Deficiency of the Mycobacterial Lipoarabinomannan Biosynthesis Glycosyltransferase MptC Enhances Antibacterial Immune Response and Rifapicin Antibiotic Susceptibility
Jiaxin Hu, Hongliang Chen, Zhongkun Li, Hao Sun, Yi-Cheng Sun, Xiao-Lian Zhang

TL;DR
Disabling a specific enzyme in mycobacteria makes them more vulnerable to antibiotics and boosts immune responses.
Contribution
The study reveals that MptC enzyme deficiency increases mycobacterial susceptibility to antibiotics and immune responses.
Findings
MptC deficiency increases LAM synthesis and reduces LM, altering cell wall properties.
MptC knockout bacteria show higher cell wall permeability and reduced hydrophobicity.
MptC-deficient mycobacteria trigger stronger T cell responses and are more susceptible to rifampicin.
Abstract
Background/Objectives: The mycobacterial complex cell envelope serves as a formidable barrier against host immunity and antibiotics. Lipomannan (LM) and lipoarabinomannan (LAM) are key structural components of the mycobacterial envelope and potent immunomodulators. The mycobacterial lipoarabinomannan biosynthesis mannosyltransferase MptC modifies the multiple α-(1→2)-linked branched mannan residues of LAM in the mycobacteria. However, the role of MptC in mycobacterial infectivity, antibiotic susceptibility and host immune regulation remains poorly understood. Methods: An mptC (also named MSMEG_4247) knockout Mycobacterium smegmatis mc2-155 (M. smeg) strain (designated as M. smegΔmptC) was generated using CRISPR–Cas12a technology. The effects of MptC on bacterial physiology, cell wall permeability, drug sensitivity, immune cell function, and survival during infection are analyzed through…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
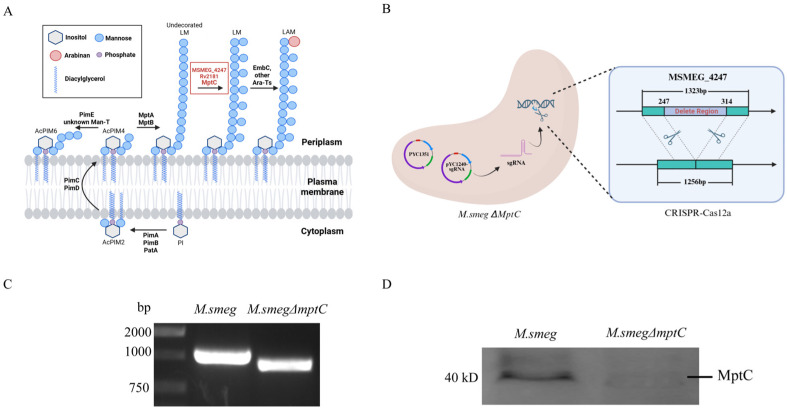 Figure 1
Figure 1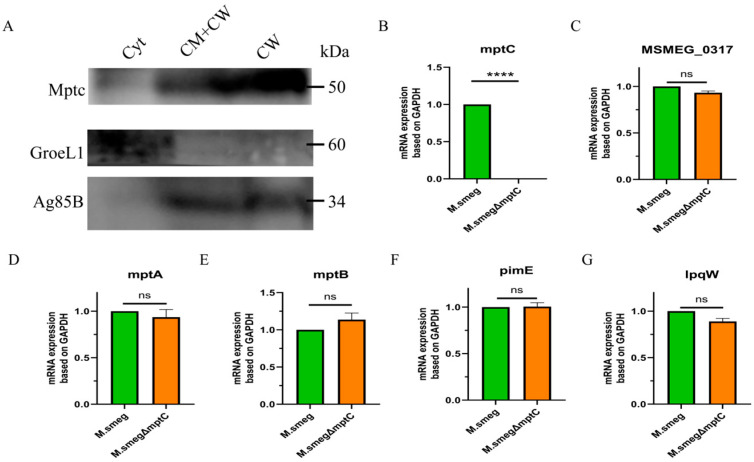 Figure 2
Figure 2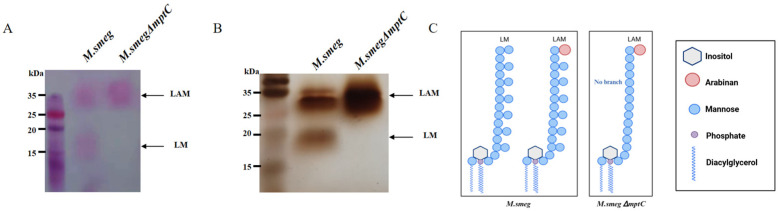 Figure 3
Figure 3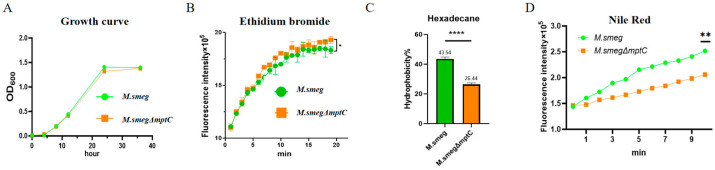 Figure 4
Figure 4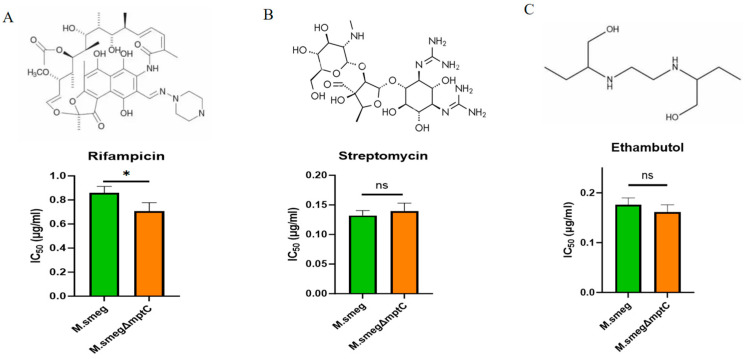 Figure 5
Figure 5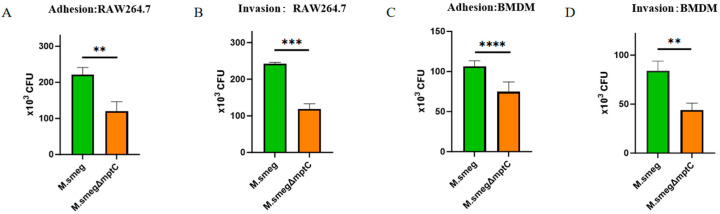 Figure 6
Figure 6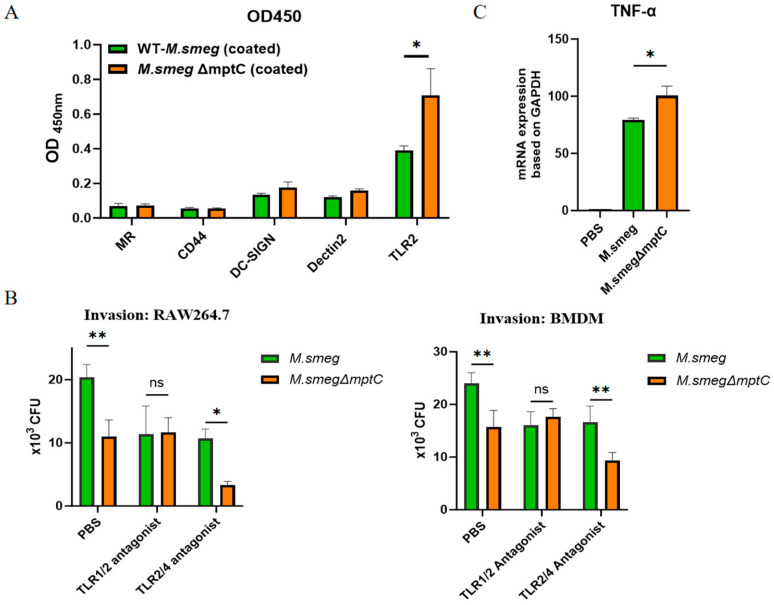 Figure 7
Figure 7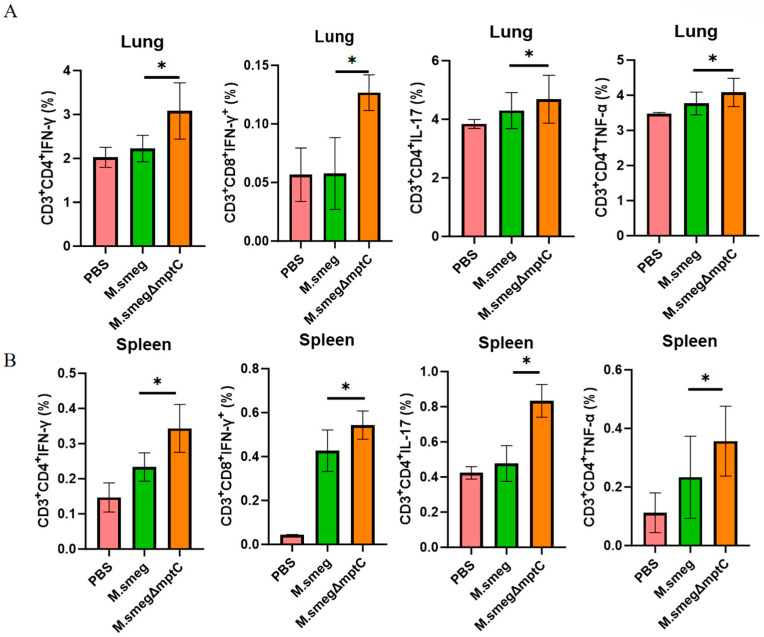 Figure 8
Figure 8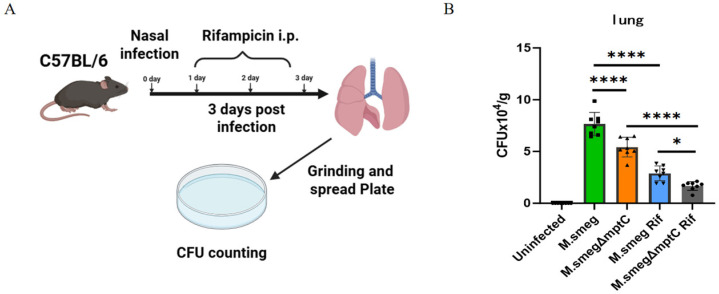 Figure 9
Figure 9- —National Natural Science Foundation of China
- —National Key R&D Program of China
- —Prevention and Control of Emerging and Major Infectious Diseases-National Science and Technology Major Project of China
- —Shenzhen Medical Research Fund
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMycobacterium research and diagnosis · Tuberculosis Research and Epidemiology · Glycosylation and Glycoproteins Research
1. Introduction
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (M. tb). It mainly affects the lungs but can also involve lymph nodes, bones, kidneys and other organs [1,2,3]. According to the 2025 report by the World Health Organization (WHO), in 2024, there were 10.7 million new cases of TB worldwide with approximately 1.23 million deaths [4]. The prevention and control of TB face significant challenges, particularly under the threat of drug-resistant TB, including multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) [4]. Therefore, an in-depth exploration of the pathogenic mechanism of M. tb, as well as the development of new treatment strategies and vaccines, have become the current research focus.
The pathogenicity of M. tb is closely related to its unique cell wall structure [5]. The cell wall serves not only as a physical barrier for the bacteria but also participates in the regulation of the host immune system [6,7,8]. Lipoarabinomannan (LAM) is one of the main components of the cell wall of M. tb, consisting of a phosphatidylinositol core, mannose (Man) side chains and arabinose (Ara) side chains [9]. The structure of LAM is complex, and it can be classified into mannose-capped LAM (ManLAM), phospho-myo-inositol-capped LAM (PILAM) and non-capped LAM (Aral AM) based on the different terminal modifications [10,11]. The biosynthesis of LM/LAM involves the coordinated action of more than twenty enzymes [12]. During the synthesis process, the mannosyltransferase PimA is responsible for transferring the first mannose to the inositol moiety of phosphatidylinositol (PI), forming PIM1. Subsequently, the mannosyltransferase PimB (Rv0557, also termed MgtA) and PimB’ (Rv2188c) catalyze the transfer of the second mannose from GDP-mannose to PIM1, yielding PIM2 [13,14]. PIM2 is then acylated by PatA on one of its mannose residues and further mannosylated by PimC/PimD to generate the tetramannosylated glycolipid (AcPIM4). The downstream fate of AcPIM4 diverges: it may be mannosylated by theα-(1→2) mannosyltransferase PimE and another uncharacterized mannosyltransferase to form AcPIM6 [15], or AcPIM4 itself may serve as the substrate. Both AcPIM6 and AcPIM4 can be further mannosylated by α-(1→6) mannosyltransferases (e.g., MptB and MptA) [16] to generate lipomannan (LM). The long mannan backbone of LM can be further extended by the mannosyltransferase MptC, which adds multiple α-(1→2)-linked branched mannose residues [17,18]. Arabinosyltransferases (such as EmbC) can then attach the arabinan domain to the α-(1→6) mannan core to produce LAM.
Functional studies support the importance of this pathway in mycobacterial physiology and virulence. An M. smegmatis (M. smeg) mptA deletion mutant producing a small LM-precursor but no mature LM or LAM grew slowly on solid LB agar medium and failed to grow at 42 °C [19]. Knocking out the gene pimB involved in LM synthesis led to an increase in the mortality rate of the macrophages [20]. A key enzyme responsible for this backbone extension is MptC. This α-(1→2)-mannosyltransferase, encoded by mptC (also named named as MSMEG_4247 in M. smeg; Rv2181 in M. tb H37Rv), is crucial for generating the full-length mannan core branching. Using the M. marinum-zebrafish model, the mannan core branching of LM/LAM (not LAM capping) mediated by MptC is demonstrated to be critical for mycobacterial virulence in innate immunity [21]. The overexpression of mptC in M. smeg resulted in the truncation of the arabinan and mannan domains of LM and LAM, and similar changes in the apparent sizes of LM and LAM were observed in M. tb [22].
Despite accumulating evidence establishing MptC as a key regulator of LAM structural formation [18,23], the precise functions of MptC on bacterial physiology and host immune response have not been fully elucidated. In this paper, we aimed to investigate the functional role of MptC by generating an mptC knockout strain of M. smeg. Using CRISPR–Cas12a-mediated gene editing technology [24,25], we disrupted the mptC homolog (MSMEG_4247) and systematically examined the effects on the LAM structure, bacterial cell wall integrity, antibiotic susceptibility and host T cell immune responses. Our findings provide new insights into the roles of MptC-mediated LM/LAM biosynthesis in mycobacterial pathogenesis and host–pathogen interactions.
2. Results
2.1. MptC Knockout Does Not Affect the Expression of Its Upstream and Downstream Genes in M. smeg
MptC modifies the multiple α-(1→2)-linked branched mannan residues of LAM in the mycobacterial LM/LAM biosynthetic pathway (Figure 1A). The MSMEG_4247 gene was deleted in M. smeg using CRISPR–Cas12a genome editing (Figure 1B). The successful gene disruption in the M. smegΔ4247 (referred to as M. smeg∆mptC) strain was confirmed by PCR (Figure 1C). Sequencing analysis reveals a 68 bp deletion (nucleotides 247–314) in the target gene (Figure S1A), resulting in both a frameshift mutation and premature termination.
To further verify the expression of mptC in M. smeg and M. smeg∆mptC, polyclonal antibodies against MptC were generated in New Zealand White rabbits. Western blot analysis confirms the successful knockout of mptC in M. smeg∆mptC (Figure 1D).
By analyzing separated fractions of the bacterial cell membrane, cell wall, and cytoplasm, we found that MptC is localized on the bacterial cell membrane and the cell wall (Figure 2A). Comparing the colony morphology of M. smeg and M. smeg∆mptC on solid media, both colonies exhibited a milky white color, and there was no significant difference in colony morphology observed with the naked eye (Figure S1B).
To detect the expression of the upstream and downstream genes of mptC in M. smeg∆mptC, bacterial RNA was extracted for RT-qPCR analysis. The results showed that the mptC (MSMEG_4247) gene was deleted successfully and mptC knockout did not affect the expression of its upstream (mptA, MSMEG_0317) or downstream (mptB, pimE, lpqW) genes (Figure 2B–G).
2.2. MptC Deficiency Affects the Production of Bacterial LM and LAM
MptC is responsible for synthesizing the mannose side chains of LM and LAM. The knocking out of mptC is likely to affect the structure of LM and LAM on the bacterial surface. Since the genes for synthesizing LAM have a certain degree of functional redundancy, it is necessary to determine whether mptC gene knockout affected its product LAM/LM. Both M. smeg and M. smeg∆mptC cell wall lipomannan components were isolated and purified. Glycogen staining shows that the positions of LM and LAM in the wild-type M. smeg are 32 kDa and 18 kDa, respectively. Meanwhile, the LAM band became thicker, and the LM band disappeared in the M. smeg∆mptC, indicating an increase in LAM content and an absence in LM content (Figure 3A). Silver staining results display similar changes in the LAM structure (Figure 3B). Consistent with a previous report [23], these results can be attributed to the fact that LM lacking mannose side chains is susceptible to degradation unless it is further synthesized as LAM (Figure 3C).
2.3. MptC Knockout Increases Bacterial Cell Wall Permeability and Reduces Hydrophobicity
Next, we examined whether the changes in MptC affected the growth rate of bacteria. We detected the changes in the OD_600_ values of the bacterial solution at different time points. We found that the deletion of mptC did not significantly affect the growth rate of bacteria (Figure 4A). The ethidium bromide (EtBr) penetration experiment proved that the permeability of the bacterial cell wall increased after the deletion of mptC (Figure 4B). The hexadecane distribution experiment (Figure 4C) and the Nile red absorption experiment (Figure 4D) demonstrated that the hydrophobicity of the bacterial cell wall significantly decreased after the deficiency of mptC.
2.4. MptC Knockout Increases Bacterial Sensitivity to Rifampicin
The issue of drug resistance of M. tb is one of the main reasons for the difficulty in treating tuberculosis. Three kinds of M. tb antibiotics, rifampicin, streptomycin and ethambutol, with different structures as shown in Figure 5, were used to treat M. smeg and M. smeg∆mptC, respectively. We used gradient concentrations of rifampicin, streptomycin and ethambutol to stimulate M. smeg and M. smegΔmptC. After three days of treatment, we detected the bacterial cell-forming units (CFUs). After knocking out mptC in M. smeg, the sensitivity of M. smeg to rifampicin increased with a significantly lower IC_50_ (half-maximal inhibitory concentration) for rifampicin. This suggests that MptC may be involved in the process of M. smeg against rifampicin (Figure 5A). The increased sensitivity is possibly due to the enhanced permeability of the cell envelope to rifampicin upon mptC deletion, allowing more drug molecules to reach their intracellular target. This suggests that MptC contributes to M. smeg resistance against rifampicin, possibly by maintaining a cell envelope barrier that limits drug permeation. In contrast, the deletion of mptC did not significantly alter bacterial sensitivity to streptomycin or ethambutol (Figure 5B,C), which was possibly due to the different structures of drugs.
2.5. MptC Knockout Decreases Mycobacterial Invasion of Macrophages
We performed the bacterial adhesion and invasion assays in macrophages at a multiplicity of infection (MOI) of 10. The results showed that M. smeg exhibited significantly greater adherence (2.22 × 10^5^ CFU of M. smeg vs. 1.20 × 10^5^ CFU of M. smegΔmptC, ** p < 0.01) and invasion (2.42 × 10^5^ CFU of M. smeg vs. 1.19 × 10^5^ CFU of M. smegΔmptC, *** p < 0.001) to RAW 264.7 macrophages (Figure 6A,B). Consistent results were observed in bacteria-infected mouse bone marrow–derived macrophages (BMDMs), where the M. smegΔmptC strain showed 29% reduced adhesion (1.06 × 10^5^ of M. smeg vs. 7.48 × 10^4^ CFU of M. smegΔmptC, **** p < 0.0001) and 47% lower invasion (8.40 × 10^4^ of M. smeg vs. 4.40 × 10^4^ CFU of M. smegΔmptC, ** p < 0.01) compared to wild-type (Figure 6C,D). These results suggest that MptC knockout impairs bacterial adhesion and the invasion of macrophages.
2.6. MptC Mediates Macrophages Binding Possibly via TLR2-TLR4 Complex
M. tb can bind to various receptors on the surface of macrophage membranes. These receptors include complement receptors (CR1, CR3, or CR4), mannose receptors (MR), DC-SIGN, Dectin-2, Toll-like receptors (TLR) and NOD-like receptors. TLR2, CLRs MR, DC-SIGN, Dectin-2 and CD44 can recognize the glycolipid ManLAM on the surface of M. tb [26,27,28]. A ligand-binding enzyme-linked immunosorbent assay (ELISA) was performed to assess bacterial interactions with host receptors. M. smeg and the M. smegΔmptC mutant were separately immobilized on ELISA plates. The plates were then incubated with a panel of His-tagged recombinant host proteins, including MR, CD44, DC-SIGN, Dectin-2, and TLR2. After washing, bound proteins were detected using an HRP-conjugated anti-His antibody, which was followed by color development with a TMB substrate (Figure S2). ELISA experiments confirmed that the glycolipids on the surface of M. smeg mainly bound to the TLR2 receptor, and knocking out MptC further increased the binding ability of bacteria to the TLR2 receptor (Figure 7A).
Next, by treatment with the TLR1/2 antagonist Cu-CRT22 and TLR2/4 antagonist Sparstolonin B, we found that the TLR2/4 antagonist could reduce the invasion of M. smeg and M. smegΔmptC with a more pronounced effect on M. smegΔmptC (Figure 7B).
RT-qPCR analysis showed that TNF-α mRNA expression in M. smegΔmptC-infected RAW264.7 cells was significantly increased compared to that in M. smeg group (Figure 7C). Thus, the enhanced TLR2 engagement by M. smegΔmptC likely contributes to stronger macrophage activation with higher levels of TNF-α mRNA expression.
2.7. MptC Knockout Increases Intracellular IFN-γ, TNF-a and IL-17 Production in T Cells in Mice
Upon invasion, the mycobacterium is directly cleared by macrophages and also activates the adaptive immune response through antigen presentation. This response relies primarily on T lymphocyte-mediated cellular immunity to combat tuberculosis. To investigate the effect of MptC on T cell immune responses, M. smeg and M. smeg∆mptC were used to infect mice intranasally, and the spleen and lung T cell responses were detected at three days post-infection (Figure S3A). Mouse lung and spleen cell lysates were stimulated with heat-inactivated M. smeg (iM. smeg) followed by flow cytometry analysis. In the knockout group, IFN-γ production was significantly increased in both lung and splenic CD8^+^ and CD4^+^ T cells (Figure 8A,B). While both lung and splenic CD4^+^ T cells also showed elevated IL-17 and TNF-α production (Figure 8A,B). IFN-γ and TNF-α can act on infected target cells to eliminate and kill mycobacteria. IL-17 is involved in protective immunity against tuberculosis [29]. These results suggest that mptC knockout promotes the host’s T cell cytokine response to M. smeg, aiding in the clearance of bacteria.
2.8. MptC Knockout Reduces M. smeg Loading in Mouse Lungs and Enhances Rifapicin Antibiotic Susceptibility
Next, we further examined the effect of mptC knockout on the bacterial loads and antibiotic resistance to rifampicin in a mouse infection model. M. smeg and M. smegΔmptC were used to infect mice intranasally (i.n.) with daily rifampicin administration by intraperitoneal injection (i.p.) (Figure 9A). After three days post-infection, the mouse lungs were ground in PBS and diluted to prepare the dilution mixture for spreading on plates. The single colonies growing on the plates for three days were counted. We observed that after knocking out mptC, the bacterial load of M. smeg in the mouse lungs significantly decreased (Figure 9B). Furthermore, MptC deficiency led to an increase in bacterial susceptibility to rifampicin (Figure 9B). In the mouse model, rifampicin treatment achieved a 14% higher bacterial inhibition rate in the M. smegΔmptC group compared to the M. smeg group. This might be due to the absence of mptC leading to an increase in both antibacterial immune response and rifapicin antibiotic susceptibility.
3. Discussion
Tuberculosis caused by M. tuberculosis is a top killer among infectious diseases. Despite the approval of several new antibiotics, including bedaquiline, delamanid, linezolid, and pretomanid, more M. tuberculosis strains resistant to these drugs have emerged. Therefore, continuous improvement and new targets against TB are urgently required. M. tuberculosis involves a complex pathogenic mechanism involving intricate interactions between the bacterium and the host immune system. The mycobacterial mannosyltransferase MptC is a key enzyme for modifying the mannan core branching of LAM in the mycobacterial wall. Until now, the role of MptC in mycobacterial characteristics, antibiotic susceptibility and host immune regulation remains poorly understood. In this paper, we demonstrate that MptC-mediated mannosylation plays an important role in maintaining the proper LAM structure and mycobacterial virulence. M. smeg∆mptC strain has less bacterial infectivity and higher susceptibility to antibiotic rifampicin in both macrophages and mice. Functionally, the MptC knockout strain increases the pro-inflammatory cytokines production of T cells in mice. To our knowledge, this is the first report showing that MptC deficiency increases the pro-inflammatory cytokines production of T cells in mouse infection models.
In this paper, we demonstrate that MptC deficiency leads to the disappearance of LM and an increase in LAM content. This result aligns with the reported function of MptC [23]—namely, its role in synthesizing the mannose side chain of LAM, whose expression directly influences LAM’s structural integrity. Alterations in the LAM structure further affect bacterial physiological traits: mptC deletion increases cell wall permeability and decreases hydrophilicity. Notably, changes in mptC expression significantly affect bacterial drug sensitivity: mptC deletion enhances bacterial sensitivity to rifampicin but not to streptomycin or ethambutol. Rifampicin targets bacterial RNA polymerase. Its penetration into mycobacteria largely depends on passive diffusion through the hydrophobic mycolic acid layer due to its apolar structure (characterized by a naphthol ring and an ansa bridge). Rifampicin is particularly sensitive to alterations in the cell wall permeability. The deletion of mptC, by disrupting the lipoarabinomannan layer and the outer membrane integrity, likely facilitates this diffusion process, thereby specifically enhancing rifampicin’s access to its intracellular target. The susceptibility to ethambutol remains not significantly changed, as its molecular target (arabinosyltransferases in arabinan synthesis) is distinct from the functional role of MptC in mannan chain extension. The persistent streptomycin susceptibility likely stems from its hydrophilic nature, which favors entry through porin channels—a pathway largely independent of the hydrophobic barrier altered by mptC deletion. This suggests that MptC-dependent LAM modifications act as a natural barrier against certain antibiotics, such as rifampicin.
Interestingly, our findings appear to contrast with a study on an M. marinum mptC mutant [21]. In that study, the mutant’s antibiotic susceptibility profile (including to streptomycin, erythromycin, isoniazid, rifampicin, polymyxin B, and chloramphenicol) was reported to be similar to that of the wild-type M. marinum, whereas our M. smegΔmptC showed increased sensitivity to rifampicin. This discrepancy might be attributed to species-specific differences or the method of mutant construction. The M. marinum mutant was generated by the transposon insertion of 286 nucleotides downstream of the start codon of mmar_3225 (an orthologue of M. tb Rv2181), which resulted in the production of smaller LAM and LM molecules. However, in another study on M. smeg, the absence of mptC led to the abolition of LM synthesis, which is consistent with our results. The absence or/and presence of LM in different species indicate that these macromolecules have complex regulation in different species, and further research is needed to explain this phenomenon.
We have confirmed that the MptC-regulated lipid-associated membrane (LAM) structure is crucial for the interaction between bacteria and host immune cells. Knockout strains exhibit significantly impaired adhesion and invasion abilities toward macrophages, and it also led to increased production of pro-inflammatory cytokines (TNF-α, IFN-γ, and IL-17) by T cells. Here, the bacterial survival in mouse infection experiments demonstrated that the M. smeg∆mptC mutant strain exhibited significantly lower lung CFU and stronger immune response compared to the M. smeg strain, indicating impaired survival and colonization capability upon mptC deletion in vivo. This finding is consistent with previous observations, where an mptC mutant also showed markedly attenuated virulence [21].
Members of the TLR family, including TLR2, TLR4, and TLR9, are involved in the immune response to M. tb infection. TLR2 recognizes surface lipoproteins and/or lipoglycans, promoting the maturation of antigen-presenting cells and the secretion of TNF-α, IL12, and IFN-γ. TLR4 recognizes the lipopolysaccharide (LPS), and TLR9 recognizes the non-methylated CpG DNA of M. tb [30]. Notably, although M. smeg∆mptC showed enhanced TLR2 binding, it exhibited reduced invasion or internalization into macrophages (Figure 6). This result might be due to the distinct roles of TLRs for M. smeg∆mptC: TLR2/4 might primarily function as a signaling receptor that promotes macrophage activation, pro-inflammatory cytokine production and immune response rather than acts as a phagocytic receptor. LAM may also induce autophagy-related processes by TLR2 and activate the expression of pro-inflammatory cytokines [31,32]. The response of B cells to LAM was shown to occur in a TLR2-dependent manner [33]. The impact of TLR2 on bacterial adhesion, invasion, and signaling pathways remains to be determined using TLR2 inhibitors or TLR2 knockout mice. Further resolving LAM–receptor complex structures via surface plasmon resonance (SPR) or cryo-electron microscopy would help to illuminate the molecular basis of their immunomodulatory effects in future studies.
Despite revealing the significant role of MptC in LAM synthesis and host immune regulation, this paper has several limitations. First, M. smeg, a non-pathogenic bacterium, might have a distinct LM/LAM composition and host interactions compared to virulent M. tb; future studies should verify MptC’s function in virulent M. tb. Second, although the transcription levels of the mptC neighboring genes were unchanged, genetic complementation of the M. smeg∆mptC mutant strain would further strengthen the causal attribution of the observed phenotypes. Third, the receptor-binding ELISA and pharmacological inhibition suggest the involvement of TLR2/TLR4-associated recognition, but genetic receptor knockout/knockdown or biophysical binding assays would be needed. Finally, the in vivo experiments were focused on an early (3-day) time point and a limited antibiotic pane; extended infection time courses and broader drug testing may further delineate the role of MptC in persistence and drug tolerance.
In summary, this paper reveals for the first time the crucial role of MptC in mediating mycobacterial tolerance to rifampicin and the pro-inflammatory cytokines production of T cells. This paper also provides a new perspective for mycobacterial cell wall biology and its interaction with host immune cells. Targeting cell wall components like MptC may thus represent a potential strategy for developing adjunctive therapies against mycobacterial infections.
4. Materials and Methods
4.1. Construction of MptC Knockout Strain of M. smeg and Observation of Bacterial Morphology
M. smeg strains [34] used in this paper were cultured on Middlebrook 7H10 agar medium (Becton, Dickinson and Company (BD) (Franklin Lakes, NJ, USA)) supplemented with 10% (v/v) OADC (oleic acid–albumin–dextrose–catalase) and 0.5% (v/v) glycerol. The plates were streaked with bacterial stocks and incubated at 37 °C under aerobic conditions for 48–72 h until single colonies appeared.
The mptC gene was disrupted in M. smeg via CRISPR–Cas12a-mediated genome editing basically according to previous publications [25,35]. The shuttle plasmid pYC1240 (pCR-hyg) was used to express sgRNA [24]. The helper plasmid pYC1351 (pNHEJ-Cas12a-recAmu) carries mycobacterial NHEJ-dependent IigD, KuC, and NrgA genes for increasing NHEJ repair efficiency [24]. First, the shuttle plasmid pYC1240 was digested with BpmI and HindIII, and the linearized backbone was gel-purified. The sgRNA sequence used in this paper is 5′-gctcgccgcgatcgcattcgctccg-3′, which targets nucleotides 258–282 of the mptC gene (MSMEG_4247). sgRNAs primers were designed to flank the MptC target region and ligated into the digested pYC1240. The pYC1240-sgRNA construct was then electroporated into the competent M. smeg harboring the helper plasmid pYC1351 and then plated on Middlebrook 7H10 agar supplemented with 10% OADC and 0.5% glycerol along with kanamycin (25 μg/mL), hygromycin (50 μg/mL), and anhydrous tetracycline (100 ng/mL), which was followed by incubation at 37 °C for 4 days. The M. smeg∆mptC mutant was confirmed by genomic DNA PCR analysis. M. smeg genomic DNA was used as a control. Primers targeting the region flanking the sgRNA were designed as follows: forward primer: 5′-ctggagatgagtaagcggcag-3′; Reverse primer: 5′-acaccggtgagaccaccaggc-3′. The expected amplicon sizes were 1000 bp for M. smeg and 932 bp for the M. smegΔmptC mutant strain. PCR products were purified and subjected to Sanger sequencing to confirm the correct gene disruption. M. smeg and the MptC knockout strain of M. smeg were applied onto the 7H10 agar containing hygromycin, respectively, and the morphological differences were observed for 3 days.
4.2. Purification and Identification of LAM/LM from M. smeg
The LAM extraction procedure was performed basically as previously described [36]. Bacterial cultures were grown to mid-log phase (OD_600_ = 0.6–0.8), harvested by centrifugation, and washed three times with PBS. The pellet was lyophilized overnight, which was followed by sequential delipidation: (1) first extraction with chloroform:methanol (2:1, vol/vol) at a 1:9 (wt/vol) ratio at 37 °C with shaking overnight and (2) second extraction with chloroform:methanol:water (10:10:3, vol/vol/vol) under the same conditions. The delipidated pellet was air-dried in a fume hood, resuspended in PBS, and subjected to enzymatic lysis using lysozyme (20 mg/mL), PMSF (1:1000 dilution), and DNase I (100 U/mL) at 37 °C for 2 h, which was followed by sonication. The lysate was mixed with 8% Triton X-114 (vol/vol), incubated at 4 °C overnight, and phase-separated at 37 °C. The aqueous phase was combined with 9 volumes of 95% ethanol, precipitated at −80 °C overnight, and the pellet was air-dried. Finally, the material was digested with proteinase K (0.24 mg/mL) at 60 °C for 2 h, and the supernatant was analyzed by glycostaining.
For visualization by silver staining, equal amounts of the purified LAM/LM preparations were resolved by SDS–PAGE and stained using a standard silver-staining procedure. Gels were fixed in 40% (v/v) ethanol and 10% (v/v) acetic acid, rinsed with water, sensitized with 0.02% (w/v) sodium thiosulfate, and incubated in 0.1% (w/v) silver nitrate. Bands were developed in 2% (w/v) sodium carbonate containing 0.04% (v/v) formaldehyde, and the reaction was stopped with 5% (v/v) acetic acid prior to imaging.
For glycogen detection, the gel was fixed and oxidized in a solution containing 30% ethanol, 10% acetic acid, and 0.7% periodic acid for 10 min with agitation. After washing with deionized water (3 × 3 min), it was stained with Schiff’s reagent for 20 min in the dark. Finally, the gel was destained with multiple washes in deionized water until clear bands were visualized.
4.3. Reverse Transcription-Quantitative Real Time PCR (RT-qPCR)
Total RNA was extracted from wild-type and knockout bacterial strains at mid-log phase (OD_600_ 0.6–0.8) using the RNeasy Mini Kit (Qiagen, Singapore, Cat. #74104) with modifications: 7 mL of bacterial culture was centrifuged, washed twice with EDTA-Tris buffer (pH 8.0), and subjected to three freeze–thaw cycles (−80 °C). Cell pellets were resuspended in RLT buffer, vortexed vigorously, and lysed by sonication. Lysates were centrifuged (12,000× g, 5 min), and supernatants were mixed with absolute ethanol before loading onto spin columns. After sequential washes with RW1 buffer (twice) and RPE buffer (once), RNA was eluted in nuclease-free water and reverse-transcribed using a SYBR Green Real-Time pCR Master Mix plus RT-pCR (Biotechnology Overseas (Osaka, Japan)).
4.4. Bacterial Physiological Characterization and Cell Wall Permeability
All experiments were performed using bacteria in the logarithmic growth phase. To assess bacterial growth kinetics, optical density measurements of bacterial culture were taken at 2 h intervals from an initial culture (~2 × 10^7^ CFU/mL, OD_600_ = 0.02). For ethidium bromide (EtBr) uptake assay, bacterial cells were resuspended in PBS supplemented with glucose, which was followed by the addition of EtBr at a final concentration of 1 μg/mL. Fluorescence intensity was monitored using an excitation wavelength of 530 nm and an emission wavelength of 590 nm [37].
Similarly, Nile red uptake assay was evaluated by resuspending bacterial cells in glucose-supplemented PBS and incubating with 10 μM Nile red. Fluorescence was measured at an excitation wavelength of 540 nm and an emission wavelength of 600 nm [30].
Bacterial hydrophobicity was determined using the hexadecane adhesion assay. Briefly, bacterial suspensions in PBS were mixed with hexadecane at a ratio of 5:4 (v/v, bacterial suspension: hexadecane). After vigorous mixing and phase separation, the aqueous phase was collected, and the OD_600_ values were measured to determine cell adhesion [38].
4.5. Antimicrobial Resistance Assay
To assess drug resistance, wild-type and knockout bacterial strains were cultured to the logarithmic growth phase (OD_600_ =0.6–0.8). Bacterial suspensions were adjusted to an initial OD_600_ of 0.02 and treated with serially diluted concentrations of the target antibiotic for 72 h. Bacterial growth was monitored by measuring OD_600_, and the IC_50_ was calculated using GraphPad Prism.
4.6. Bacterial Adhesion and Invasion Assays in Macrophages
BMDMs were isolated from the femurs and tibias of C57BL/6 mice. Mice were euthanized by an intraperitoneal injection of sodium pentobarbital, and hind limbs were aseptically removed. Bone marrow cavities were flushed with ice-cold RPMI 1640 medium (containing 10% FBS and 1% penicillin–streptomycin) to obtain bone marrow cells. After centrifugation at 600× g for 5 min at 4 °C, cells were treated with ACK lysis buffer for 5 min at room temperature to remove red blood cells. Lysis was stopped by adding excess RPMI 1640 medium, which was followed by two washes with the same medium. Cells were resuspended in DMEM supplemented with 10% FBS, 1% penicillin–streptomycin, and 40 ng/mL recombinant murine M-CSF, and then they were seeded into 12-well plates at 1 × 10^6^ cells/well. Cells were cultured at 37 °C with 5% CO_2_. After three days incubation, non-adherent cells were removed, and fresh DMEM containing 40 ng/mL M-CSF was added. BMDMs were obtained for experiments on day 6–7 upon reaching confluence.
Mouse Raw264.7 macrophages or BMDMs were seeded in a 12-well plate, at a density of 1 × 10^6^ cells per well, and cultured in 1 mL of 1640 cell culture medium for 20 h prior to infection. The cells were infected at an MOI of 10. For adhesion assay, after 1 h of infection, cells were washed twice with PBS to remove non-adherent bacteria. Macrophages were lysed with 0.2% Triton X-100 for 5 min, and serial dilutions of the lysates were plated to quantify adherent bacterial CFUs. For invasion assay, following 1 h of infection, extracellular bacteria were removed by PBS washing. Cells were incubated for 1 h in gentamicin-containing medium to kill remaining extracellular bacteria. The medium was replaced with fresh antibiotic-free medium, and macrophages were further incubated for 4 h to allow intracellular bacterial replication. Cells were lysed with 0.2% Triton X-100, and serial dilutions were plated to determine invaded bacterial CFUs.
4.7. ELISA-Based Ligand-Binding Assay
Wells of a 96-well plate were coated with 100 µL of 0.1 mg/mL poly-L-lysine (Merck KGaA, Darmstadt, Germany) and incubated at 4 °C for 1 h. Following this, either M. smeg or M. smeg∆mptC was then immobilized onto the coated wells. After blocking, recombinant His-tagged pattern recognition receptors (PRRs), including MR, CD44, DC-SIGN, Dectin-2, and TLR2 (IPODXI, Wuhan, China), were added to the wells and incubated at 37 °C for 2 h. The wells were then washed to remove unbound proteins. Bound His-tagged receptors were detected by incubation with an HRP-conjugated anti-His antibody (ABclonal, Wuhan, China) at 37 °C for 1 h. Finally, the reaction was developed with TMB substrate, stopped with sulfuric acid, and the absorbance was measured at 450 nm.
4.8. Mouse Infection and Lung CFU Enumeration
C57BL/6 mice (6–8 weeks old) were used for infection experiments. C57BL/6 mice were selected for this paper due to their documented use in tuberculosis research and their association with a Th1-skewed response, which is protective against intracellular pathogens [39,40]. Mice were lightly anesthetized with isoflurane and intranasally inoculated with WT M. smeg or M. smeg∆mptC at a dose of 1 × 10^8^ CFU in 50 µL PBS. Control mice received PBS only. Where indicated, rifampicin was administered by intraperitoneal injection according to the experimental design (dose and schedule described elsewhere/see figure).
At 3 days post-infection, mice were euthanized and lungs were aseptically collected and weighed. Lung tissues were homogenized in sterile PBS, and homogenates were serially diluted and plated on Middlebrook 7H10 agar for CFU determination. Plates were incubated at 37 °C for 4 days before colony counting. Bacterial burden is reported as CFU per gram of lung tissue (CFU/g). The inhibition rate was calculated as follows: [1 − (mean CFU of treated group/mean CFU of untreated control)] × 100%.
Group sizes (for lung CFU counting: n = 8 per group) were chosen based on sample sizes commonly used in comparable murine mycobacterial infection to minimize animal use in accordance with the 3R (Replacement, Reduction, and Refinement) principles [41].
All animal experiments were conducted in accordance with international and national guidelines for the care and use of laboratory animals. This paper was approved by the Institutional Animal Care and Use Committee (IACUC) of Wuhan University (Approval No.: WP20240336; Date of approval: 5 July 2024).
Specific-pathogen-free (SPF) female C57BL/6 mice (aged 6–8 weeks) were purchased from Skobees Biotechnology Co., Ltd. (Anyang, Henan, China). All mice were healthy and without genetic modifications. At the end of each experiment, euthanasia was performed on the mice by an intraperitoneal injection of sodium pentobarbital, and every effort was made to minimize their suffering.
4.9. Flow Cytometry Detection of T Cell Cytokines Production in a Mouse Infection Model
Groups of mice were inoculated intranasally with 10^8^ CFU of either M. smeg or M. smeg∆mptC mutant in a volume of 30 µL PBS; control mice received PBS only. Three days post-infection, mice were euthanized. Spleen and lung organs were aseptically harvested and prepared into single-cell suspensions. For intracellular cytokine staining, cells were restimulated ex vivo with heat-inactive M. smeg (iM. smeg) at an MOI of 10 for 6 h in the presence of GolgiPlug (protein transport inhibitor). Following stimulation, cells were stained for surface markers (CD3, CD4, CD8), fixed, permeabilized, and then stained intracellularly for cytokines production in T cells (IFN-γ, TNF-α, IL-17). Data were acquired on a flow cytometer and analyzed to determine the frequency of cytokine-positive cells within CD4^+^ and CD8^+^ T cell populations.
Group sizes (for flow cytometry analysis: n = 3 per group) were chosen based on sample sizes commonly used in immunophenotyping studies and to minimize animal use in accordance with the 3R principles [41].
4.10. Statistical Analysis
Data were analyzed using GraphPad Prism (version 9.0.2). Normality was assessed using the Shapiro–Wilk test, and the homogeneity of variances was assessed using the Brown–Forsythe/Levene test (for ANOVA) or F test (for t test). For two-group comparisons, an unpaired two-tailed Student’s t-test was used when assumptions were met (for data with normal distribution but unequal variances, Welch’s t test was used); otherwise, the Mann–Whitney U test was applied. For comparisons among more than two groups, one-way ANOVA with Tukey’s multiple-comparison test was used when assumptions were met (for data with normal distribution but unequal variances, Welch and Brown–Forsythe tests were used); otherwise, the Kruskal–Wallis test with Dunn’s post hoc test was applied. For time-course or two-factor designs, two-way ANOVA with appropriate multiple-comparison correction was used when assumptions were met (for data with normal distribution but unequal variances, Geisser–Greenhouse correction was used). Statistical significance was set at p < 0.05. Asterisks in the figures indicate the following levels of significance: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
5. Conclusions
In summary, MptC deletion alters LAM structure, thereby affecting bacterial cell wall traits, drug sensitivity, and interactions with host immune cells. These findings deepen our understanding of mycobacterial infection and immune mechanisms, and they provide a theoretical basis for developing novel anti-TB drugs or vaccines. Future research should further clarify the precise relationship between LAM structure and immune regulation to drive innovative progress in TB prevention and treatment.
While our findings highlight the roles for MptC in LM/LAM homeostasis, envelope properties, host immune responses and rifampicin susceptibility, the conclusions are currently based on a non-pathogenic M. smeg model and an early infection time point. Future work should validate these observations in pathogenic mycobacteria (e.g., M. tb), including genetic complementation, and structural and genetic approaches to define the receptor pathways involved, as well as evaluating a broader range of antibiotics and longer infection courses.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Moule M.G. Cirillo J.D. Mycobacterium tuberculosis Dissemination Plays a Critical Role in Pathogenesis Front. Cell. Infect. Microbiol.2020106510.3389/fcimb.2020.0006532161724 PMC 7053427 · doi ↗ · pubmed ↗
- 2Du J. Qin W. Zhang Y. Yang Z. Li J. Yang J. Deng Q. Topical streptomycin irrigation of lesions to prevent postoperative site infections in spinal tuberculosis: A retrospective analysis J. Orthop. Surg. Res.20231859210.1186/s 13018-023-04059-y 37563683 PMC 10413591 · doi ↗ · pubmed ↗
- 3Fontanet S. Gallioli A. Baboudjian M. Huguet J. Territo A. Gaya J.M. Gavrilov P. Izquierdo P. Verri P. Algaba F. Topical instillation of BCG immunotherapy for biopsy-proven primary upper urinary tract carcinoma in situ: A single institution series and systematic review Urol. Oncol.20234127428310.1016/j.urolonc.2022.11.00136526527 · doi ↗ · pubmed ↗
- 4Global Tuberculosis Report 2025 Available online: https://www.who.int/teams/global-programme-on-tuberculosis-and-lung-health/tb-reports/global-tuberculosis-report-2025(accessed on 12 November 2025)
- 5Mainardi J.L. Fourgeaud M. Hugonnet J.E. Dubost L. Brouard J.P. Ouazzani J. Rice L.B. Gutmann L. Arthur M. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway J. Biol. Chem.2005280381463815210.1074/jbc.M 50738420016144833 · doi ↗ · pubmed ↗
- 6Blanc L. Gilleron M. Prandi J. Song O.R. Jang M.S. Gicquel B. Drocourt D. Neyrolles O. Brodin P. Tiraby G. Mycobacterium tuberculosis inhibits human innate immune responses via the production of TLR 2 antagonist glycolipids Proc. Natl. Acad. Sci. USA 2017114112051121010.1073/pnas.170784011428973928 PMC 5651758 · doi ↗ · pubmed ↗
- 7Hessel M.v.D.J. van der Marel G.A. Codée J.D.C. Developments in the Synthesis of Mycobacterial Phenolic Glycolipids Chem. Rec.2021213295331210.1002/tcr.20210020034581501 · doi ↗ · pubmed ↗
- 8Rao V. Fujiwara N. Porcelli S.A. Glickman M.S. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule J. Exp. Med.200520153554310.1084/jem.2004166815710652 PMC 2213067 · doi ↗ · pubmed ↗