A Novel Frameshift Mutation in SLC20A2 in a Korean Patient with Primary Brain Calcification, Parkinsonism and Memory Impairment
Eva Bagyinszky, Minju Kim, Young Ho Park, Danyeong Kim, Seong Soo A. An, SangYun Kim

TL;DR
A new mutation in the SLC20A2 gene is linked to brain calcification and neurological symptoms in a Korean patient.
Contribution
A novel frameshift mutation in SLC20A2 is identified as a cause of primary brain calcification.
Findings
A c.1152_1153delCA frameshift mutation in SLC20A2 was found in a patient with brain calcification and neurological symptoms.
Reduced SLC20A2 protein levels in plasma suggest a loss-of-function mechanism due to the mutation.
The patient showed increased amyloid-beta oligomerization, hinting at a potential link between SLC20A2 and amyloid pathways.
Abstract
Objectives: The patient presented various neurological symptoms in her 50s, such as memory issues, insomnia, depression, and motor impairment. Diverse investigations were performed to identify the underlying causes on her neurological symptoms and understand her neuro- deteriorations. Methods: Clinical neurological and brain imaging analyses: CT, MRI and PET were performed on the patient. Blood was drawn for the whole-exome sequencing and functional studies with biomarker for amyloid-beta oligomers and SLC20A2 protein in plasma. Results: Brain imaging revealed calcifications in multiple regions, including the subcortical white matter, basal ganglia, thalami, and dentate nuclei. Genetic analysis revealed a c.1152_1153delCA, p.Asn384Lysfs*30 variant in SLC20A2 gene. The decreased SLC20A2 protein levels in plasma in comparison to healthy controls suggested a loss-of-function mechanism from…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
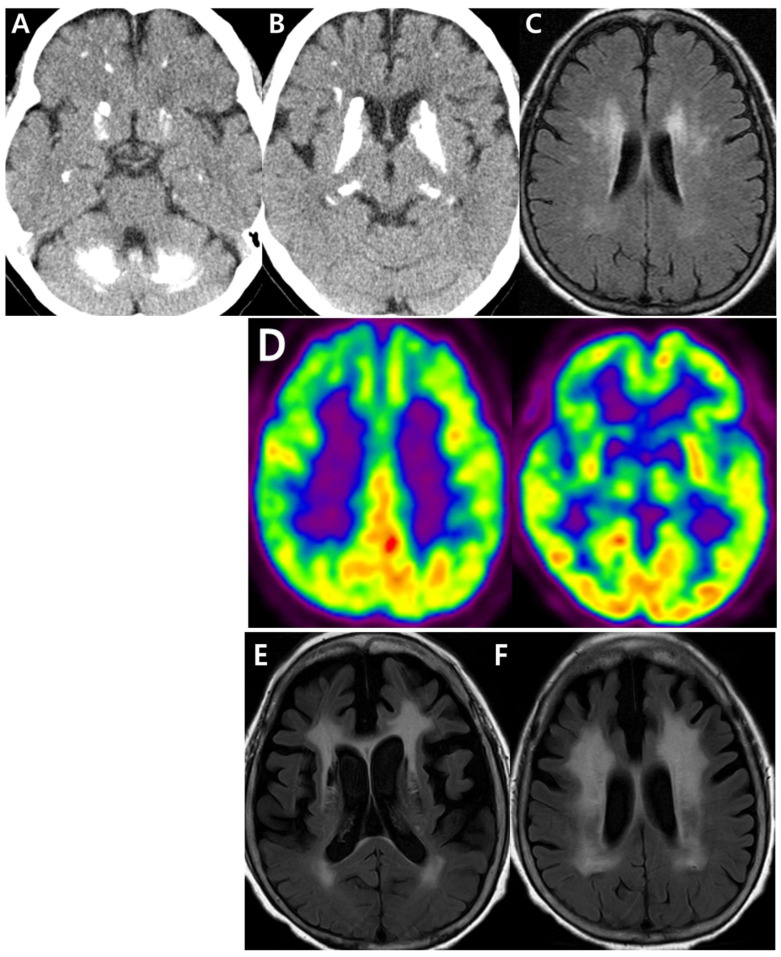 Figure 1
Figure 1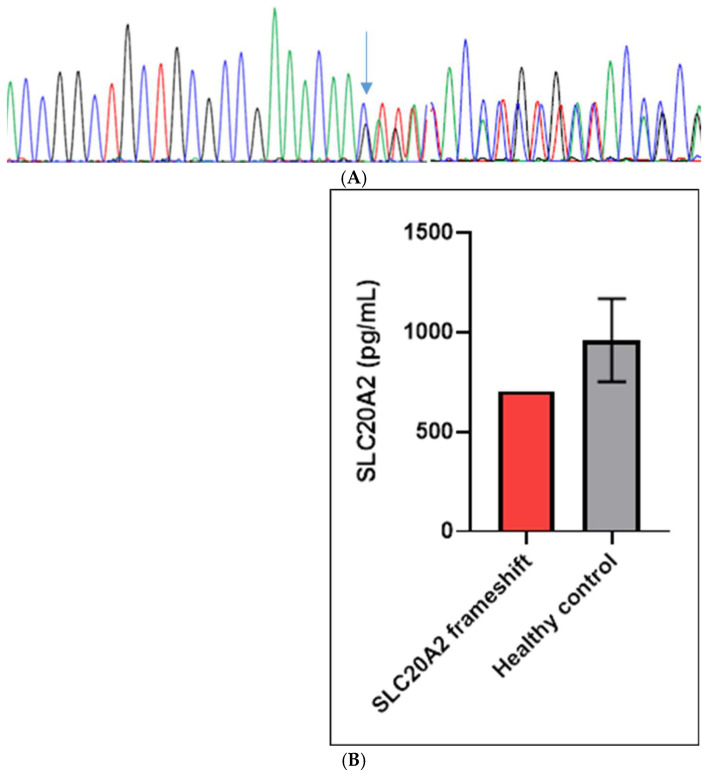 Figure 2
Figure 2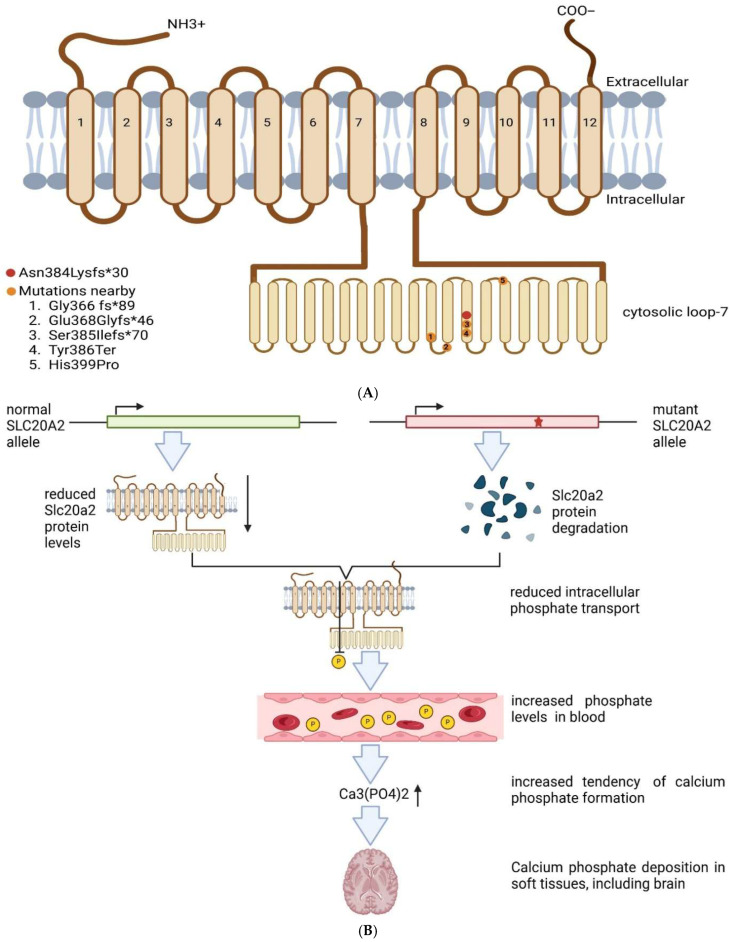 Figure 3
Figure 3- —National Research Foundation of Korea (NRF)
- —Ministry of Education
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsThyroid and Parathyroid Surgery · Parathyroid Disorders and Treatments · Dermatological and Skeletal Disorders
1. Introduction
Primary brain calcification (PBC) is a brain disease characterized by calcium deposits, called calcifications, in various brain regions. Calcifications primarily affect the basal ganglia, but they may occur in other areas, including the thalamus, subcortical white matter, or dentate nucleus. Disease age of onset may have a wide range, since it can occur in childhood, young adulthood or later adulthood. Clinical symptoms may be similar to other adult-onset neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) or frontotemporal dementia (FTD). Diverse clinical phenotypes were reported in patients with PBC, including motor dysfunctions (such as clumsiness, muscle cramping or unstable gait), difficulties in concentration, memory impairments, personality changes, dementia, psychosis or headache [1,2,3,4,5].
Several genes were identified as causative factors for PBC, and they may act through loss-of-function mechanisms. Both autosomal dominant and recessive forms of PBC were reported. Autosomal dominant PBC was associated with mutations in the SLC20A2, PDGFRB (platelet-derived growth factor receptor-β), PDGFB (platelet-derived growth factor subunit B), or XPR1 (xenotropic and polytropic retrovirus receptor 1) genes. Autosomal recessive PBC was linked to mutations in MYORG (myogenesis regulating glycosidase), JAM2 (junctional-adhesion-molecule-2), CMPK2 (uridine monophosphate-cytidine monophosphate kinase 2) genes, or N-alpha-acetyltransferase 60 (NAA60) [1,2,3,4,5,6]. Furthermore, potential candidates for PBC were also reported, including Cytidine/Uridine Monophosphate Kinase 2 (CMPK2) and ribosomal RNA Processing Protein Coding (RRP12) genes. Both genes were reported to be inherited in autosomal recessive form; however, their impact in PBC should be verified by independent replication studies [7,8].
SLC20A2 gene, located on chromosome 8, encodes a type-III sodium-dependent phosphate transporter, PiT2, involved in phosphate homeostasis in neurons, astrocytes, and endothelial cells. Deficiency of SLC20A2 may result in motor, cognitive, and behavioral dysfunctions, and calcium deposits in the brain [1,2,3,4]. In this study, a frameshift mutation in SLC20A2, Asn384Lysfs*30, was found in a Korean patient with PFC, parkinsonism, memory dysfunctions, insomnia and depression. Reduced SLC20A2 levels were detected in plasma, suggesting loss-of-function mechanisms from the mutation.
2. Materials and Methods
2.1. Patient Information
The 61-year-old female patient visited Seoul National University Bundang Hospital with a ten-year history of subjective memory problems, depressive mood and insomnia. Physical examination during her first visit revealed no motor impairments (e.g., rigidity, tremor, weakness). Brain computed tomography (CT) revealed calcifications in the bilateral subcortical white matter, basal ganglia, thalami, and dentate nuclei (Figure 1A,B). Magnetic resonance imaging (MRI) of the brain indicated severe diffuse periventricular white matter hyperintensities on Fluid-Attenuated Inversion Recovery (FLAIR) sequences (Figure 1C). Positron Emission Tomography with 18F-Fluorodeoxyglucose (F-18 FDG-PET) imaging showed hypometabolism in the bilateral frontal, temporal, and parietal cortices, basal ganglia, and right thalamus (Figure 1D). The total calcification score (TCS) was measured by the TCS tool, based on the full CT scan sequence (https://pbc-tcstool.github.io/ accessed on 2 March 2026). The TCS score was 43, which suggested a mild to moderate degree of calcification (Supplementary Table S1). Laboratory tests showed that serum calcium, phosphate, thyroid hormone, and parathyroid hormone levels were all normal.
Thirteen years later, the patient developed hand and foot tremors, decreased voice volume, and postural instability. The patient was treated with levodopa (100 mg) and benderizine (25 mg) under the suspicion of PD or atypical parkinsonism. No definite clinical improvement was observed. Follow-up brain MRI FLAIR images obtained during outpatient visits showed progression of periventricular white matter hyperintensities, which had become extensive (Figure 1E,F). The patient’s Clinical Dementia Rating score increased from 0.5 at her first visit to 1 at the most recent evaluation, with the sum of boxes score rising from 2.5 to 6 and the Mini-Mental State Examination score declining from 26 to 21. UPDRS part III was not performed, but it will be performed in a follow-up study.
Despite the absence of lacunar infarcts typically associated with cerebral small vessel disease (SVD) and the lack of vascular risk factors, the patient exhibited a progressively worsening clinical course, specifically indicating progression of SVD, accompanied by extensive and atypical calcifications. Consequently, whole-exome sequencing was conducted to investigate the possibility of underlying genetic mutations. There was no relative affected by similar symptoms; however, family members refused to go under genetic testing. This study was approved by the Institutional Review Board of Seoul National University Bundang Hospital (B-2508-988-701).
2.2. Methods
Total genomic DNA was extracted from white blood cells (WBCs) using the Qiagen blood kit (Seoul, Republic of Korea). Whole-exome sequencing was performed by Macrogen using the Illumina platform (https://www.macrogen.com, Seoul, Republic of Korea). The patient was screened for several genetic factors associated with neurodegenerative diseases, including Alzheimer’s disease (AD), frontotemporal dementia, motor diseases (PD, amyotrophic lateral sclerosis), and vascular diseases [9]. Whole-exome sequencing data was uploaded to the Franklin Genoox tool, which was an online platform designed for classifying genetic variants. This tool was used to analyze variants in multiple public and proprietary databases, including ClinVar, dbSNP, gnomAD, and HGMD, and provides comprehensive variant information (https://franklin.genoox.com/clinical-db/home, accessed on 1 March 2024) [6]. Candidate variants were confirmed by Sanger sequencing. Furthermore, functional verification of the SLC20A2 frameshift mutation in plasma was performed in comparison to healthy controls using an ELISA kit (MyBiosource; MBS9338960, San Diego, CA, USA). The blood was tested for amyloid-beta (Aβ) oligomerization tendency using AlzOn (PeopleBio Inc., Sungnam, Republic of Korea).
3. Results
3.1. Genetic Analysis
Franklin Genoox analysis revealed a rare frameshift mutation in the SLC20A2 gene (g. 8:42437358:CTG>C; c.1152_1153delCA, p.Asn384Lysfs30 variant, rs775204334). This mutation was predicted to result in a premature stop codon 30 residues downstream of the frameshift, and it was confirmed by Sanger sequencing (Figure 2A). SLC20A2 Asn384Lysfs30 was absent in the 1000Genomes database, but it was found in the GnomAD database (https://gnomad.broadinstitute.org) in one American and two non-Finnish European individuals; it was not observed in East Asians or South Asians. No additional pathogenic or likely pathogenic variants were found in other PBC-related genes or PBC candidates [1,2,3,4,5,6,7,8]. Furthermore, no pathogenic or likely pathogenic mutations were observed when analyzing the patient using our gene panel for several neurodegenerative diseases, including other neurodegenerative risk factors such as AD, FTD, or PD [9] (Supplementary Table S2).
3.2. Biomarker Studies
The concentrations of SLC20A2 protein in the plasma were lower in the patient compared to healthy controls (Figure 2B, Supplementary Table S3, Supplementary Figure S1). Furthermore, the patient showed an increased tendency for beta-amyloid oligomerization. The AlzOn assay yielded a positive result (value: 1.001), indicating an increased tendency for Aβ oligomerization.
4. Discussion
A frameshift mutation, Asn384Lysfs*30 in the SLC20A2 gene, was discovered in a 61-year-old Korean patient with probable de novo PBC. CT analysis revealed calcifications in different brain areas, including the bilateral subcortical white matter, basal and ganglia. Her initial symptoms were insomnia and depression, followed by foot tremors and postural instability.
The levels of SLC20A2 protein were reduced in the patient compared to age-matched controls, suggesting the presence of a loss-of-function mechanism. Interestingly, AlzOn findings indicated positive results for amyloid oligomers. However, potential AD pathology was not proven because the cerebrospinal fluid (CSF) could not be obtained for measuring biomarkers, including the Ab42/Ab40 ratio, p-Tau, or t-Tau. Furthermore, amyloid PET imaging was not performed. The positive AlzOn results suggested a possible involvement of SLC20A2 in amyloid pathology. It may be possible that SLC20A2 dysfunction indirectly impacted the amyloid metabolism. One explanation could be that abnormal phosphate transport could disrupt calcium metabolism, leading to calcium deposition in the brain, leading to increased amyloid accumulation [10,11]. Frentz et al. (2024) [12] revealed that patients with arterial calcification and amyloid biomarkers presented worse cognition, suggesting that SLC20A2 may impact amyloid metabolism and cognitive decline in proband patients. However, additional investigations would be needed to clarify the relationship between SLC20A2 dysfunctions and amyloid metabolism [12].
The Asn384Lysfs30 mutation was located in the large loop-7 of the SLC20A2 protein, which may impact PiT2 protein transport to the cell surface. This region was suggested to be involved in neuronal overgrowth and PiT2 interactions with microtubule-associated protein 1B (MAP1B). Abnormal SLC20A2 function was reported to reduce phosphate transport into cells and the elevate phosphate levels in the bloodstream, leading to risk of calcium phosphate formation and calcium deposition in the brain [13,14,15,16,17,18,19,20] (Figure 3A,B). Several mutations were reported near Asn384Lysfs30 (Table 1). The Gly366fs mutation was found in a 57-year-old male patient, who developed motor and language impairment. CT revealed calcifications in several brain areas, e.g., the bilateral caudate nucleus, bilateral thalamus, bilateral cerebellum, and bilateral parietal lobes [15]. The Glu368Glyfs46 mutation was identified in a familial case of PBC, where segregation was proven among affected relatives. The proband developed behavioral issues in her 40s, followed by motor and vocal dysfunctions in her 50s and cognitive decline in late disease stages. Calcifications were detected in her basal ganglia, thalamus, occipital cortex and dentate nuclei. The patient’s son was diagnosed with bipolar disorder and excessive eye blinking. His first CT showed calcifications in the basal ganglia and thalamic nuclei, which later spread to cerebellar dentate nuclei [16]. A patient with Ser385Ilefs70 was found in sporadic PBC and presented motor impairments, including involuntary movement of the left limbs and bradykinesia in his 40s. Brain CT revealed calcification in multiple brain areas, including the caudate nuclei, lentiform nuclei, thalami, and cerebellar dentate nuclei [17]. The Tyr386Ter mutation was observed in a 33-year-old patient with migraine and mild functional impairment. CT revealed calcifications in different areas, including the globus pallidus and pulvinar region of the thalamus and dentate nucleus. RT-qPCR revealed a mild decrease (−10%) in SLC20A2 expression [18]. The His399Pro mutation was reported in a 41-year-old female patient with migraine and aura. CT showed calcifications in the bilateral putamen, and mild calcifications were found in the bilateral globus pallidus and dentate nuclei. CT performed 4 years later showed an approximately 30% elevation in calcification volume [19]. Table 1 compared the symptoms of mutations located near the novel SLC20A2 Asn384fs*30 mutation.
In conclusion, a rare variant of Asn384Lysfs30 in SLC20A2 was identified in a Korean patient with PBC. Phenotypes of PBC with SLC20A2 mutations may be similar to other neurodegenerative diseases, including AD, PD, depression or migraine [1,2,3,4,5,20,21]. Plasma analysis revealed reduced SLC20A2 protein levels from the plasma of the patient with SLC20A2 Asn384Lysfs30 mutation, leading to pathogenic pathways through haploinsufficiency. According to ACMG–AMP criteria [5], the SLC20A2 c.1152_1153delCA (p.Asn384Lysfs30) variant was classified as probably pathogenic, supporting a predicted loss-of-function effect, functional protein reduction, rarity in population databases, and a highly specific PBC phenotype. Interestingly, the co-existence of parkinsonism and a high tendency for amyloid oligomerization in the patient could explain the poor response to Levidopa and progressive white matter changes. These results suggested that further research may be needed to investigate the association between amyloid-related pathways and phosphate–calcium dysregulation, as well as the role of SLC20A2 in these pathways. In the future, cell studies with CRISPR-CAS9 should be performed on SLC20A2 Asn384Lysfs30 mutation to verify its loss-of-function mechanisms.
A limitations of this study was that we could not obtain enough WBCs from the patient for qPCR analysis. We also could not get obtain to confirm AD pathology from brain pathology, even though the amyloid oligomers were positive. Furthermore, all living family members refused the genetic test, hence the segregation analysis could not be carried out. Animal models were performed to examine the effects of antisense oligonucleotides (ASOs) in the case of SLC20A2 haploinsufficiency. These mouse models suggested that ASO administration resulted in elevated inorganic phosphate levels in CSF of mice, resulting in reduced levels of brain calcification. In the case of patients with SLC20A2 haploinsufficiency, treatment in ASO may be promising, since it could slow down the disease progression [21,22,23,24]. Furthermore, astrocyte-mediated expression of SLC20A2 may also improve inorganic phosphate homeostasis inside the brains of SLC20A2 knock-in mice, leading to a reduced degree of brain calcification [25].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Xu X. Sun H. Luo J. Cheng X. Lv W. Luo W. Chen W.J. Xiong Z.Q. Liu J.Y. The Pathology of Primary Familial Brain Calcification: Implications for Treatment Neurosci. Bull.20233965967410.1007/s 12264-022-00980-036469195 PMC 10073384 · doi ↗ · pubmed ↗
- 2Donzuso G. Mostile G. Nicoletti A. Zappia M. Basal ganglia calcifications (Fahr’s syndrome): Related conditions and clinical features Neurol. Sci.2019402251226310.1007/s 10072-019-03998-x 31267306 PMC 6817747 · doi ↗ · pubmed ↗
- 3Quintáns B. Oliveira J. Sobrido M.J. Primary familial brain calcifications Handb. Clin. Neurol.20181473073172932562010.1016/B 978-0-444-63233-3.00020-8 · doi ↗ · pubmed ↗
- 4Guo X.X. Zou X.H. Wang C. Yao X.P. Su H.Z. Lai L.L. Chen H.T. Lai J.H. Liu Y.B. Chen D.P. Spectrum of SLC 20A 2, PDGFRB, PDGFB, and XPR 1 mutations in a large cohort of patients with primary familial brain calcification Hum. Mutat.20194039240310.1002/humu.2370330609140 · doi ↗ · pubmed ↗
- 5Luo W. Cen Z. Koek H. Carecchio M. Hozumi I. Chen W.J. Batla A. Balck A. Magrinelli F. Yang D. Primary Brain Calcification: An International Consensus on Nomenclature, Diagnosis, Evaluation, and Management Mov. Disord.20254131533610.1002/mds.7014041346103 · doi ↗ · pubmed ↗
- 6Chelban V. Aksnes H. Maroofian R. La Monica L.C. Seabra L. Siggervåg A. Devic P. Shamseldin H.E. Vandrovcova J. Murphy D. 6 Biallelic NAA 60 variants with impaired n-terminal acetylation capacity cause autosomal recessive primary familial brain calcifications Nat. Commun.202415226910.1038/s 41467-024-46354-038480682 PMC 10937998 · doi ↗ · pubmed ↗
- 7Zhao M. Su H.Z. Zeng Y.H. Sun Y. Guo X.X. Li Y.L. Wang C. Zhao Z.Y. Huang X.J. Lin K.J. Loss of function of CMPK 2 causes mitochondria deficiency and brain calcification Cell Discov.2022812810.1038/s 41421-022-00475-236443312 PMC 9705363 · doi ↗ · pubmed ↗
- 8Monfrini E. Rinchetti P. Anheim M. Klingseisen A. Lagha-Boukbiza O. Cen Z. Yang D. Chen X. Maroofian R. Houlden H. RRP 12 Variants Are Associated With Autosomal Recessive Brain Calcifications Mov. Disord.2025402792280310.1002/mds.7005841059649 PMC 13001700 · doi ↗ · pubmed ↗