Mitochondrial Homeostasis in Diabetic Cardiomyopathy: From Dysfunction to Therapeutic Strategies
Yafei Huang, Wenyu Zou, Xindi Jiang, Jing Cheng, Jia Zheng

TL;DR
This paper reviews how mitochondrial dysfunction contributes to heart disease in diabetes and suggests new ways to manage it.
Contribution
The paper provides a novel perspective on diabetic cardiomyopathy by focusing on mitochondrial homeostasis and its signaling pathways.
Findings
Mitochondrial homeostasis is disrupted in diabetic cardiomyopathy, leading to heart dysfunction.
Oxidative stress, inflammation, and apoptosis are key factors in the disease's progression.
Understanding mitochondrial dynamics and mitophagy could lead to new therapeutic strategies.
Abstract
Diabetic cardiomyopathy is a specific form of heart dysfunction that occurs in diabetic patients independent of other cardiomyopathies such as coronary artery disease. It significantly contributes to heart failure and mortality in this population. The pathogenesis of diabetic cardiomyopathy mainly includes oxidative stress, inflammatory response, apoptosis and disrupted mitochondrial homeostasis. Mitochondrial homeostasis, encompassing mitochondrial dynamics, mitochondrial oxidative metabolism and mitophagy, is regulated by a variety of signaling pathways and plays a pivotal role in maintaining the normal function of cardiomyocytes. At present, the exact mechanisms underlying diabetic cardiomyopathy pathogenesis remain unclear, and effective prevention and treatment methods are lacking. This review therefore expounds the pathogenesis of diabetic cardiomyopathy from the perspective of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
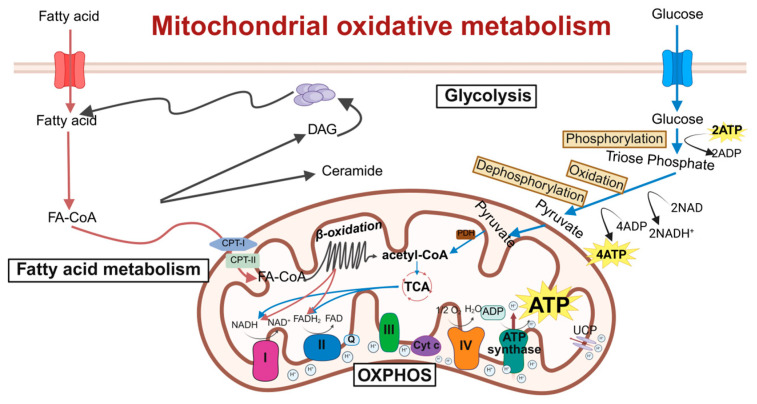 Figure 1
Figure 1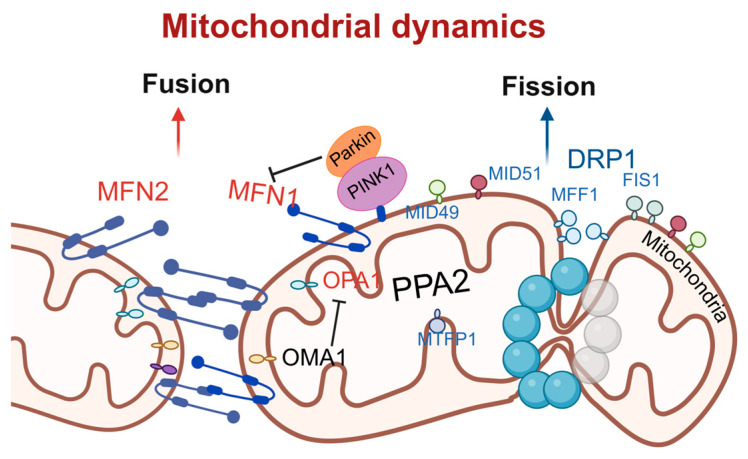 Figure 2
Figure 2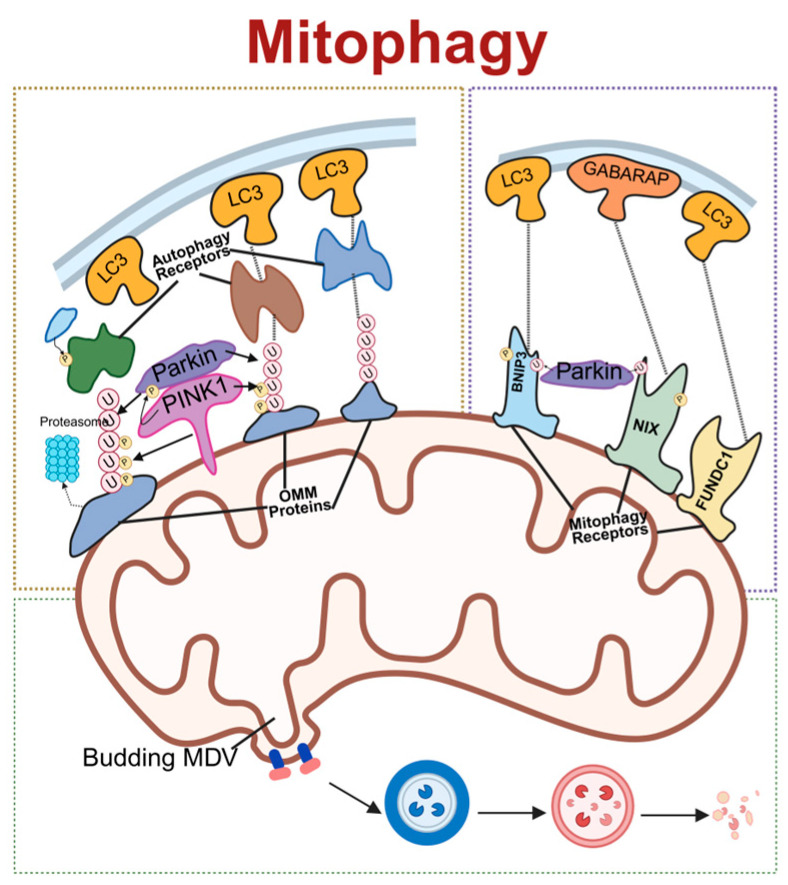 Figure 3
Figure 3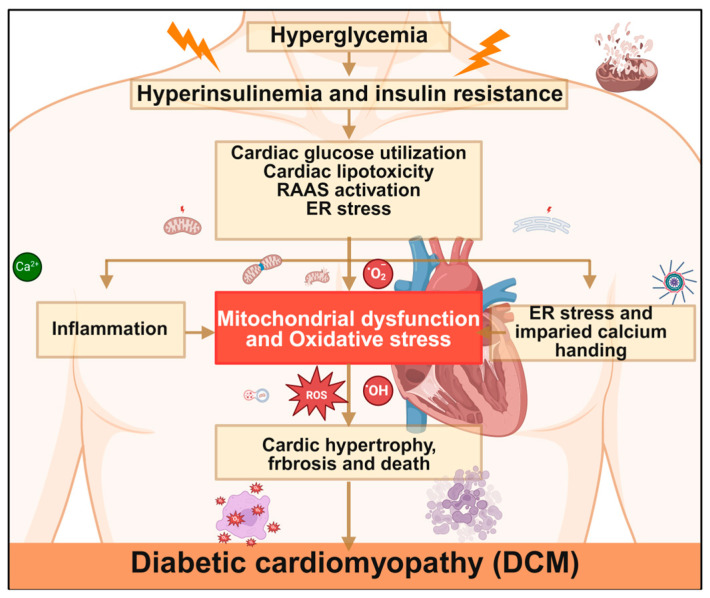 Figure 4
Figure 4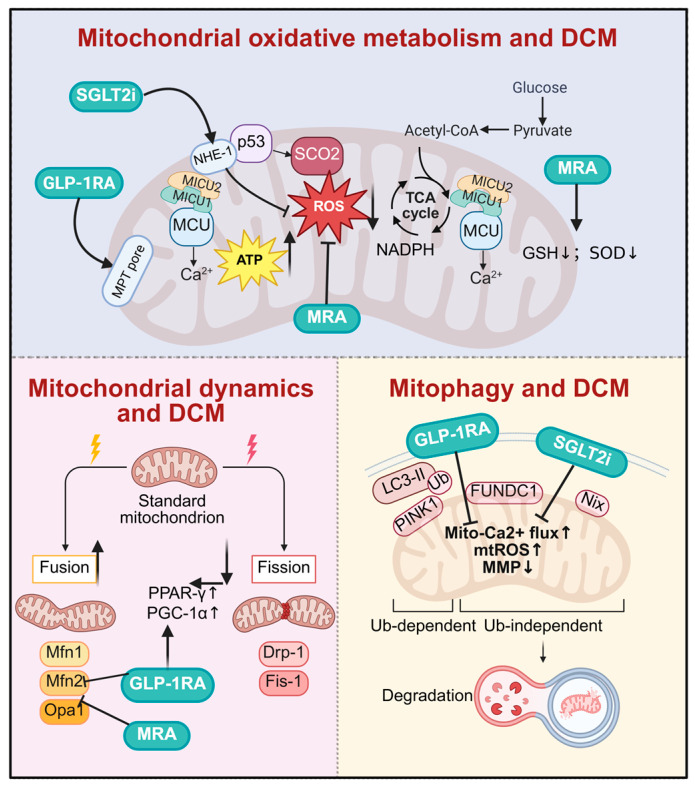 Figure 5
Figure 5- —Beijing Municipal Natural Science Foundation
- —National Science and Technology Major Project
- —National Natural Science Foundation of China
- —Beijing Physician-Scientist Training Program
- —National High Level Hospital Clinical Research Funding
- —Chinese Cardiovascular Association-Natural lipid-lowering drugs fund
- —China Endocrine and Metabolism Young Scientific Talent Research Project
- —China Diabetes Young Scientific Talent Research Project
- —Bethune-Merck Diabetes Research Fund of Bethune Charitable Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiovascular Function and Risk Factors · Mitochondrial Function and Pathology · GDF15 and Related Biomarkers
1. Introduction
The prevalence of type 2 diabetes mellitus (T2DM) has been rising steeply and persistently, with the disease now recognized as a global epidemic [1,2,3,4]. In 2021, 529 million individuals had diabetes globally, with projections indicating the number of patients will reach 1.31 billion by 2050 [5,6]. Uncontrolled diabetes leads to a variety of long-term health complications, including cardiovascular disease (CVD), neuropathy, vision problems, and amputation [3,7]. Among these complications, CVD is the leading cause of mortality and morbidity in diabetic patients, accounting for nearly 70% of heart failure cases [8,9,10]. Compared with non-diabetic individuals, diabetic patients face a 2- to 8-fold higher risk of CVD events, primarily due to microvascular and macrovascular atherosclerosis exacerbated by concomitant CVD risk factors including hypertension, dyslipidemia, and the activation of neurohormonal–inflammatory mechanisms [6,8,11].
Diabetic cardiomyopathy (DCM) denotes cardiac dysfunction—initially characterized by diastolic dysfunction with preserved ejection fraction, with potential progression to systolic dysfunction with reduced ejection fraction—observed in patients with diabetes mellitus in the absence of concomitant cardiovascular diseases, including but not limited to coronary artery disease, hypertension, valvular abnormalities, and congenital heart disease [12,13]. Its high prevalence, diagnostic challenges, and poor prognosis profoundly impact patients’ quality of life while being a heavy burden on families and society, driving extensive research into the treatment and prevention of DCM [14,15].
The current understanding of DCM pathogenesis mainly implicates the occurrence of inflammatory responses, changes in calcium signaling, and renin–angiotensin–aldosterone system hyperactivity [16,17,18,19,20]. Recent studies have demonstrated that mitochondrial dysfunction contributes to DCM pathology by disrupting myocardial metabolic and non-metabolic signals, ultimately inducing myocardial dysfunction [21]. This article explores the role of mitochondrial homeostasis in DCM occurrence and progression, reviews current research advances, summarizes its mechanistic involvement in DCM, and establishes a theoretical framework for the clinical treatment of DCM.
2. Mitochondrial Homeostasis
Mitochondria, acting as the “powerhouses” of the cell, primarily supply energy for various cellular activities and serve as the central and principal site for cellular metabolism and oxidative respiration. Abnormalities in mitochondrial morphology and function are closely associated with the development of numerous diseases. Mitochondrial homeostasis represents a dynamic regulatory process involving mitochondrial dynamics (fusion and fission), mitochondrial oxidative metabolism, and mitophagy. These coordinated mechanisms maintain both the quantity and structural integrity of “healthy” mitochondria to meet the various energy demands of the cell [22]. Disruption in mitochondrial homeostasis constitutes a key pathological factor in the progression of many diseases. Therefore, maintaining mitochondrial homeostasis is crucial for the normal growth and development of cells and organisms [23].
2.1. Mitochondrial Oxidative Metabolism
Mitochondrial oxidative metabolism is a critical process by which cells generate energy in the form of adenosine triphosphate (ATP) through the oxidation of nutrients, which primarily takes place within the mitochondria [24]. Pyruvate, derived from glycolysis, enters the mitochondria and is converted into acetyl-CoA by the pyruvate dehydrogenase complex. Acetyl-CoA then enters the tricarboxylic acid (TCA) cycle, also known as the Krebs cycle, where it is oxidized to produce high-energy electron carriers including nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH_2_), while releasing carbon dioxide as a waste product [25]. These high-energy electron carriers donate their electrons to the electron transport chain (ETC), which is embedded in the inner mitochondrial membrane. The ETC consists of four protein complexes (Complex I to IV) that transfer electrons sequentially, generating a proton gradient across the inner mitochondrial membrane [26]. As protons flow back into the mitochondrial matrix through ATP synthase (Complex V), the resulting energy drives the synthesis of ATP from adenosine diphosphate (ADP) and inorganic phosphate, thereby coupling the electron transport chain with ATP production.
However, during oxidative metabolism, a small percentage of electrons may leak from the ETC and react with oxygen, forming reactive oxygen species (ROS) such as superoxide radicals. While physiological levels of ROS function as signaling molecules, excessive ROS production induces oxidative stress and damages cellular components, including proteins, lipids, and DNA [26,27].
Mitochondrial oxidative metabolism is precisely regulated by multiple factors, such as substrate availability, hormonal signals, and cellular energy demands. For instance, during fasting or exercise, the expression of oxidative metabolism-related genes increases to meet elevated energy requirements. The dysregulation of mitochondrial oxidative metabolism is linked to numerous diseases, including metabolic disorders, neurodegenerative diseases, cardiovascular diseases, and aging-related conditions [28,29]. Therefore, understanding the mechanisms and regulation of mitochondrial oxidative metabolism is essential for developing therapeutic interventions to restore cellular energy balance and mitigate oxidative stress (Figure 1).
2.2. Mitochondrial Dynamics
Mitochondria are highly dynamic organelles that undergo continuous cycles of fusion and fission to modulate their morphology, size, and location. This physiological process is known as mitochondrial dynamics [30]. The relative balance between mitochondrial fission and fusion is crucial for maintaining mitochondrial quality and function, which is of great significance to normal cellular activities [31,32].
Mitochondrial fission is primarily regulated by dynamin-related protein 1 (DRP1) [33]. DRP1, a member of the dynamin superfamily, is typically localized in the cytoplasm and comprises a GTP hydrolysis (GTPase) domain, a middle domain, and a GTPase effector domain. Upon stimulation by fission factors, DRP1 is recruited to the mitochondria, where it oligomerizes to form ring-like structures. These structures, utilizing the GTPase activity of DRP1, induce the scission of the mitochondrial outer and inner membranes, resulting in mitochondrial fission [34]. The receptors for DRP1 include mitochondrial fission factor (MFF), fission1 (FIS1), mitochondrial dynamics proteins of 49 kDa (MiD49), and mitochondrial dynamics proteins of 51 kDa (MiD51), which mediate both the recruitment of DRP1 and the process of mitochondrial fission [35]. Among these, MFF is a predominant receptor for DRP1 in mammalian cells. The overexpression of MFF promotes mitochondrial fission, whereas its knockdown leads to mitochondrial elongation [36]. Inorganic pyrophosphatase phosphatase 2A (PPA2), a protein localized to the mitochondrial matrix, activates the downstream fission signaling pathway by directly interacting with the mitochondrial inner-membrane protein mitochondrial fission process 1 (MTFP1). Specifically, the overexpression of PPA2 significantly promotes the phosphorylation of DRP1 at serine 616 and enhances the recruitment of phosphorylated DRP1 to mitochondria, thereby initiating the fission process. When MTFP1 is knocked down, even the overexpression of PPA2 cannot induce the activation of DRP1 and subsequent fission, confirming the necessity of MTFP1 in this pathway [37]. It has also been reported that MTFP1 itself is not a pro-fission factor; instead, it functions by inhibiting fusion, thereby “isolating” damaged or dysfunctional inner-mitochondrial-membrane (IMM) subdomains from the healthy mitochondrial network. Subsequently, these isolated subdomains are separated into small MTFP1-enriched mitochondria (SMEM) via peripheral fission and are ultimately degraded through the autophagy pathway [38].
Mitochondrial fusion is a multistep process that includes mitochondrial tethering, outer-membrane fusion, and inner-membrane fusion [39]. The fusion process is primarily regulated by mitochondrial fusion proteins, including mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy 1 protein (OPA1) [40]. MFN1 and MFN2 mainly mediate the fusion of the mitochondrial outer membrane by forming homodimers or heterodimers through cis-dimerization, thereby facilitating the tethering and fusion of the outer membranes of adjacent mitochondria [41,42]. OPA1 is primarily involved in the fusion of the mitochondrial inner membrane. Its gene deletion leads to mitochondrial fragmentation, while overexpression results in mitochondrial elongation [43]. OPA1 induces inner-membrane fusion in a manner dependent on MFN1 but not MFN2, indicating potential communication and interaction between the outer and inner mitochondrial membranes [43]. However, the exact mechanism by which OPA1 induces inner-membrane fusion remain incompletely understood and requires further investigation. Recent studies have revealed a synergistic role of two classic mitochondrial stress response systems, the PTEN-induced kinase 1 (PINK1)/E3 ubiquitin–protein ligase parkin (Parkin) pathway and the metalloprotease overlapping with the M-AAA protease 1 homolog (OMA1), in cooperatively inhibiting excessive mitochondrial fusion under physiological conditions, thereby maintaining mitochondrial structural and genomic integrity. The PINK1/Parkin pathway primarily targets the outer-membrane fusion protein MFN1, promoting its ubiquitination and degradation to inhibit outer-membrane fusion. OMA1 primarily targets the inner-membrane fusion protein OPA1, inactivating it through proteolytic cleavage to inhibit inner-membrane fusion. The individual deletion of either Parkin or OMA1 does not affect mitochondrial integrity, but the combined deletion of both leads to excessive mitochondrial fusion, forming megamitochondria, which are particularly prominent in the brainstem and heart [44] (Figure 2).
2.3. Mitophagy
Mitophagy is a selective process by which cells remove damaged or dysfunctional mitochondria through autophagy, playing a crucial role in maintaining cellular homeostasis and mitochondrial quality control. The main pathways of mitophagy include ubiquitin-mediated mitophagy, receptor-mediated mitophagy and mitochondrial-derived vesicles (MDVs) pathways.
Receptor-mediated mitophagy is initiated by the direct interaction of the outer-mitochondrial-membrane (OMM) proteins with microtubule-associated protein 1 light chain 3 (LC3)/gamma–aminobutyric acid receptor-associated protein (GABARAP) proteins on the autophagosome membrane through the LC3 interaction region (LIR) motif [45]. PINK1 and Parkin, two proteins associated with Parkinson’s disease, have recently been identified as regulators of mitophagy [46]. PINK1 functions as an upstream regulator of Parkin, collaborating with it to promote the autophagic degradation of damaged mitochondria via the polyubiquitination of mitochondrial surface proteins. Following mitochondrial depolarization or damage, PINK1 accumulates extensively on OMM, triggering the translocation of Parkin from the cytosol to the OMM [47]. Parkin, leveraging its E3 ligase activity, conjugates ubiquitin to substrate proteins, forming polyubiquitin chains [46,48]. Subsequently, adaptor proteins such as p62/sequestosome-1 are recruited to the mitochondria through their ubiquitin-binding domains and interact with LC3, facilitating the aggregation of ubiquitinated proteins into autophagosomes for degradation [48]. Moreover, the PINK1/Parkin pathway is involved in an unconventional autophagic process, where specific proteins are transported to lysosomes for degradation via MDVs [49].
Receptor-mediated mitophagy is initiated by the direct interaction of mitochondrial OMM proteins with LC3/GABARAP proteins on the autophagosome membrane through the LIR motif [45]. NIP3-like protein X (NIX) and BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), as members of the B cell lymphoma 2 (Bcl-2) family, are OMM-localized proteins that mediate mitophagy through their N-terminal WXXL-like sequences that bind to LC3. NIX is involved in mitochondrial clearance during reticulocyte maturation, whereas BNIP3 participates in hypoxia-induced mitophagy and plays a key role in the induction and maintenance of pluripotency [50,51]. Under hypoxic conditions, hypoxia-inducible factor-1 upregulates the expression of NIX and BNIP3, disrupting the interaction between Bcl-2 and Beclin-1 and activating mitophagy [52]. FUN14 domain containing 1 (FUNDC1), another receptor protein for hypoxia-induced mitophagy, also interacts with LC3 via its WXXL-like sequence, with its phosphorylation status precisely controlling mitophagy initiation [53]. When mitophagy is induced, FUNDC1 dissociates from OPA1 and binds to DRP1, subsequently leading to mitochondrial fission and mitophagy. It has been reported that FUNDC1 plays a role in cardiac progenitor cell differentiation by remodeling the mitochondrial network and exerts cardioprotective effects during ischemia–reperfusion injury [54]. Collectively, these proteins orchestrate the removal of mitochondria damaged by hypoxia, thereby maintaining oxygen homeostasis and mitigating ROS accumulation [55].
MDV formation is a key pathway in mitochondrial quality control. MDV biogenesis complements mitophagy and serves to recycle damaged mitochondria [56]. When the primary mitophagy mechanism is impaired due to aging or other conditions, mitochondrial Rho GTPase1/2 proteins located on the mitochondrial outer membrane sense signals and recruit the dynamin-related protein DRP1, driving local constriction and budding of the mitochondrial membrane to initiate MDV formation [57]. Meanwhile, a decrease in damaged mitochondrial membrane potential can activate the PINK1/Parkin pathway: PINK1 stabilizes on the outer mitochondrial membrane, and recruits and activates the E3 ubiquitin ligase Parkin, which then ubiquitinates outer membrane proteins to tag damaged components [58]. The adaptor protein Tollip recognizes these ubiquitination signals, assists in recruiting autophagy-related proteins, and sorts specific cargo (such as oxidatively damaged proteins, lipids, and mitochondrial DNA) into the forming MDVs [54,59]. Finally, mature MDVs fuse with late endosomes/lysosomes via sensitive factor attachment protein receptors proteins such as syntaxin-17 on their membrane, delivering their contents to the lysosome for degradation [60]. This process is independent of canonical macroautophagy/mitophagy and represents an important supplementary mechanism for cells to clear oxidatively damaged mitochondrial components and maintain mitochondrial network integrity (Figure 3).
3. Mitochondrial Homeostasis and DCM
In the pathogenesis and progression of DCM, mitochondrial dysfunction constitutes a core pathological mechanism involving multiple levels such as impaired oxidative metabolism, kinetic abnormalities, and disrupted autophagy (Figure 4 and Table 1). The following provides a systematic elaboration on key molecular events.
3.1. Mitochondrial Oxidative Metabolism and DCM
Mitochondrial oxidative metabolism refers to the central mitochondrial pathway where sugars, fats and amino acids undergo oxidative phosphorylation through the tricarboxylic acid cycle, ultimately generating ATP through redox reaction to meet energy demands. Ji et al. [62] demonstrated that mitochondrial calcium uptake 1 (MICU1) serves as a key regulator of mitochondrial Ca^2+^ uptake and critically modulates mitochondrial oxidative phosphorylation and redox homeostasis by elevating mitochondrial Ca^2+^. This mechanism activates the antioxidant system, thereby inhibiting the apoptosis of cardiomyocytes induced by high sugar and high lipids, and exerts a protective effect on myocardium.
As highly energy-dependent cells, cardiomyocytes require continuous oxidative metabolism to generate ATP necessary for maintaining cardiac output and contractile function [77]. Emerging evidence suggests that Ca^2+^ homeostasis is closely related to mitochondrial energy metabolism, functioning as an essential second messenger to maintain proper cardiac function [78,79]. Notably, abnormal Ca^2+^ signaling can be observed in type 2 diabetic DCM, with Ca^2+^ mishandling being the main reason for an interference in energy production. Studies reveal that significantly elevated pyruvate dehydrogenase phosphorylation in diabetic cardiomyocytes alters the mitochondrial energy metabolism and counteracts mitochondria-associated membrane Ca^2+^ signaling. Its correction could restore cardiac function and prevent DCM progression.
Mitochondrial oxidative metabolism dysfunction is accompanied by a large amount of ROS production, mitochondrial peroxidative damage, and myocardial injury due to energy insufficiency, constituting a key pathological mechanism in DCM. Li et al. [80] reported that T2DM exacerbates transient receptor potential vanilloid 1 (TRPV1) blockade and ROS overload, leading to cardiac microvascular injury. Notably, inhibiting TRPV1/Ca^2+^-mediated oxidative/nitrifying stress response effectively protected cardiac microvessels from diabetic damage. Excess ROS can, in turn, activate the p53/synthesis of cytochrome c oxidase 2 (SCO2) signaling, increasing mitochondrial oxygen consumption, and perpetuating ROS production and lipid accumulation, thereby driving diabetic myocardial injury [81].
Berthiaume et al. [82] identified that diabetic cardiomyocytes display “metabolic rigidity” with pathologically enhanced fatty acid uptake and mitochondrial oxidation, despite the fact that the healthy adult heart already relies primarily on fatty acids for energy (following the normal developmental switch from glucose that occurs after birth). This rigidity is characterized by impaired mitochondrial electron transport chain function, insulin resistance-mediated suppression of glucose transporters, and, consequently, markedly reduced glucose utilization. The transformation of energy metabolism changes the transcriptional and redox status of NAD and metabolite signaling of key enzymes, resulting in a decrease in glycolysis and mitochondrial oxidative metabolism that can cause cardiac energy deficiency and cardiac insufficiency [83]. Furthermore, A-kinase-anchored protein 121 increases cardiomyocyte apoptosis by enhancing ROS production, and deficiency of this protein impairs mitochondrial respiration, reduces ATP production, and induces mitochondrial oxidative metabolism dysfunction, along with cardiomyocyte apoptosis [84]. Studies have demonstrated that reduced NADPH oxidase 5, a Ca^2+^-sensitive, pro-contractile NADPH oxidase isoform, can be mediated to participate in oxidative stress regulation, resulting in an increase in ROS levels. Activation of the mitogen-activated protein kinase pathway leads to cardiac hypertrophy and systolic dysfunction [85]. Collectively, Ca^2+^ affects myocardial oxygen consumption by regulating mitochondrial energy metabolism in cardiomyocytes, induces oxidative stress, and changes cardiac structure and function. In summary, mitochondrial oxidative metabolic dysfunction plays a pivotal role in DCM. The dysregulation of mitochondrial oxidative metabolism leads to excessive production of ROS and peroxidative damage to mitochondria, thereby causing myocardial energy deficiency and myocardial injury. Moreover, the disruption of Ca^2+^ homeostasis further interferes with mitochondrial energy metabolism, exacerbating myocardial dysfunction and ultimately leading to alterations in cardiac structure and function.
Importantly, the aged myocardium provides a vulnerable substrate for diabetic metabolic insults [86]. Cardiac aging is characterized by mitochondrial metabolic inflexibility, including a diminished capacity for fatty acid oxidation and compromised glucose utilization, which predisposes the heart to energetic deficiency [87]. Concurrently, aged mitochondria exhibit increased electron leak and a decline in antioxidant defense systems, creating a state of basal oxidative stress [88]. When diabetic conditions—such as glucotoxicity, lipotoxicity, and insulin resistance—are superimposed on this aged milieu, they act synergistically to exacerbate mitochondrial ROS production and overwhelm cellular antioxidant capacity [89]. This age-aggravated metabolic dysregulation and oxidative stress not only accelerate ATP depletion but also inflict more severe damage on proteins, lipids, and mtDNA, thereby propelling the progression of diastolic dysfunction and myocardial fibrosis characteristic of advanced DCM [90,91].
3.2. Mitochondrial Dynamics and DCM
Mitochondrial dynamics refers to the process that shape the mitochondrial network, regulate mitochondrial function, and maintain quality control. This dynamic process integrates mitochondrial fusion and fission with diverse cellular functions while responding to cellular pathophysiological changes. Strategically balancing these two processes demonstrates therapeutic potential for DCM improvement [92].
Previous studies have shown that increased fission and decreased fusion can aggravate mitochondrial fragmentation, in which fission-mediated mitochondrial fragmentation directly contributes to high-glucose-induced ROS overproduction, whereas fusion counteracts this damage by facilitating complementation between damaged mitochondria [93]. Dai and Jiang [94] found that mitochondrial fusion is controlled by three guanosine triphosphate (GTP) enzymes, and fission is mainly regulated by GTPase kinetics-related protein 1. The decrease in GTPase activity will inhibit mitochondrial dynamics and cellular metabolism, predisposing to metabolic diseases such as DCM.
In diabetic conditions, increased mitochondrial fission can lead to myocardial damage and systolic dysfunction. Hu et al. [69] found that diabetic myocardium exhibits excessive mitochondrial fission, which can be suppressed by mitochondrial fusion, thereby promoting the recovery of mitochondrial membrane potential and oxidative stress, and arresting DCM progression. Similarly, Ding et al. [95] found that diabetic myocardial systolic dysfunction is associated with increased mitochondrial fission, and that melatonin regulates the protein 1/peroxisome proliferator-activated receptor γ coactivator 1-alpha (PGC-1α) pathway by silencing, reduces the expression of GTPase kinetics-related protein 1, prevents mitochondrial fission, and alleviates diabetes-induced cardiac dysfunction. Li et al. [96] reported that Ophiopogonin D alleviates mitochondrial damage and dysfunction by inhibiting mitochondrial fission in cardiomyocytes, improving cell survival, and preventing and treating diabetic myocardial injury. In summary, the targeted modulation of mitochondrial dynamics reduces pathological ROS release, alleviates myocardial oxidative stress, inflammatory response and fibrosis, and ultimately improves myocardial injury and dysfunction in diabetes.
The regulation of mitochondrial dynamics is further compromised by the aging process itself [88]. In the senescent heart, a baseline shift towards excessive fission (elevated DRP1 activity) and impaired fusion (reduced MFN2 expression) is commonly observed, leading to a progressive fragmentation of the mitochondrial network even in the absence of diabetes [97,98]. The diabetic milieu, with its associated oxidative stress and activation of kinases like cyclin-dependent kinases (CDK1)/Cyclin B, further amplifies DRP1-mediated fission and impairs OPA1-mediated inner membrane fusion [89,99]. Thus, aging and diabetes converge on the same molecular machinery, creating a feed-forward cycle that drives the mitochondrial network toward a highly fragmented, dysfunctional state.
3.3. Mitophagy and DCM
Mitophagy refers to the selective removal of damaged and dysfunctional mitochondria, which helps to improve energy metabolism while regulating the quantity of mitochondria so as to maintain the basic quality of cardiac mitochondria. Mitophagy is affected by misfolded protein accumulation, mitochondrial dysfunction, and the deletion of gene fragments [100]. There is a large body of evidence that mitophagy can improve cardiac function in diabetic patients, and the loss of mitophagy is strongly correlated to DCM development [101].
Mu et al. [76] found that the upregulation of bromodomain-containing protein 4 in the heart of diabetic mice suppresses PINK1 expression, inhibiting PINK1/Parkin-mediated mitophagy and causing damaged mitochondria accumulation, resulting in impaired cardiac structure and function that aggravate cardiomyopathy. It also reduces the therapeutic effect of bromodomain-containing protein 4 inhibitors, which restores mitophagy by inhibiting the upregulation of bromodomain-containing protein 4 and repairs cardiac structure and function in diabetic hearts. Metformin alleviates low-grade inflammatory responses through blocking mitochondrial complex 1, inhibiting inflammasome activation, increasing autophagy and improving mitochondrial bioenergy metabolism. It also inhibits the mammalian target protein pathway of rapamycin, alleviates pyroptosis of DCM, and exerts cardioprotective effects [102,103].
Tong et al. [75] revealed that impaired mitophagy induces mitochondrial dysfunction and lipid deposition to aggravate DCM, while mitophagy activation prevents high-fat-diet-induced DCM. The inhibition of mitophagy leads to the excessive accumulation of malformed and inefficient mitochondria, disrupting mitochondrial quality control and redox balance, and ultimately compromising myocardial function [104]. Tahrir et al. [105] further established that mitophagy isolates unhealthy mitochondria and maintains cardiac homeostasis by selectively eliminating damaged or dysfunctional mitochondrial proteins. Meanwhile, Chen et al. [106] identified Ca^2+^ as a key modulator of DCM pathogenesis through directly or indirectly involved in the regulatory mechanisms. Collectively, mitophagy is involved in various physiological and pathological processes such as cardiomyocyte metabolic activity, cell differentiation, and apoptosis, playing an important role in the regulation of cardiovascular diseases [107].
The efficiency of mitophagy, a critical line of defense, is susceptible to the dual assault of both aging and diabetes [108,109]. Aging is associated with a global decline in autophagic flux, attributed to reduced lysosomal function and impaired autophagosome formation. This results in an accumulation of dysfunctional, ROS-producing mitochondria at baseline [86]. Diabetic stress, through mechanisms such as the oxidative modification of Parkin or inhibition of the PINK1/Parkin pathway, directly suppresses the activation of targeted mitophagy. The synergistically impaired mitophagy allows for the unchecked proliferation of damaged mitochondria, leading to overwhelming oxidative stress, activation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, and the release of pro-apoptotic factors [108]. This axis represents a pivotal convergence point where aging exacerbates diabetic injury, and restoring mitophagy may hold particular therapeutic promise for the elderly DCM population [109].
In summary, mitochondrial damage exerts regulatory effects in multiple links of DCM. For example, it leads to abnormal mitochondrial energy metabolism, increased ROS production, mitochondrial fission and fusion disorders, cardiolipin changes, and calcium disorders. These collectively contribute to cardiac dysfunction, coronary microvascular disease, and cardiac structural changes, thereby accelerating the progression of DCM. Conversely, DCM perpetuate mitochondrial functional damage, creating a vicious cycle where these processes mutually reinforce disease progression.
4. Pharmacological Treatment
DCM is a complication of diabetes mellitus that develops independently of coronary artery disease and hypertension. It is characterized by alterations in myocardial structure and function, progressing inevitably to heart failure. The underlying pathogenesis of DCM is complex, with no specific therapies currently available. Pharmacological treatment focuses on glycemic control, improvement in cardiac function, prevention of complications, and retardation of disease progression (Figure 5 and Table 2).
4.1. Sodium–Glucose Co-Transporter 2 (SGLT2) Inhibitors
SGLT2 inhibitors are a new class of glucose-lowering drugs primarily acting by inhibiting renal glucose reabsorption, thereby promoting urinary glucose excretion and lowering blood sugar [122,123,124,125,126]. Empagliflozin exerts cardioprotective effects in DCM by enhancing mitochondrial ketone body metabolism (upregulating 3-hydroxybutyrate dehydrogenase 1(BDH1)/3-Oxoacid CoA-Transferase 1 (OXCT1)) and attenuating oxidative stress via nuclear factor erythroid 2-related factor 2 (NRF2) activation, thereby reducing ROS production and apoptosis in db/db mouse hearts [110,127,128,129]. Additionally, dapagliflozin exerts anti-cardiac fibrosis effects independent of its hypoglycemic action by inhibiting the endothelial-to-mesenchymal transition (EndMT) to block fibroblast recruitment and directly suppressing cardiac fibroblast proliferation, activation, and collagen synthesis. Its cardioprotective efficacy is comparable to that of metformin [111,130,131]. Canagliflozin improves DCM by activating PINK1/Parkin-dependent mitochondrial autophagy. This study systematically elucidates its protective mechanism from the cellular to animal levels and validates key targets using PINK1 knockdown, providing new insights into the cardioprotective effects of SGLT2 inhibitors. However, its dose-dependent nature and long-term efficacy require further clinical validation [112,132].
SGLT2 inhibitors exert direct cardioprotective effects through a variety of mechanisms, including improved mitochondrial function, anti-inflammatory actions, anti-fibrotic effects, and the mitigation of oxidative stress [133,134,135]. Among these, the impact of SGLT2 inhibitors on mitochondrial function is particularly noteworthy [136]. Pioglitazone exerts its cardioprotective effects through the peroxisome proliferator-activated receptor γ (PPAR-γ)/PGC-1α signaling axis. The knockdown of PGC-1α completely abolished its ability to upregulate Mn Superoxide dismutase (Mn-SOD) and stabilize mitochondrial membrane potential (MMP), confirming that PGC-1α is a key and essential molecular target mediating pioglitazone’s improvement in mitochondrial oxidative stress and prevention of diabetic atrial remodeling [113,137]. Chen et al. (2024) [138] demonstrated that empagliflozin (Empa) ameliorates cardiac dysfunction through inhibiting sodium–hydrogen exchanger 1 (NHE-1), thereby reducing intracellular sodium and calcium overload, decreasing mitochondrial ROS production, and preserving nitric oxide (NO) bioavailability. These effects ultimately improve cardiac contractile function and vascular endothelial metabolic homeostasis, protecting cardiomyocytes from oxidative stress and energy metabolic imbalance, with the underlying mechanism involving the NHE-1/protein kinase B (AKT)/endothelial nitric oxide synthase (eNOS) signaling pathway.
In recent years, SGLT2 inhibitors have shown potential in the treatment of DCM. The results of the EMPA-REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) trial showed that patients with T2DM at high risk for cardiovascular events who received empagliflozin had a lower rate of the primary composite cardiovascular outcome and a lower rate of death from any cause when the study drug was added to standard care [139,140]. In EMPA-HEART CardioLink-6 clinical trial, among people with T2DM and coronary artery disease, SGLT2 inhibition with empagliflozin was associated with a significant reduction in left ventricular mass indexed to body surface area after 6 months [141]. Clinical trials have unequivocally demonstrated benefits in heart failure across the ejection fraction spectrum. Empagliflozin reduced the composite risk of cardiovascular death or hospitalization for heart failure (HF) in patients with heart failure and preserved ejection fraction (HFpEF) in the EMPEROR-Preserved trial (Empagliflozin Outcome Trial in Patients with Chronic Heart Failure with Preserved Ejection Fraction) [142]. Similarly, dapagliflozin improved outcomes in patients with HFpEF and mildly reduced ejection fraction in the DELIVER (Dapagliflozin Evaluation to Improve the Lives of Patients With Preserved Ejection Fraction Heart Failure) trial [143], and reduced mortality and HHF in patients with heart failure and reduced ejection fraction (HFrEF) in the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial [144]. The consistency of benefit irrespective of diabetes status underscores that SGLT2 inhibitor effects extend beyond glycemia control.
Looking ahead, SGLT2 inhibitors are in a position to play a significant role in the treatment of DCM through their ability to enhance mitochondrial function, mitigate oxidative stress and regulate inflammatory responses. These multifaceted effects establish them as a therapeutic strategy that protects cardiac function, improves vascular endothelial metabolism, and reduces cardiovascular risk.
4.2. Glucagon-like Peptide-1 (GLP-1) Receptor Agonists
GLP-1 receptor agonists are a class of medications that lower blood glucose by mimicking the action of incretin hormones [145]. In addition to effectively controlling blood sugar levels, GLP-1 receptor agonists can reduce insulin dosage and improve postprandial glycemic variability. GLP-1 receptor agonists have also demonstrated potential cardioprotective effects in DCM management [146,147].
GLP-1 receptor agonists exert crucial protective effects in DCM by safeguarding mitochondrial function and modulating oxidative stress pathways [148]. Under the hyperglycemic conditions in DCM pathogenesis, mitochondrial dysfunction and the excessive generation of ROS trigger oxidative stress and inflammatory responses, ultimately resulting in myocardial fibrosis, ventricular remodeling, and impaired cardiac function. GLP-1 receptor agonists such as Exenatide intervene in the progression of DCM through multiple mechanisms, including autophagy activation to clear damaged mitochondria, caspase-1 inhibition to reduce ROS accumulation, and the optimization of energy metabolism via promoted glucose oxidation [149,150,151,152,153].
Exenatide exerts cardioprotective effects at the animal level through a GLP-1 receptor-dependent, multi-target mechanism: it regulates the miR-29b-3p/sarcolemma associated protein (SLMAP) axis to correct abnormal myocardial protein expression and reduce lipid deposition and fibrosis; it simultaneously activates adenosine 5′-monophosphate AMP-activated protein kinase (AMPK)/AKT and inhibits the mammalian target of rapamycin (mTOR) signaling pathways to restore the imbalance in mitochondrial dynamics and autophagy dysfunction [154,155]. This reverses myocardial structural remodeling and cardiac dysfunction in diabetic mice, though its efficacy and safety in clinical patients require further validation [115]. Exendin-4 promotes mitochondrial biogenesis by activating the sirtuin 1 (SIRT1)/PGC-1α/nuclear respiratory factor 1 (NRF-1) pathway, restores the NADPH oxidase 1 (NOX1)/superoxide dismutase (SOD1) antioxidant balance, improves myocardial structural remodeling in diabetic mice, reduces cardiac load markers (atrial natriuretic peptide/brain natriuretic peptide), and inhibits fibrosis (transforming growth factor beta 1), thereby exerting multidimensional cardioprotective effects. However, its efficacy, safety, and mechanism of action in humans require further clinical validation [116,156,157,158,159,160,161,162,163,164,165].
GLP-1 receptor agonists, exemplified by semaglutide, have also shown significant cardiovascular risk reduction. In patients with T2DM and established CVD, semaglutide reduced the risk of major adverse cardiovascular events (MACE) [166]. The recent SELECT (Semaglutide Effects on Cardiovascular Outcomes in People with Overweight or Obesity) trial further demonstrated that semaglutide reduced MACE risk by 20% in adults with overweight or obesity and pre-existing CVD but without diabetes, indicating cardioprotective effects independent of its glucoregulatory actions [167]. These clinical outcomes align with preclinical evidence of GLP-1 receptor agonists-mediated anti-inflammatory, anti-apoptotic, and anti-fibrotic effects in the diabetic heart. Furthermore, the FLOW (Research Study To See How Semaglutide Works Compared to Placebo in People With Type 2 Diabetes and Chronic Kidney Disease) trial revealed that semaglutide significantly reduces the risk of major kidney disease events and cardiovascular outcomes in patients with T2DM and chronic kidney disease, highlighting its organ-protective pleiotropy [168].
Emerging evidence suggests GLP-1 receptor agonists may play an increasingly important role in DCM treatment by restoring mitochondrial function and modulating oxidative stress pathways, becoming an effective strategy for controlling blood sugar, reducing insulin dosage, and improving cardiovascular health.
4.3. Mineralocorticoid Receptor Antagonist (MRA)
In DCM, chronic hyperglycemia induces insulin resistance, shifting the myocardial energy substrate’s preference towards free fatty acids. The upregulated cluster of differentiation 36 (CD36) facilitates excessive free fatty acid uptake, activating peroxisome proliferator-activated receptor α (PPARα) and accelerating mitochondrial β-oxidation, which increases mitochondrial reactive oxygen species (mtROS) production and exacerbates myocardial dysfunction. Aldosterone binding to mineralocorticoid receptor (MR) in cardiomyocytes promotes detrimental remodeling. MRAs block cardiac MR, reducing morbidity and mortality in advanced heart failure, with nonsteroidal MRAs like finerenone potentially offering better cardiac protection than traditional steroidal MRAs. Finerenone antagonizes MR, reduces ROS generation, downregulates PPARα and CD36, limits free fatty acids’ entry into mitochondria, and improves ATP/O_2_ coupling efficiency. Furthermore, it modulates mitochondrial dynamics, promotes fusion and biogenesis, reduces oxidative injury, and impedes maladaptive hypertrophy and fibrosis in DCM [121,169,170,171].
Finerenone regulates mitochondrial dynamics by reducing DRP1-S616 phosphorylation and enhancing MFN2 and OPA1 expression, promoting mitochondrial fusion and network integrity. It restores the AMPK-PGC-1α pathway, stimulates mitochondrial biogenesis, and selectively eliminates damaged mitochondria via PINK1/Parkin-mediated mitophagy. Additionally, finerenone augments SIRT3 activity, which activates MnSOD and isocitrate dehydrogenase (IDH2) to detoxify mtROS. Collectively, these mechanisms re-establish mitochondrial quality control, attenuate cardiomyocyte oxidative injury, and impede maladaptive hypertrophy and fibrosis in DCM [172,173]. Finerenone effectively alleviates DCM by antagonizing MR, reducing ROS generation, and optimizing mitochondrial function. Future research should prioritize its long-term effects on DCM progression and its potential in combination therapies to maximize clinical benefits [117].
Eplerenone selectively antagonizes mineralocorticoid receptors to block aldosterone-mediated pathological pathways, providing multi-target protection against DCM: it enhances glutathione (GSH)/SOD ratios and reduces malondialdehyde (MDA) levels to alleviate oxidative stress; downregulates glucose regulated protein78kD (GRP78) and spliced X-box binding protein-1 (XBP1s) to relieve endoplasmic reticulum stress; and inhibits NLRP3 inflammasomes and interleukin-1β (IL-1β) to mitigate inflammation. Its cardioprotective effects are independent of glycemic control, reducing troponin I levels and improving myocardial injury with fewer side effects than spironolactone, offering a new option for combination therapy in DCM [118].
Animal studies demonstrate that spironolactone confers cardioprotection through a tripartite mechanism: mitochondrial restoration (preserving cristae integrity and upregulating ATP synthase F1 subunit alpha (ATP5A1)/cytochrome C oxidase subunit 5B (COX5b)/SIRT1 to enhance bioenergetics), oxidative stress mitigation (activating NRF-1/glutamate–cysteine ligase catalytic subunit (GCLC) antioxidant defenses while suppressing Nox4-driven ROS production), and inflammation–fibrosis suppression (attenuating tumor necrosis factor-α (TNF-α)/monocyte chemoattractant protein-1 (MCP-1) release, macrophage infiltration, and transforming growth factor (TGF-β1)/collagen I deposition). Importantly, these benefits occur independently of glycemic and blood pressure control. Nevertheless, the translation of these preclinical findings to clinical practice warrants validation through large-scale randomized controlled trials [119].
However, the translation of other promising mechanistic targets from preclinical studies to clinical success is not guaranteed, highlighting the complexity of DCM. The ARISE-HF (Aldose Reductase Inhibition for Stabilization of Exercise Capacity in Heart Failure) trial, which evaluated the selective aldose reductase inhibitor AT-001 in patients with DCM and impaired exercise capacity, found that 15 months of treatment did not result in a statistically significant improvement in peak oxygen uptake (VO_2_) compared to placebo [174]. The successful translation of SGLT2 inhibitors and GLP-1 receptor agonists from mechanistic preclinical discovery to proven clinical benefit provides a powerful paradigm. It confirms that interventions targeting core DCM pathophysiological axes—such as metabolic dysregulation, inflammation, and fibrosis—can effectively improve hard clinical endpoints. This bridge from bench to bedside underscores the value of the preclinical research landscape mapped in Table 2. Future therapeutic strategies should focus on combining these foundational therapies with other promising mechanistic approaches (e.g., targeting mitochondrial dysfunction) identified in preclinical studies and employing more clinically representative models (e.g., aged, comorbid) to enhance the predictive validity and translational success of future drug development for DCM.
As a new generation of antidiabetic agents, SGLT2 inhibitors, GLP-1RAs, and finerenone not only provide excellent glycemic control, but also exhibit cardioprotective effects against DCM. These drugs improve mitochondrial function, attenuate oxidative stress and inflammatory responses through various mechanisms, thereby alleviating myocardial damage and improving cardiac function. Future studies will clarify the specific molecular targets in DCM and strengthen the scientific evidence base for clinical application.
5. Conclusions
In summary, this article systematically explores the pathogenic link between DCM and mitochondrial homeostasis. Disrupted mitochondrial function and homeostasis contribute to a variety of the pathological features of DCM, including cardiomyocyte apoptosis, inflammatory response, fibrosis, etc., which make mitochondrial homeostasis an important part of DCM’s pathophysiology. Mechanisms such as mitochondrial dynamics, mitochondrial oxidative metabolism, and mitophagy alleviate DCM-related myocardial injury by regulating ATP synthesis, optimizing myocardial energy expenditure caused by impaired mitochondrial decoupling, controlling ROS release from mitochondrial oxidative stress, and removing excess free fatty acids and damaged mitochondria from the heart. Therefore, future investigations into the biological mechanism of mitochondria, particularly through reducing oxidative stress, strengthening free fatty acid metabolism, regulating mitochondrial dynamics and autophagy, and improving mitochondrial oxidative metabolism to restore mitochondrial homeostasis, will provide effective therapeutic approaches to DCM treatment.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Liu L. Zhang J. Cheng Y. Zhu M. Xiao Z. Ruan G. Wei Y. Gut microbiota: A new target for T 2DM prevention and treatment Front. Endocrinol.20221395821810.3389/fendo.2022.95821836034447 PMC 9402911 · doi ↗ · pubmed ↗
- 2Jaacks L.M. Siegel K.R. Gujral U.P. Narayan K.M. Type 2 diabetes: A 21st century epidemic Best Pract. Res. Clin. Endocrinol. Metab.20163033134310.1016/j.beem.2016.05.00327432069 · doi ↗ · pubmed ↗
- 3American Diabetes Association Diagnosis and classification of diabetes mellitus Diabetes Care 201437 S 81S 9010.2337/dc 14-S 08124357215 · doi ↗ · pubmed ↗
- 4Sung K.C. Lee M.Y. Kim Y.H. Huh J.H. Kim J.Y. Wild S.H. Byrne C.D. Obesity and incidence of diabetes: Effect of absence of metabolic syndrome, insulin resistance, inflammation and fatty liver Atherosclerosis 2018275505710.1016/j.atherosclerosis.2018.05.04229860108 · doi ↗ · pubmed ↗
- 5Ong K.L. Stafford L.K. Mc Laughlin S.A. Boyko E.J. Vollset S.E. Smith A.E. Dalton B.E. Duprey J. Cruz J.A. Hagins H. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021 Lancet 2023402203234 Erratum in Lancet 2023, 402, 1132. Erratum in Lancet 2025, 405, 20210.1016/S 0140-6736(23)01301-637356446 PMC 10364581 · doi ↗ · pubmed ↗
- 6Zimmet P. Alberti K.G. Shaw J. Global and societal implications of the diabetes epidemic Nature 200141478278710.1038/414782 a 11742409 · doi ↗ · pubmed ↗
- 7Deshpande A.D. Harris-Hayes M. Schootman M. Epidemiology of diabetes and diabetes-related complications Phys. Ther.2008881254126410.2522/ptj.2008002018801858 PMC 3870323 · doi ↗ · pubmed ↗
- 8Farmaki P. Damaskos C. Garmpis N. Garmpi A. Savvanis S. Diamantis E. Complications of the Type 2 Diabetes Mellitus Curr. Cardiol. Rev.20201624925110.2174/1573403 X 160420122911553133407062 PMC 7903505 · doi ↗ · pubmed ↗