Establishment and Maintenance of Repressed Chromatin States on Dosage-Compensated Sex Chromosomes
Joshua Eduful, Lily LeSarge, Györgyi Csankovszki

TL;DR
This paper compares how different organisms silence one X chromosome to balance gene dosage, focusing on chromatin regulation in nematodes and mammals.
Contribution
The paper contrasts mechanisms of X-chromosome repression in C. elegans and mammals, highlighting differences in chromatin regulation and structure.
Findings
C. elegans uses a condensin-based complex to downregulate X chromosomes in hermaphrodites.
Mammals use XIST RNA and associated proteins to inactivate one X chromosome in females.
Both systems maintain repressed chromatin states through distinct topological and architectural mechanisms.
Abstract
Sex chromosome imbalance is a genetic challenge in species with unequal X-chromosome numbers. Organisms have developed distinct strategies to control this imbalance through a process called dosage compensation. These strategies include X-chromosome inactivation in mammals mediated by the XIST long noncoding RNA and proteins recruited by XIST, and X-linked hypertranscription in male Drosophila driven by the Male-Specific Lethal (MSL) complex. In Caenorhabditis elegans, gene expression is downregulated from each of the two X chromosomes of hermaphrodites by half, thereby matching the levels in XO males. This is mediated by a specialized condensin-containing protein complex, the Dosage Compensation Complex (DCC). In all cases, the chromatin states on the sex chromosomes must be first established and then maintained for the entire lifetime of the organism. Although mammals and nematodes…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
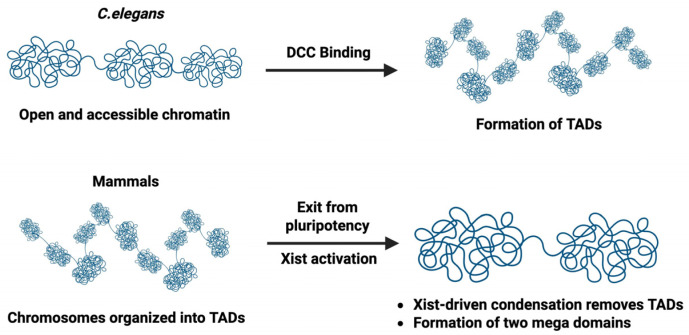 Figure 4
Figure 4- —National Institutes of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Genomics and Chromatin Dynamics · Chromosomal and Genetic Variations
1. Introduction
In organisms that use chromosome-based sex determination, the number and composition of sex chromosomes play a key role in sex determination. For instance, in mammals, males have XY and females have XX chromosomes, and sex is determined by the presence or absence of the Y-linked SRY gene [1]. In Drosophila melanogaster, males have one X-chromosome and a Y chromosome, and females have two X chromosomes. In C. elegans, males have one X (and no Y), and hermaphrodites have two X chromosomes. In Drosophila and C. elegans, sex is determined by the ratio of X chromosomes to autosomes [2]. The difference in the number of X chromosomes between sexes of organisms creates a chromosomal imbalance between the two sexes. Without compensating for the difference in the number of X chromosomes, it results in potentially deleterious differences in gene dosage and disruption in cellular homeostasis and development. For example, in C. elegans, the differences in X-chromosome number results in X-linked gene expression which is two-fold higher in hermaphrodites compared to males. To solve this problem, organisms employ diverse mechanisms to control gene expression levels between their sexes in a process called dosage compensation. Dosage compensation is a fundamental biological process that ensures balanced expression of X-linked genes between individuals with different numbers of X chromosomes.
Different organisms use distinct mechanisms of dosage compensation. In 1961, Mary Lyon formulated the X chromosome inactivation hypothesis, proposing that one X chromosome in female mammals is transcriptionally silenced early in development, thereby equalizing X-linked gene expression between XX females and XY males [3]. Mammals therefore undergo dosage compensation by transcriptionally silencing one of the two X chromosomes in females [3,4]. Mammalian X-chromosome inactivation (XCI) is driven by the long noncoding RNA XIST expressed exclusively from the inactive X-chromosome [4,5,6,7]. Silencing is mediated by proteins recruited to the X, including SPEN, hnRNP-K, and Polycomb repressive complexes (PRC), and SmcHD1 [4]. XCI is tightly coupled to loss of pluripotency and cellular differentiation [8]. In pluripotent embryonic stem cells and early embryos, core pluripotency factors such as OCT4, NANOG, and SOX2 repress the Xist long noncoding RNA and maintain both X chromosomes in an active state; upon differentiation and exit from pluripotency, repression of Xist is relieved, leading to its upregulation and initiation of XCI [4,9,10,11,12]. Complete silencing of X-linked genes also requires differentiation-dependent mechanisms [8].
In contrast, Caenorhabditis elegans uses a different dosage compensatory strategy in which XX hermaphrodites reduce transcription from each X chromosome by approximately half, thereby matching the gene expression output of XO males [13]. This is accomplished by a group of proteins referred to as Dosage Compensation Complex (DCC). The members of DCC were first identified through the discovery of sex-specific lethal mutations which result in hermaphrodite-only lethality and have no impact on males. It was observed that mutations in six genes including sdc-2, sdc-3, dpy-26, dpy-27, dpy-28, and dpy-30 were associated with hermaphrodite-specific lethality [14,15,16]. Some of the DCC proteins form a complex related to condensin. Condensins are evolutionarily conserved protein complexes that associate with chromosomes and play key roles in chromosome compaction and segregation in mitosis and meiosis [17,18]. Higher eukaryotes have two main types of condensin: condensin I and condensin II [19,20,21] (Figure 1A), which play key but distinct roles in mitosis and meiosis [22]. Interestingly, C. elegans has a third condensin called condensin I^DC^ which plays an exclusive role in dosage compensation in C. elegans hermaphrodites [23] (Figure 1B). Condensin I and condensin I^DC^ share some common protein subunits, including the Structural Maintenance Chromosome 2 (SMC2) protein MIX-1, chromosome-associated polypeptide G (CAPG-1), DPY-26, and DPY-28 [23,24]. The difference between condensin I and condensin I^DC^ stems from the presence of DPY-27 in condensin I^DC^, whose paralog is SMC-4 in condensin I. The only subunit unique to condensin I^DC^, DPY-27, therefore, ensures the dosage compensation-specific function of condensin I^DC^ [23,25]. Condensin I^DC^, together with additional proteins, including SDC-1, -2, and -3, as well as DPY-30 and DPY-21, form the complete DCC [26] (Figure 1B). As in mammals, initiation of dosage compensation in C. elegans is linked to loss of pluripotency and differentiation [27].
Mammals and Caenorhabditis elegans are the best studied examples of chromosome-wide repressive mechanisms used to achieve sex chromosome dosage compensation. It is interesting that different evolutionary lineages adapted such fundamentally different underlying molecular mechanisms to achieve the same goal. While in female mammals, one X chromosome is shut down and the other stays active, in hermaphroditic C. elegans, the mechanism involves chromosome-wide transcriptional repression of both Xs without fully silencing the X chromosomes. From an epigenetic perspective, dosage compensation raises two central questions. First, how is a chromosome-wide regulatory state initially established in a developmentally controlled and sex-specific manner? Second, once established, how is this state maintained through successive rounds of DNA replication, mitosis, and cellular differentiation? Not only do mammals and C. elegans use widely different molecular machinery to establish repression on the target chromosome(s) (long non-coding RNA versus a condensin-like complex), the fundamental logic behind mechanisms that sustain dosage compensation over time are distinct as well. In mammals, the initiating machinery triggers additional mechanisms that render maintenance of X-inactivation largely independent of the initial trigger [4,9,28,29], and thus is a phenomenon truly “epigenetic” in nature. By contrast, in C. elegans, maintenance of dosage compensation continues to rely on its initiating machinery, the DCC, to maintain dosage compensation over time [30].
In this review, we synthesize recent advances in understanding how repressive chromatin states are established and maintained, with a particular focus on contrasting Caenorhabditis elegans dosage compensation with XIST-mediated X-chromosome inactivation in mammals. We examine how specialized chromosome topology, repressive chromatin modifications, and higher-order nuclear architecture cooperate to achieve stable, sex-specific regulation of X-linked gene expression, and we highlight key unresolved questions and important directions for future research.
2. Mechanism of Dosage Compensation Initiation and Establishment in Mammals
Mammals undergo dosage compensation through XCI, a process that transcriptionally silences one X chromosome in XX females. Mammalian dosage compensation relies on long noncoding RNA-mediated recruitment of chromatin regulators. Recent work has emphasized that chromatin modifications and higher-order chromosome architecture is not just a consequence of XCI but a central driver of both initiation and establishment of the inactive X chromosome [4,31].
2.1. Developmental Timing and Context of Mammalian Dosage Compensation
In mammals, dosage compensation through X chromosome inactivation (XCI) is tightly coupled to cellular state. In mouse embryos, an initial form of imprinted XCI occurs during early cleavage stages, but this silencing is transient and restricted to extraembryonic lineages [32,33]. Cells of the inner cell mass subsequently reactivate the inactive X-chromosome, returning to a state with two active X chromosomes [34,35,36]. Canonical, random XCI is then initiated after implantation, coincident with exit from pluripotency and onset of differentiation [4,37]. Indeed, studies have shown that complete and stable X-linked gene silencing in mammals requires differentiation [8].
Recent work has highlighted that mammalian dosage compensation is not a single, uniform process, but rather proceeds through distinct developmental stages. Human preimplantation embryos employ the mechanism of X-chromosome dampening, in which transcription from both X chromosomes is reduced without full inactivation of either chromosome [4,38,39,40,41]. This is similar to the dosage compensation mechanism used by C. elegans, suggesting that partial, reversible dampening may represent an evolutionarily conserved solution that mammals adopt briefly before committing to stable XCI. While both early human embryos and C. elegans use chromosome dampening, the molecular machineries behind these processes are different. At these early stages in humans, XIST RNA and additional non-coding RNA XACT are expressed from both X chromosomes [42]. Although not fully understood, dampening seems to require XIST and one of its interacting protein partners SPEN [41], similar to the machinery involved in XCI (see below), and different from the machinery used in C. elegans.
2.2. Genetic and Developmental Signals in Establishment of Mammalian Dosage Compensation
In mammals, initiation of XCI is controlled by developmental state, and this regulation occurs through a bona fide pluripotency network. Central to mammalian dosage compensation is the activation of the long noncoding RNA XIST, which is transcribed from the X-inactivation center and acts as the primary initiator of chromosome-wide silencing [5,6,7]. In mammals, regulation of XIST expression is closely tied to pluripotency and early differentiation. In the early stages of embryogenesis, cells of the inner cell mass (ICM) and epiblast exist in a pluripotent state, defined by the activity of core transcription factors such as OCT4, SOX2, and NANOG, which maintain pluripotency and prevent premature differentiation [43,44,45]. They also actively repress Xist expression in undifferentiated cells through multiple mechanisms. At this stage, pluripotency factors bind regulatory elements around the Xist locus and cooperate to suppress Xist transcription, thereby ensuring that XCI initiation does not occur while a cell is pluripotent [10,37,46,47]. One of the pluripotency factors which regulate XCI is OCT4. OCT4 interacts with chromatin architectural proteins such as CTCF and YY1 and binds to regulatory regions of the X-inactivation center, including Tsix and Xite, antagonizing Xist expression [10,47,48,49] (Figure 2). This binding enhances Tsix expression and facilitates X-chromosome pairing and counting, which is required for proper choice of the inactive X, while preventing inappropriate Xist upregulation in pluripotent cells [10]. Xist therefore remains repressed in pluripotent cells; hence, XCI initiation is blocked until cells start to exit the pluripotent state (Figure 2).
PRC1 and PRC2 control the onset and progression of cellular differentiation in mammals, acting as epigenetic “brakes” to silence developmental genes in pluripotent stem cells (PSCs) [50,51]. Through deposition of H3K27me3 at developmental gene loci, PRC2 helps silence pluripotency programs and create a chromatin environment permissive for Xist expression and XCI initiation. As cells differentiate and pluripotency networks collapse, repression of Xist is relieved, enabling Xist upregulation and initiation of XCI [12]. This promotes the upregulation of Xist by one of the X chromosomes, leading to the initiation of XCI (Figure 2). Thus, the decline in pluripotency factor activity constitutes a molecular permissive signal for Xist activation and the onset of X-chromosome silencing.
2.3. Establishment of X-Chromosome Silencing Through Xist-Dependent Protein Recruitment
Mammals use Xist RNA as the chromosome-recognition and recruitment platform. Upon activation, Xist is expressed from the inactive X chromosome and spreads in cis across the X chromosome from which they originate, remaining strictly bound to that chromosome [7,8]. This spreading is not guided by linear DNA sequence but by three-dimensional chromatin architecture. Xist initially associates with genomic regions that are spatially proximal to its transcription site, using existing nuclear organization as a scaffold for chromosome-wide coverage [4,52]. This process results in the formation of discrete Xist RNA granules within the X-chromosome territory.
Mammalian XCI is established through RNA-guided assembly of a multi-protein repressive machinery. During this stage, SPEN, a transcriptional repressor, binds to the A-repeat region of Xist RNA (Figure 3A). SPEN is required for silencing most X-linked genes and acts predominantly by recruiting histone deacetylases, leading to rapid loss of active chromatin marks at promoters and gene bodies [53,54,55,56,57] and disruption of either the Xist A-repeat or SPEN results in a near-complete failure of transcriptional repression. Xist recruits additional factors that promote stable chromatin remodeling. The B-repeat region of Xist binds hnRNP-K, which functions as a scaffold for recruitment of Polycomb repressive complexes PRC1 and PRC2 [58,59,60,61,62,63]. PRC1 catalyzes monoubiquitination of histone H2A at lysine 119 (H2AK119ub), while PRC2 deposits trimethylation of histone H3 at lysine 27 (H3K27me3) (Figure 3A). These modifications spread across the X chromosome and reinforce transcriptional repression. An additional, less well-understood, chromatin mark, histone H4 lysine 20 monomethylation (H4K20me1) appears on the inactive X chromosome at about the same time [64,65].
As XCI progresses, other structural regulators are recruited to the inactive X chromosome. Among these, SmcHD1 helps in consolidating gene silencing, particularly at genes that are repressed later during differentiation [8,58,66] (Figure 3A). SmcHD1 recruitment occurs after initial Xist spreading and Polycomb deposition. The delayed recruitment of SmcHD1 demonstrates that the establishment of XCI is multistep, with distinct classes of genes silenced at different stages. Together, these findings reveal a model of the establishment of dosage compensation in mammals: Xist RNA first recruits transcriptional repressors to dampen gene expression, then recruits Polycomb complexes to impose chromatin-based repression, and finally recruits architectural proteins that stabilize the inactive state (Figure 3B).
2.4. Reorganization of X-Chromosome Architecture: Xist Granules, Protein Condensation, and Compartment Formation
Beyond recruitment of transcriptional repressors and chromatin modifiers, establishment of mammalian dosage compensation relies on a reorganization of three-dimensional chromosome architecture. XCI is associated with the collapse of active regulatory architecture and formation of a repressive nuclear compartment driven by Xist RNA. Xist does not coat chromatin uniformly. Instead, Xist accumulates in discrete RNA granules within the X chromosome territory, with approximately 50 granules per inactive X chromosome, each containing a small number of Xist transcripts [4,67,68]. Given the scale of the X-chromosome and the number of genes subject to silencing, this organization promotes the amplification of silencing signals beyond direct RNA–chromatin interactions.
This amplification occurs through protein condensation. Xist recruits a large cohort of interacting proteins via its conserved repeat elements. These proteins engage in protein–protein interactions, many of which are low-affinity but highly multivalent. As a result, Xist-associated factors form highly dynamic supramolecular assemblies, often referred to as supramolecular complexes (SMACs) [68]. These generate regions of high local protein density surrounding Xist granules, leading to the concentration of repressive and architectural regulators within the X-chromosome territory. Disruption of Xist-mediated condensation impairs transcriptional silencing, even when Xist RNA remains chromosome-associated. This demonstrates that compartment formation is not a secondary consequence of repression but a mechanistic requirement for it [68]. Within these condensed regions, transcriptional activators and components of the transcription machinery are depleted, largely due to reduced chromatin binding capacity. This creates a chromatin environment in which transcription is inhibited not only by epigenetic marks but also by physical exclusion of activating factors.
Following protein condensation formation, the large-scale chromatin architecture of the X chromosome is remodeled. The inactive X-chromosome loses conventional topologically associated domains (TADs) and long-range enhancer–promoter interactions, which are a characteristic of active chromosomes. Instead, the inactive X adopts a simplified, compartmentalized state of two megadomains with reduced regulatory complexity [69,70,71] (Figure 4). Disruption of this structure by deleting the Dxz4 locus at the megadomain border does not lead to widespread X-chromosome reactivation, suggesting that the structure may contribute to silencing, in a manner that is redundant with other silencing mechanisms, but it is not strictly required [69,72,73].
Different structural changes are induced on the X chromosomes in C. elegans and mammals. In C. elegans, the DCC establishes new TADs to modulate transcription while in mammals, the inactive X-chromosome loses conventional TADs instead, and adopts a simplified, compartmentalized state of two megadomains.
3. Initiation and Establishment of Dosage Compensation in C. elegans
3.1. Developmental Timing and Biological Context of Dosage Compensation Onset
Genetic and developmental signals which trigger the onset of dosage compensation in mammals and C. elegans are different, although in both cases, the onset of dosage compensation is linked to loss of plasticity and the onset of differentiation. In C. elegans, dosage compensation is initiated as cells lose their ability to adopt alternative fates [27]. This loss of developmental plasticity in C. elegans is regulated by Polycomb repressive complex 2 (PRC2) [74]. The PRC2 catalytic subunit MES-2, the worm homolog of mammalian EZH2, promotes the transition from a plastic embryonic state to differentiation by repressing genes associated with alternative developmental programs. In mes-2 mutants, embryonic cells retain an extended capacity for fate reprogramming, demonstrating that PRC2 is required for timely exit from developmental plasticity [74]. In these mutants, dosage compensation onset is also delayed [27]. Thus, although C. elegans does not possess a pluripotent stem cell state analogous to mammalian embryonic stem cells [75,76], PRC2 nonetheless plays a conserved role in coordinating developmental progression toward differentiation.
Together, the onset of dosage is regulated by exit from a highly plastic developmental state in both mammals and in C. elegans. In C. elegans, this transition is regulated by PRC2-mediated restriction of developmental plasticity and permits DCC recruitment to the X chromosomes, whereas in mammals, dissolution of pluripotency networks enables Xist activation and initiation of XCI. Despite this similarity, the molecular mechanisms and regulatory outcomes remain distinct.
The onset of dosage compensation marks the earliest phase at which organisms respond to sex chromosome imbalance by initiating chromosome-wide regulatory control. In C. elegans, initiation of dosage compensation occurs early in embryogenesis, with the recruitment of DCC onto X chromosome occurring around the 30–40 cell stage [77]. This timing ensures that dosage compensation is established before the onset of high transcriptional activity. One of the key proteins in the DCC is SDC-2. SDC-2 plays a critical role in both sex determination and dosage compensation. It is negatively regulated by a protein called XOL-1 in the sex determination pathway (Figure 5). XOL-1, XO lethal, is the master switch gene whose activity is determined by the X: A ratio [2,78,79,80,81]. Unlike in mammals, where sex determination and dosage compensation are regulated by distinct mechanisms, in C. elegans, these two processes are linked and regulated by the activity of XOL-1. When the X:A ratio is 0.5, XOL-1 levels become high, which then represses the expression of SDC-2. With SDC-2 expression being low, HER-1 expression then becomes high, and the embryo develops to become a male, and dosage compensation is not initiated. In xol-1 mutant XO animals, SDC-2 is expressed, which leads to repression of the single male X and lethality [78]. On the other hand, when X:A ratio is 1, the expression levels of XOL-1 become low and SDC-2 expression levels become high. SDC-2 then triggers the onset of dosage compensation and blocks the expression of HER-1, and the embryo develops into a hermaphrodite (Figure 5). Other DCC members are maternally loaded into oocytes. SDC-2, however, is not maternally loaded and it is only produced in XX hermaphrodite embryos. This ensures that DCC assembly only occurs in XX embryos and reveals that SDC-2 is the key protein required for the assembly of DCC on X chromosome at the onset of dosage compensation.
3.2. Recruitment of DCC on the X Chromosome in XX Hermaphrodites
DCC can distinguish between X chromosomes and autosomes. X chromosomes have distinct DNA motifs, called rex (recruitment element on X), which serve as an initial recruitment site for DCC binding. The rex site is a 12 bp consensus motif enriched on the X chromosome [82,83,84]. Chromatin immunoprecipitation analysis revealed that these sites correspond to the highest peaks of DCC binding on the X [84,85]. Recruitment of DCC to the X chromosomes occurs through a hierarchical and ordered assembly pathway that ensures sex-specific and chromosome-specific regulation (Figure 6). While in mammals, the lncRNA XIST plays a pivotal role in recruiting proteins to the Xs; no such RNA has been identified in C. elegans. Instead, the earliest limiting factor in this process is SDC-2, a hermaphrodite-specific protein whose expression is restricted to XX embryos, and which serves as the master initiator of dosage compensation [77]. SDC-2 is therefore the first DCC component to associate with the X-chromosome, although whether it can directly bind DNA is unknown. SDC-2 can bind to the X-chromosome even in the absence of other DCC members [77,86]. This further affirms that SDC-2 is the key protein which first binds to the X chromosomes and triggers DCC assembly.
Following SDC-2 binding to the rex sites, additional regulatory proteins are recruited in an SDC-2-dependent manner. SDC-3 and DPY-30 are among the earliest factors to associate with the X after SDC-2 and function to stabilize DCC assembly and facilitate recruitment of downstream components. These proteins do not independently recognize the X chromosome, instead, their localization requires prior SDC-2 binding, placing them downstream in the recruitment hierarchy [77,86]. SDC-3 is a zinc-finger protein which assists SDC-2 in the recruitment of other DCC members on the X chromosomes. DPY-30 contributes to the stabilization of early DCC assembly and recruitment to the X.
Once the SDC proteins are in place, the condensin I^DC^ complex is recruited to the X chromosome. This complex includes the dosage-compensation-specific SMC subunit DPY-27, its shared SMC partner MIX-1, and the non-SMC subunits DPY-26, DPY-28, and CAPG-1. Condensin I^DC^ loading depends on the prior action of SDC-2 and SDC-3 and cannot occur independently of these factors [23,26]. After initial loading at rex sites, condensin I^DC^ spreads along the length of the X chromosome, occupying both recruitment sites and transcriptionally active regions, thereby enabling chromosome-wide repression of X-linked gene expression [84,85,86]. DPY-21 later localizes to the DCC on the X at around the bean-to-comma stage (200–500 cells) [83]. Together, these observations support a model in which DCC binds to the X chromosome proceeds in a strictly ordered sequence, beginning with SDC-2, followed by stabilization through SDC-3 and DPY-30, the loading and spreading of condensin I^D^, and culminating in the binding of DPY-21.
3.3. Establishment of Dosage-Compensated State in C. elegans
Dosage compensation initiation is characterized by the binding of the DCC to the X chromosome. After this, dosage compensation is established through changes in chromosome topology, histone modification, chromatin accessibility, transcriptional activity and nuclear organization. Establishment of dosage compensation begins immediately after DCC recruitment to the X and continues through embryogenesis. In this subsection, we have discussed major events which lead to the establishment of dosage compensation.
3.3.1. Formation of a Condensin I DC Structural Scaffold
DCC recruitment is followed by the assembly of a condensin-like structural scaffold along the X chromosomes. Following recruitment to rex sites, condensin I^DC^ spreads along the X chromosomes to occupy a broader set of secondary sites that lack autonomous recruitment activity. DCC activity in hermaphrodites leads to X-chromosome compaction in interphase nuclei [87]. At higher resolution, Hi-C analysis demonstrated that recruitment of the DCC induces large-scale remodeling of X-chromosome topology in XX hermaphrodites [88,89]. In contrast to autosomes, the compensated X chromosomes adopt a distinctive three-dimensional organization characterized by regularly spaced self-interacting domains with strengthened boundary insulation. These self-interacting domains resemble topologically associated domains (TAD) in mammals. Many of these domain boundaries coincide with high-affinity rex sites, indicating that DCC recruitment not only targets the X-chromosome but also defines its topological architecture [88,90,91,92].
The involvement of condensin in C. elegans dosage compensation raises the question of whether condensin plays similar roles in other organisms. Although condensin complexes are broadly conserved as drivers of loop extrusion and chromosome compaction, the activity of condensin I^DC^ in dosage compensation is specific. Rather than inducing uniform compaction as in mitosis, condensin I^DC^ establishes a stable interphase architecture composed of insulated domains and altered long-range interaction frequencies [88,91]. Mammalian condensin I is cytoplasmic during interphase, but condensin II is nuclear [20,21], suggesting that condensin II may be able to contribute to shaping interphase chromosome architecture, similar to C. elegans condensin I^DC^. However, condensin II activity is inhibited in interphase by the binding of MCPH1, and at the onset of mitosis, a phosphorylation-dependent switch triggers the displacement of MCPH1, the binding of M18BP1, and condensin’s chromosome compaction activity [93,94]. This contrasts with the activity of condensin I^DC^, which remains associated with chromosomes throughout the cell cycle. Whether its localization is regulated by phosphorylation is not known. Although there is no evidence for condensin’s involvement in XCI in mammals, the related complex cohesin has been linked to chromosome topology changes in the inactive X [95,96]. Notwithstanding, condensin has been implicated in interphase gene regulation and chromosome territory formation in other contexts, for example [97,98,99].
Even though dosage compensation involves repression in both mammals and C. elegans, the structural changes induced on the X chromosomes are very different. In C. elegans dosage compensation, the DCC establishes new loop domains to modulate transcription while preserving overall chromosome accessibility. Thus, in C. elegans, DCC actively creates chromosome topology across an otherwise active chromosome. In mammals, Xist-driven condensation removes active architecture and builds a facultative heterochromatin compartment that enforces near-complete transcriptional repression. In both cases, chromosome architecture is important in dosage compensation output, but the directionality and molecular mechanisms of architectural change are fundamentally distinct (Figure 4).
In C. elegans, genetic evidence suggests that this structural scaffold contributes to the establishment of dosage compensation. In SDC-2-depleted XX hermaphrodites, the TAD boundary formation is lost. In these mutants, the insulation profile of the X chromosome was greatly reduced and was accompanied by elevated X-linked gene expression, suggesting a link between TAD formation and gene repression [91]. However, deletion of the eight strongest rex sites leads to disruption of TAD formation, but only minimal changes in gene expression, implying that these topological changes are not required for gene repression. Hermaphrodite worms with rex site deletions are shorter-lived, which suggests that proper X-chromosome topology promotes longevity even if it is not strictly required for repression [89]. These observations suggest that these topological changes are not the major drivers of gene expression changes in either system.
3.3.2. Chromatin Modifications and Nuclear Architecture
In parallel with large-scale architectural reorganization, establishment of dosage compensation on the C. elegans X chromosomes is accompanied by changes in chromatin state. As in mammals, the X-chromosome acquires a unique set and distribution of histone modifications, but the actual modifications are different between the species. Mammals almost completely silence the inactive X, while C. elegans only dampens expression, requiring a different set of chromatin marks. While there is a general reduction in active chromatin marks on the dosage compensated for X in C. elegans, the degree of depletion does not reach the levels seen on the mammalian inactive X [90,100]. The best characterized modification associated with the C. elegans dosage-compensated X is the repressive mark monomethylation of histone H4 at lysine 20 (H4K20me1), a mark also seen on the mammalian inactive X. Immunofluorescence microscopy and genome-wide chromatin profiling demonstrated that H4K20me1 is selectively enriched across X chromosomes in XX C. elegans hermaphrodites and that this enrichment depends on a functional Dosage Compensation Complex [100,101,102].
Deposition of H4K20me1 onto unmethylated histone H4 is catalyzed by SET-1, the C. elegans ortholog of mammalian SETD8/PR-Set7, whereas conversion of monomethylation to di- and trimethylation of H4K20 (H4K20me3) is mediated by SET-4, the ortholog of Suv4-20 [100,102]. During dosage compensation, the DCC subunit DPY-21, a member of the Jumonji C family of demethylases, converts me2/me3 back to H4K20me1 selectively on the X [101]. H4K20me1 reduction due to set-1 or dpy-21 null mutation led to a significant increase in X chromosome expression compared to autosomes in L3 larvae or embryos [30,101,103]. Importantly, DCC localization to the X-chromosome remains intact in set-1, set-4 and dpy-21 mutants [100,103,104], indicating that H4K20me1 acts downstream of DCC recruitment and contributes specifically to establishment of transcriptional repression rather than to targeting of the complex. H4K20me1 is thought to reinforce the structural changes imposed by condensin I^DC^ [101]. Importantly, DCC begins to repress X chromosomes in early embryogenesis (40- cell stage) before DPY-21-mediated H4K20me1 enrichment on the X happens between 100-cell to comma stage [27,102,103]. This may suggest that condensin I^DC^-mediated loop structures gain enhanced stability when embedded within an H4K20me1-enriched chromatin environment, increasing resistance to transcriptional activation. H4K20me1 is required for mitotic chromosome condensation in mammals, suggesting that H4K20me1 enrichment on the X may reduce the access of transcription machinery to X-linked genes by inducing chromatin compaction [102,105].
Chromatin modification and chromosome architecture are tightly coupled during establishment of dosage compensation. Depletion of H4K20me1 results in weakened repression, reduced chromatin compaction, and impaired domain insulation, despite continued presence of condensin I^DC^ on the X [30,87,101]. DPY-21 regulates the dynamics of condensinI^DC^ binding, which is important for transcription repression [106]. These results suggest that establishment of dosage compensation relies on reciprocal reinforcement between chromosome structure and chromatin state. Condensin I^DC^-mediated topology provides a spatial framework that promotes selective chromatin modification, while DPY-21-mediated H4K20me1 enrichment stabilizes this architecture and enhances transcriptional repression.
In addition to chromosome topology and histone modification, nuclear organizations also support establishment of dosage compensation. CEC-4 is a nuclear tethering protein which binds H3K9-methylated chromatin and anchors heterochromatic regions to the nuclear lamina [107]. CEC-4-mediated tethering and the DCC cooperate to compact the X chromosomes and anchor them to the nuclear lamina [108]. Loss of CEC-4 disrupts X-chromosome condensation and subnuclear localization, but this loss does not affect DCC recruitment or H4K20me1 enrichment on X. This indicates that nuclear tethering acts downstream of DCC binding and independently of DCC localization. Also, loss of CEC-4 function led to a modest but significant derepression of X-linked genes, suggesting that CEC-4-mediated nuclear tethering of X chromosomes stabilizes repression but it is not strictly required. Mutations in cec-4 lead to X decompaction comparable to DCC mutants, but only minimal gene expression changes [108]. This provides evidence that chromosome compaction and gene repression can be uncoupled.
3.3.3. Transcriptional Dampening via Reduced Pol II Binding
Although condensin I^DC^-mediated architectural remodeling and DPY-21-dependent chromatin modification provides the physical and epigenetic framework for dosage compensation, transcriptional analyses demonstrate that reduction in X-linked gene expression is itself a progressive and dynamic process. The transition from a fully active X chromosome to a dosage-compensated state does not occur instantaneously upon DCC recruitment. Instead, transcriptional dampening unfolds in stages, with gene-specific and region-specific kinetics that reflect the interplay between chromosome topology, chromatin state, and transcriptional regulation.
Genome-wide chromatin immunoprecipiation studies mapping RNA Pol II occupancy provided early insight into how transcription is modulated during dosage compensation by limiting Pol II binding to the X chromosomes [86]. DCC-mediated repression can be detected by RNA-seq in early embryos, shortly after DCC recruitment to the X chromosome [103]. This initial reduction is detectable even before full establishment of chromatin modifications, indicating that architectural changes can influence transcriptional engagement at early stages. GRO-seq experiment tracking nascent RNA transcripts also show that dosage compensation reduces the levels of engaged Pol II across X-linked gene bodies in XX hermaphrodites [109]. As embryogenesis proceeds, Pol II depletion becomes more pronounced, particularly in regions with strong condensin I^DC^ binding. Mid-embryonic stages show separation between X chromosomes and autosomes in Pol II occupancy profiles, with the most robust DCC-bound regions exhibiting the earliest and strongest decreases. Thus, C. elegans equalizes X-chromosome-wide gene expression between the sexes by reducing Pol II recruitment to the promoters of X-linked genes in XX [109].
4. Maintenance of Dosage Compensation
4.1. Maintenance of X-Chromosome-Inactivated State in Mammals
Once established, dosage compensation must be maintained for the lifetime of the organism. In mammals, maintenance of dosage compensation through X-chromosome inactivation (XCI) is mechanistically distinct from its initiation and establishment. It relies on epigenetic memory rather than continuous activity of the initiating factor, XIST or the X inactivation center (XIC). Early experiments analyzing derivatives of the human inactive X chromosome in mouse/human somatic cell hybrids suggested that XIC is not required for maintenance of X-chromosome inactivation in somatic cells [28]. Thus, once dosage compensation is established, the inactive X chromosome can be maintained independently of XIST. Even though the conditional deletion of Xist in somatic cells led to the disruption of the localization of macroH2A on the inactive X chromosome, it did not cause global reactivation of X-linked genes [110]. This further reveals that maintenance of XCI in differentiated cells does not rely on XIST. Similarly, Xist-mediated repression in embryonic stem (ES) cells and during early XCI is reversible and requires Xist expression [9]. However, after 48–72 h of differentiation, XCI becomes irreversible and independent of Xist. This further implies that XIST is required for XCI initiation and establishment but is dispensable for maintenance.
In mouse embryonic fibroblasts, loss of Xist in the maintenance phase resulted in limited reactivation of X-linked genes [29]. Inhibition of DNA demethylation or suppression of histone deacetylation further exacerbated gene derepression. This implies that Xist interacts with other epigenetic factors to contribute to gene silencing, even though it is not strictly necessary for XCI maintenance [8]. Although XCI can be maintained without Xist in differentiated cells under controlled conditions, long-term loss of Xist in vivo destabilizes the inactive X chromosome and leads to pathological consequences, linking XCI maintenance to genome integrity and cancer suppression [111].
Mammals rely on epigenetic inheritance systems that are linked to cell cycles to maintain XCI. Later in the X inactivation process, as silencing becomes Xist-independent, additional mechanisms are recruited to the Xi. CpG islands are methylated on cytosine residues on the Xi [112]. This methylation takes place after silencing has been established [113,114]. The methylation marks are placed by the de novo methyltransferase Dnmt3b, and at least at some loci, the process depends on SmcHD1 [113]. SmcHD1 plays important roles in establishing the higher-order structure of the Xi, as described above, but it was originally described as a protein required for X inactivation maintenance [115].
Once established, DNA methylation can be maintained without the initiating trigger. DNA methylation is copied during the S phase by the maintenance DNA methyltransferase Dnmt1 [113]. These methyltransferases recognize hemimethylated DNA and restore full methylation on newly synthesized strands. This provides a heritable means of preserving transcriptional repression across cell divisions.
In addition, histone modifications associated with repressive chromatin may play important roles in long-term maintenance. In addition to the H3K27me3 mark placed by PRC2 described above, the inactive X is also enriched for H3K9 trimethylation (H3K9me3), placed by Setdb1, in the intergenic regions [116]. SmcHD1 is also necessary for the establishment of these blocks of H3K9me3 enrichment on the inactive X. Lack of SmcHD1 leads to the loss of these H3K9me3 blocks and redistribution of the H3K27me3 mark [117]. Importantly, H3K9 methylation can also be inherited through mitosis and can contribute to the maintenance of silent chromatin states [118]. Similarly, Polycomb-mediated H3K27 trimethylation is maintained when PRC2 recognizes pre-existing H3K27me3 marks and catalyzes methylation of newly incorporated histones during DNA replication and across cell divisions [119,120]. Therefore, establishment of these silencing marks sets up a heritable repressed state. Apf7ip, a protein that coordinates DNA methylation and H3K9 methylation and is known to interact with Mbd1 and Setdb1, is also necessary for maintenance of silencing in differentiated cells [121], thus linking H3K9 methylation and DNA methylation. The nuclear matrix protein CIZ1 localizes to the inactive X in earlier stages but only appears to be required for maintenance [122,123].
Finally, the RNA-binding proteins PTBP1, MATR3, CELF1 and TDP-43 bind to the E-repeat region of Xist [124]. The proteins form a condensate which is nucleated by Xist but that can be maintained in its absence. This transition occurs at the time when silencing becomes Xist-independent. E-repeat mutants can establish silencing, but not maintain it, suggesting that condensate formation by these proteins is yet another mechanism that contributes to maintenance [124]. Together, these studies establish that maintenance of mammalian XCI is epigenetic in nature. Although Xist helps preserve long-term stability and genome integrity in vivo once established, the XCI is maintained by self-propagating DNA and histone modifications and nuclear compartmentalization and does not require ongoing XIST activity.
4.2. Maintenance of Dosage-Compensated Repressed State in C. elegans
In contrast to mammals, dosage compensation in C. elegans does not transition into a self-sustaining, epigenetically locked state after establishment [30]. Instead, maintenance of X-chromosome repression in C. elegans requires the continuous presence of condensin I^DC^, ongoing chromatin modification, and sustained nuclear architecture. By the end of embryogenesis in C. elegans, dosage compensation is fully established, and most somatic cells have exited mitosis and have become post-mitotic [125]. From this stage onward, the dosage-compensated state must be maintained to promote proper growth, tissue function, and overall fitness. However, unlike in mammals, maintenance in this case does not involve epigenetic inheritance through mitotic divisions. While the molecular mechanisms governing dosage compensation establishment during embryogenesis have been extensively studied, comparatively, less is known about how dosage compensation is maintained after its establishment. Early genetic studies using cold-sensitive dpy-27 alleles showed that condensin I^DC^ activity is required for survival during mid-embryogenesis, whereas loss of dpy-27 function at later stages has a much milder impact on viability [16]. These initial observations suggested that DPY-27 is required for dosage compensation establishment, but perhaps not for maintenance. Recent work by Trombley et al. provides a detailed and systematic analysis of dosage compensation maintenance in C. elegans. Depletion of DPY-27 using an auxin-inducible degron (AID) system during embryogenesis resulted in nearly complete embryonic lethality, confirming that condensin I^DC^ is indispensable for the establishment of dosage compensation. In contrast, depletion of DPY-27 during larval or adult stages did not compromise survival, although animals exhibited pronounced developmental defects [30]. However, detailed analysis revealed that these larvae survived not because dosage compensation was maintained in the absence of DPY-27, but rather that they survived even though dosage compensation was not maintained. Loss of DPY-27 was associated with dissociation of condensin I^DC^ from the X chromosomes, decondensation of X-chromosome territories, loss of peripheral nuclear positioning, and loss of H4K20me1 enrichment [30]. These changes were accompanied by a significant increase in X-linked gene expression. These results demonstrate that maintenance of dosage compensation is not a passive epigenetic memory but requires continuous and cooperative action of multiple repressive mechanisms [30]. Thus, continuous condensin I^DC^ activity is required to preserve the repressive architecture of the X chromosome and maintain dosage compensation in post-mitotic cells. These findings also reveal that while dosage compensation establishment is required for embryonic viability, maintenance of dosage compensation in larvae and adults is critical for normal development and for overall fitness, but not viability.
Maintenance of dosage compensation in C. elegans also requires the continuous activity of DPY-21, which enriches H4K20me1 on X chromosomes [101]. In post-mitotic cells, H4K20me1 enrichment on X chromosomes is gradually lost in DPY-27-depleted worms, despite having been deposited earlier during embryogenesis [30]. Thus, H4K20me1 is not a long-term epigenetic memory mark and hence requires continuous replenishment, possibly through ongoing recruitment of DPY-21 by condensin I^DC^. The loss of H4K20me1 enrichment is associated with X-chromosome decompaction and transcriptional derepression. This suggests that chromatin modification and chromosome architecture are tightly coupled during maintenance of dosage compensation.
Additionally, the nuclear lamina protein, CEC-4, contributes to maintaining repressed chromatin state. CEC-4 tethers H3K9me3-enriched chromatin to the nuclear periphery and contributes to maintaining the compact and peripheral localization of the dosage-compensated X chromosome [108]. While loss of CEC-4 alone causes only mild defects in X-chromosome repression, loss of CEC-4 function mutation exacerbated X-linked gene derepression [30]. Together, these findings support a model in which maintenance of dosage compensation in C. elegans depends on the continuous presence of condensin I^DC^, DPY-21 and CEC- 4. Yet, it is important to note that the defects observed in cec-4 mutants following DPY-27 depletion cannot be attributed solely to a direct role for CEC- 4 in dosage compensation maintenance, as the CEC- 4 function was lacking at all stages of development. It will therefore be interesting in future to use the AID system to study the function of CEC- 4 exclusively in post-mitotic cells to assess its potential role in establishment versus maintenance of dosage compensation. It is also unknown whether the non-condensin subunits of DCC, especially SDC-2, are required for maintaining the already established dosage-compensated state in post-mitotic cells. It will also be interesting to investigate whether temporal depletion and recovery of DCC might result in re-establishment and maintenance of dosage compensation in post-mitotic cells.
5. Conclusions
Studies of dosage compensation enable us to understand how chromosome-wide gene repression is established and maintained in organisms. In this review, we highlight that although both mammals and Caenorhabditis elegans achieve dosage compensation through repression of X-linked gene expression, the mechanisms of initiation, establishment, and maintenance are different. These comparisons highlight that different lineages evolved to make use of distinct molecular machinery to solve the same issue: unequal gene expression from the sex chromosomes in the different sexes. In mammals, X-chromosome inactivation is initiated by XIST, and once established, it is maintained through DNA methylation, heritable histone modifications, and nuclear compartmentalization, with XIST contributing primarily to long-term stability and genome integrity. In contrast, C. elegans dosage compensation maintenance requires the continuous activity of the initiating machinery itself, including condensin I^DC^, enrichment of H4K20me1, and reinforcement through CEC-4-mediated nuclear lamina tethering. Thus, maintenance of dosage compensation in C. elegans is an active, dynamic process rather than passive epigenetic memory. There are still gaps in understanding how dosage compensation is maintained once established in C. elegans. This includes whether SDC-2, the protein that triggers DCC assembly on the X, is required for maintenance. Also, the role of CEC-4 in establishment and maintenance has not been decoupled. Future experiments in these areas would enhance our understanding of how the repressed chromatin state is maintained in C. elegans.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Koopman P. Gubbay J. Vivian N. Goodfellow P. Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry Nature 199135111712110.1038/351117 a 02030730 · doi ↗ · pubmed ↗
- 2Meyer B.J. Mechanisms of sex determination and X-chromosome dosage compensation Genetics 2022220 iyab 19710.1093/genetics/iyab 19735100381 PMC 8825453 · doi ↗ · pubmed ↗
- 3Lyon M.F. Gene Action in the X-chromosome of the Mouse (Mus musculus L.)Nature 196119037237310.1038/190372 a 013764598 · doi ↗ · pubmed ↗
- 4Dror I. Tan T. Plath K. A critical role for X-chromosome architecture in mammalian X-chromosome dosage compensation Curr. Opin. Genet. Dev.20248710223510.1016/j.gde.2024.10223539053028 PMC 11317216 · doi ↗ · pubmed ↗
- 5Borsani G. Tonlorenzi R. Simmler M.C. Dandolo L. Arnaud D. Capra V. Grompe M. Pizzuti A. Muzny D. Lawrence C. Characterization of a murine gene expressed from the inactive X chromosome Nature 199135132532910.1038/351325 a 02034278 · doi ↗ · pubmed ↗
- 6Brown C.J. Ballabio A. Rupert J.L. Lafreniere R.G. Grompe M. Tonlorenzi R. Willard H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome Nature 1991349384410.1038/349038 a 01985261 · doi ↗ · pubmed ↗
- 7Brockdorff N. Ashworth A. Kay G.F. Cooper P. Smith S. Mc Cabe V.M. Norris D.P. Penny G.D. Patel D. Rastan S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome Nature 199135132933110.1038/351329 a 02034279 · doi ↗ · pubmed ↗
- 8Bowness J.S. Nesterova T.B. Wei G. Rodermund L. Almeida M. Coker H. Carter E.J. Kadaster A. Brockdorff N. Xist-mediated silencing requires additive functions of SPEN and Polycomb together with differentiation-dependent recruitment of Smc HD 1Cell Rep.20223911083010.1016/j.celrep.2022.11083035584662 PMC 9637994 · doi ↗ · pubmed ↗