlatchev A. Kyriakis J.M. Luo Z. Tzivion G. Vavvas D. Zhang X.-F. Ras activation of the Raf kinase: Tyrosine kinase recruitment of the MAP kinase cascade Recent Prog. Horm. Res.20015612715510.1210/rp.56.1.12711237210 · doi ↗ · pubmed ↗
- 5Naoki K. Chen T.-H. Richards W.G. Sugarbaker D.J. Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma Cancer Res.2002627001700312460919 · pubmed ↗
- 6Caunt C.J. Keyse S.M. Dual-specificity MAP kinase phosphatases (MK Ps): Shaping the outcome of MAP kinase signalling FEBS J.201328048950410.1111/j.1742-4658.2012.08716.x 22812510 PMC 3594966 · doi ↗ · pubmed ↗
- 7Jones D.T. Kocialkowski S. Liu L. Pearson D.M. Backlund L.M. Ichimura K. Collins V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas Cancer Res.2008688673867710.1158/0008-5472.CAN-08-209718974108 PMC 2577184 · doi ↗ · pubmed ↗
- 8Tiacci E. Trifonov V. Schiavoni G. Holmes A. Kern W. Martelli M.P. Pucciarini A. Bigerna B. Pacini R. Wells V.A. BRAF mutations in hairy-cell leukemia N. Engl. J. Med.20113642305231510.1056/NEJ Moa 101420921663470 PMC 3689585 · doi ↗ · pubmed ↗