Primary Cilia Are Required for Efficient BMP Signaling in Traumatic Heterotopic Ossification
Xinyuan Yuan, Saman Toutounchi, Susan F. Law, David Achudhan, Abhishek Chandra, Kai He, Yingshu Cao, Jinghua Hu, Robert J. Pignolo, Haitao Wang

TL;DR
This study shows that primary cilia are essential for BMP signaling in traumatic heterotopic ossification, suggesting they could be a target for treatment.
Contribution
The study reveals a novel role of primary cilia in traumatic HO development through BMP signaling.
Findings
Disrupted primary cilia function attenuates BMP signaling.
Deleting Ift88 suppressed pathological BMP signaling and HO formation.
Ciliation frequency and length were reduced in genetically modified tenocytes.
Abstract
Background/Objectives: Heterotopic ossification (HO), the aberrant formation of bone within soft tissues, arises either from rare genetic mutations or more commonly from traumatic insults. It is a major cause of morbidity not only in individuals harboring causative mutations, but also in those undergoing musculoskeletal surgery or trauma and in soldiers sustaining blast or burn injuries. Bone morphogenetic protein (BMP) signaling is a central driver of both hereditary and acquired forms of HO. Primary cilia are nonmotile, antenna-like organelles that extend from the cell surface and serve as crucial sensory and signaling hubs by concentrating key pathway components within a confined volume at the ciliary tip. However, their functional role in the pathogenesis of traumatic HO remains poorly understood. Methods: We investigate the role of primary cilia in traumatic HO using a genetically…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
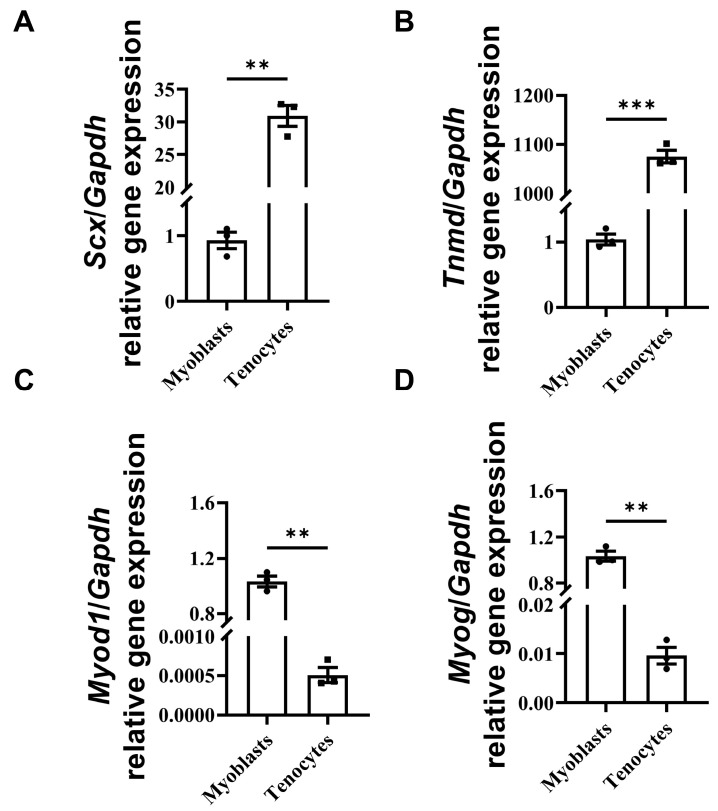 Figure 1
Figure 1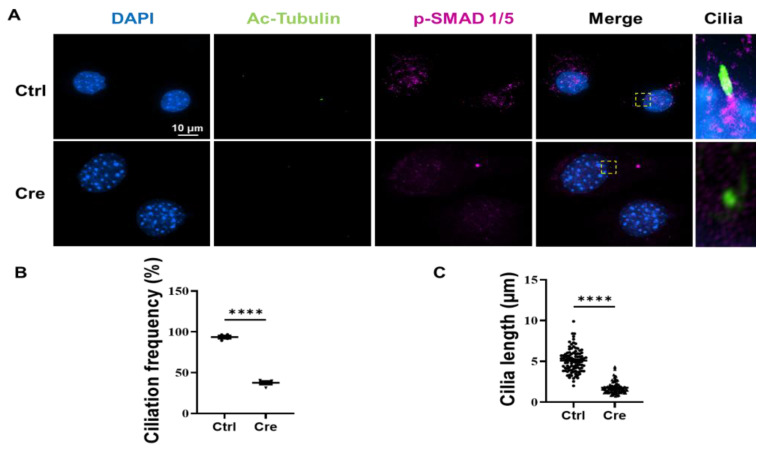 Figure 2
Figure 2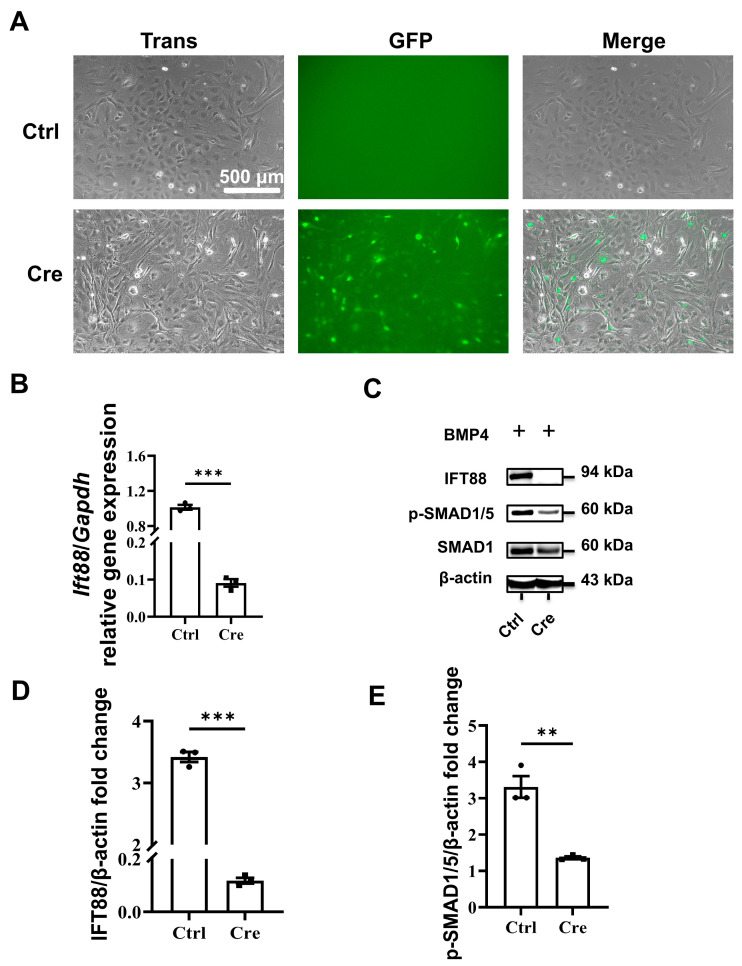 Figure 3
Figure 3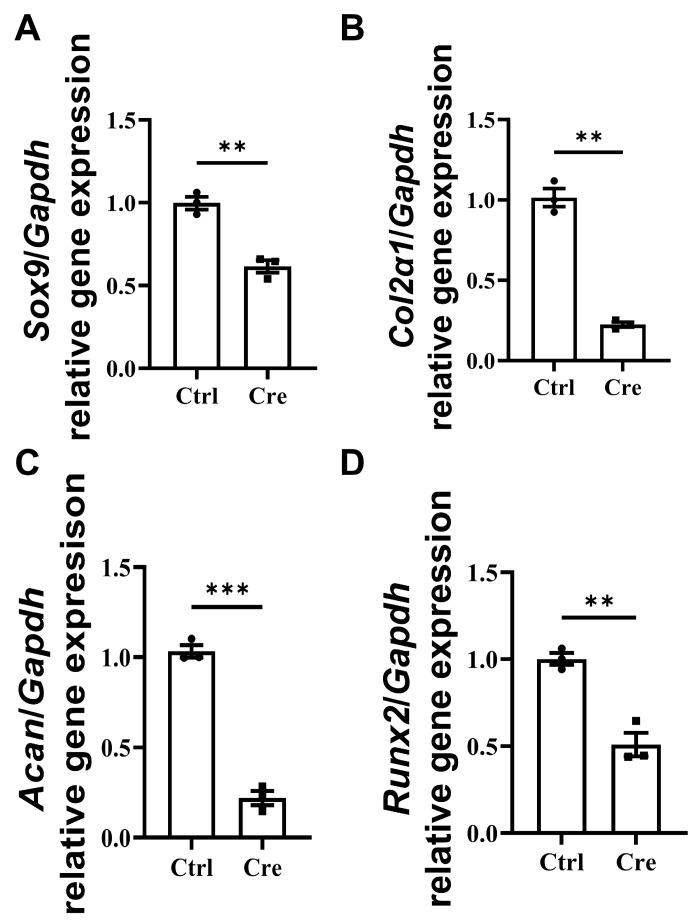 Figure 4
Figure 4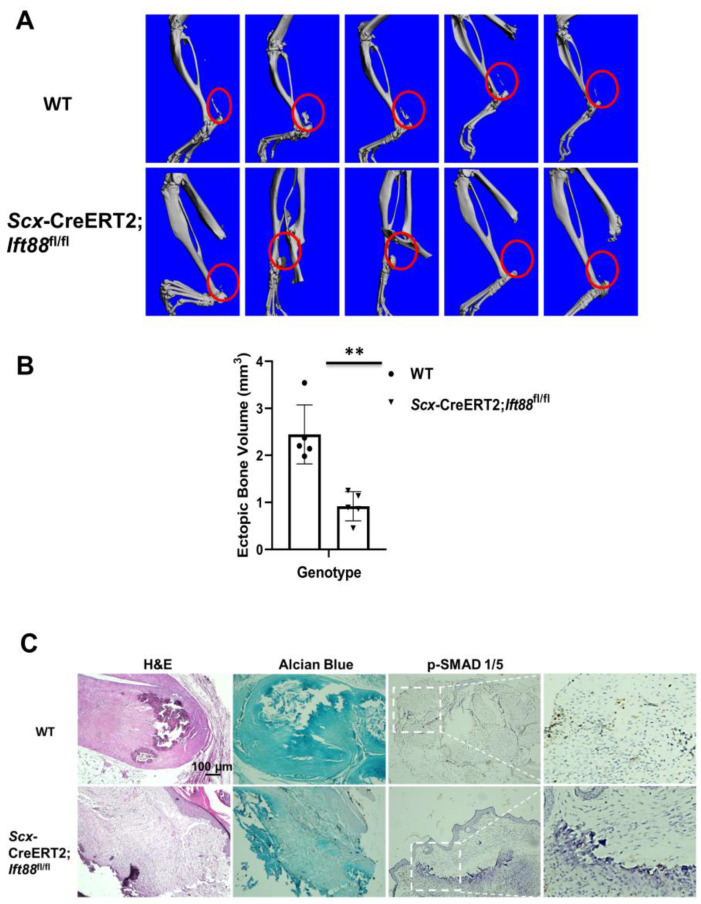 Figure 5
Figure 5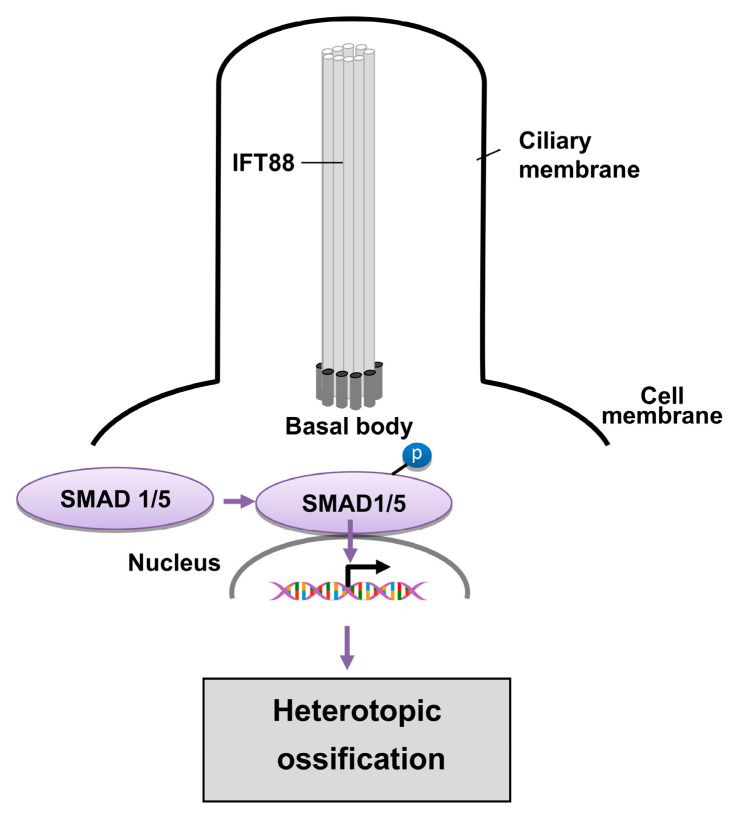 Figure 6
Figure 6- —National Institutes of Health
- —International Fibrodysplasia Ossificans Progressiva Association (IFOPA)
- —Robert and Arlene Kogod Professorship
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHeterotopic Ossification and Related Conditions · Genetic and Kidney Cyst Diseases · Medical Imaging and Pathology Studies
1. Introduction
Heterotopic ossification (HO) is a pathological condition characterized by the formation of mature bone within soft tissues outside the normal skeletal framework. It can arise from rare genetic mutations or be triggered by more common forms of tissue injury and trauma [1]. HO poses a substantial risk of functional impairment, affecting individuals with inherited predisposition as well as patients undergoing invasive musculoskeletal procedures. In addition, HO is highly prevalent among military personnel exposed to modern combat injuries, particularly those involving high-energy blast trauma and extensive burns.
Based on etiology, HO is broadly divided into hereditary and nonhereditary forms. Hereditary HO is exemplified by fibrodysplasia ossificans progressiva (FOP), an extremely rare congenital disorder caused by a gain-of-function point mutation (R206H) in the Activin receptor type I gene (Acvr1, also known as ALK2) [2]. Clinically, FOP is marked by episodic heterotopic bone formation in skeletal muscles, tendons, and other connective tissues, ultimately leading to progressive and irreversible loss of mobility [1,3]. Mechanistically, FOP mutations result in constitutive activation of ACVR1 and enhanced sensitivity to Bone Morphogenetic Protein (BMP) signaling [4,5,6,7,8,9]. Although Activin A (Act A), a member of the transforming growth factor-beta (TGF-β)/BMP superfamily, normally binds ACVR1 to inhibit BMP signaling [10,11,12], pathogenic ACVR1 mutations in FOP convert this inhibitory signal into aberrant activation of downstream BMP effectors SMAD 1/5/8 [10,13]. This dysregulated BMP signaling drives inappropriate chondro-osseous differentiation of progenitor cells during tissue repair, culminating in the replacement of soft tissues with structurally normal bone [14,15].
In contrast, nonhereditary or acquired HO is far more common and typically develops in muscles, tendons, or ligaments following tissue injury [16]. Acquired HO frequently occurs in association with orthopedic procedures—most notably hip arthroplasty, which accounts for approximately 40% of cases [17,18,19]—as well as fractures or dislocations (~30%) [20], elbow trauma [21], high-energy extremity trauma [22], traumatic brain or spinal cord injury (with an incidence of up to 50% in spinal cord injury patients) [23], and severe burns (∼20% of third-degree burns) [24]. Among patients with severe combat-related traumatic amputations, the incidence of HO exceeds 90% [25]. Surgical excision remains the primary treatment for established lesions; however, it is associated with substantial morbidity, and the biological mechanisms underlying acquired HO remain incompletely understood [26]. Consequently, effective therapeutic options for non-genetic HO are limited.
BMP signaling has emerged as a central pathway in both hereditary and acquired forms of HO. In FOP, the canonical R206H mutation and all reported ACVR1 variants lead to loss of autoinhibitory control and persistent activation of BMP signaling [2,27]. Similarly, in trauma-induced HO, the injury-associated stem cell niche has been shown to be BMP-dependent [28], suggesting a partially shared mechanistic basis across HO subtypes.
Primary cilia are solitary, non-motile, microtubule-based organelles that extend from the surfaces of most mammalian cells and function as critical sensory and signaling centers. By concentrating signaling components within a confined subcellular domain, primary cilia enable cells to respond efficiently to low levels of extracellular ligands [29,30]. They play essential roles in regulating key developmental and homeostatic pathways, including Hedgehog, Wnt, and TGF-β signaling [31,32]. Intraflagellar transport protein 88 (Ift88) is a core component of the intraflagellar transport machinery and is indispensable for ciliogenesis across species [33]. Conditional Ift88 knockout mouse models have therefore provided a powerful approach for interrogating cilia-dependent signaling mechanisms in disease contexts [34]. In addition to BMP signaling, other cilia-dependent pathways, including Hedgehog and Wnt signaling, have been implicated in injury-induced HO and in genetic disorders such as progressive osseous heteroplasia [35]. We previously demonstrated that primary cilia regulate skeletogenic BMP and Hedgehog signaling in FOP-associated HO [36]; however, the contribution of primary cilia to BMP signaling in tenocytes during traumatic HO remains poorly defined. Elucidating the spatial and temporal regulation of BMP signaling in injury-responsive tenocytes is therefore critical for understanding the pathogenesis of HO following trauma.
2. Materials and Methods
2.1. Animal Models
All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee. Mice were maintained in a temperature-controlled environment (23–25 °C) under a 12 h light/dark cycle and were provided standard laboratory chow (PicoLab Rodent Diet 20 #5053; LabDiet, St. Louis, MO, USA) and water ad libitum. Scx-CreERT2 mice [B6(129S4)-Scx^tm2.1(cre/ERT2)Stzr^/J, Jackson Laboratory] express a tamoxifen-inducible Cre recombinase (CreERT2) under the control of the endogenous Scleraxis (Scx) promoter, which drives expression predominantly in tendon and tenocyte lineages [37,38]. Ift88^fl/fl^ mice carry loxP sites flanking essential exons of the Ift88 gene, enabling conditional deletion upon Cre-mediated recombination. Scx-CreERT2;Ift88^fl/fl^ mice were obtained by crossing Scx-CreERT2 transgenic mice with Ift88 floxed mice. Thus, Scx-CreERT2;Ift88^fl/fl^ mice were used as a genetic background to obtain tendon-lineage cells, while Ift88 deletion was induced in vitro to allow for controlled analysis of cilia loss in primary tenocytes.
2.2. Mouse Burn–Tenotomy-Induced HO
Seven–nine-week-old male WT and Scx-CreERT2;Ift88^fl/fl^ mice (~20 g) were used for all experiments. Animals were anesthetized with 2.5% inhaled isoflurane, and the left hindlimb (knee to ankle), as well as a 2 cm × 3 cm region along the left paraspinal area, were shaved and disinfected. A longitudinal incision was made over the Achilles tendon to expose the posterior tibial tendon, which was transected transversely. The incision was closed using one or two interrupted 5-0 sutures. To induce burn injury, a 60 °C aluminum block (2 cm × 3 cm) was applied to the left side of the spinal midline for approximately 17 s without added pressure. Eight weeks after surgery, the entire left hindlimb was harvested and analyzed by micro-computed tomography (μCT) to assess heterotopic bone formation.
2.3. Isolation of Primary Tail Tenocytes
Primary tenocytes were isolated from the tails of one-month-old mice using a two-step enzymatic digestion protocol. Tissues were first incubated with 0.25% trypsin for 30 min to remove surface-associated cells. The remaining tendon tissue was then digested with collagenase type I and dispase II (0.2% and 0.3%, respectively) for 2 h to degrade the extracellular matrix and release resident tenocytes. Isolated cells were cultured in high-glucose DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Because donor mice were not treated with tamoxifen prior to tenocyte isolation, Cre-mediated recombination was induced in vitro using adenoviral Cre; this approach was employed solely to delete Ift88, and potential nonspecific effects of viral exposure cannot be entirely excluded.
2.4. Western Blotting
Cultured cells were washed with cold PBS and lysed in 1.5× RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentrations were quantified, and equal amounts of lysates were resolved by SDS–PAGE (Thermo Fisher Scientific, Waltham, MA, USA, 1862495) and transferred onto PVDF membranes. Membranes were probed with antibodies against β-actin, p-SMAD 1/5 (Cell Signaling Technology, Danvers, MA, USA, 3700) for β-actin, total SMAD 1 (Cell Signaling Technology, 6944), and IFT88 (Proteintech, Rosemont, IL, USA, 13967-1-AP), followed by appropriate HRP-conjugated secondary antibodies. Protein signals were detected using enhanced chemiluminescence and visualized using a ChemiDoc™ MP Imaging System (Bio-Rad Laboratories, Hercules, CA, USA).
2.5. RT-qPCR
Total RNA was extracted from cultured cells using TRIzol reagent according to the manufacturer’s instructions. RNA concentration and purity were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). First-strand cDNA was synthesized from 500 ng of total RNA using a high-capacity reverse transcription kit. Quantitative real-time PCR was performed using SYBR Green chemistry on a QuantStudio™ 7 Pro Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). Primer sequences are listed in Table 1. All reactions were performed in triplicate, and gene expression levels were normalized to GAPDH. Relative expression was calculated using the 2^−ΔΔCt^ method, and data were analyzed using GraphPad Prism 10.
2.6. Immunofluorescence
Primary tail tenocytes isolated from Scx-CreERT2;Ift88^fl/fl^ mice were transduced overnight with Ad5-CMV-Cre-eGFP (Baylor College of Medicine) at a multiplicity of infection (MOI) of 1000 when cells reached approximately 80% confluence. Transduction efficiency was assessed using a fluorescence microscope (EVOS M5000 Imaging System; Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA). Following transduction, Ift88-deficient cells were serum starved for 48 h and subsequently treated with recombinant BMP4 (50 ng/mL; R&D Systems, Minneapolis, MN, USA) for 1 h. Cells were then fixed in cold methanol for 30 min at −20 °C and permeabilized with 0.1% Triton X-100 for 10 min at room temperature. After blocking with 3% bovine serum albumin (BSA) for 2 h, cells were incubated with primary antibodies at 4 °C overnight, followed by incubation with fluorophore-conjugated secondary antibodies (Goat anti-rabbit IgG Alexa Fluor 647 (Cat. No. A21246, Invitrogen, Carlsbad, CA, USA) and goat anti-mouse IgG Alexa Fluor 488 (Cat. No. A10667, Invitrogen, Carlsbad, CA, USA) for 2 h at room temperature. Primary antibodies were used at the following dilutions: acetylated α-tubulin (1:500; Cat. T7451, Sigma-Aldrich, St. Louis, MO, USA) and phosphorylated SMAD 1/5 (1:500; Cat. 6944, Cell Signaling Technology, Danvers, MA, USA). Fluorescence images were acquired using a Nikon ECLIPSE Ti microscope, and quantitative analysis was performed using NIS-Elements software 5.21.03 (Nikon Corporation, Tokyo, Japan). Only clearly identifiable, intact primary cilia labeled by acetylated α-Tubulin were included in the analysis. For each experimental condition, ciliary length was quantified from 100 cilia obtained from 3 independent experiments, and measurements were performed in a blinded manner when applicable.
2.7. Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue specimens were sectioned at a thickness of 5 µm. Hematoxylin and eosin (H&E) staining, Alcian Blue staining, and p-SMAD 1/5 staining were performed as previously described [39].
2.8. Micro-Computed Tomography
Hindlimbs harvested from WT or Scx-CreERT2;Ift88^fl/fl^ mice following burn–tenotomy were analyzed by micro–computed tomography (µCT) using a VivaCT 40 micro-CT system (Scanco Medical AG, Brüttisellen, Switzerland). Heterotopic bone volume and two-dimensional medial sagittal plane images were obtained for each limb. Scanning parameters included a source voltage of 55 kV, a current of 142 μA, and an isotropic voxel size of 10.5 μm. Mineralized tissue was distinguished from non-mineralized tissue using a lower threshold of 200 and an upper threshold of 1000 Hounsfield units.
2.9. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 10 (GraphPad Software, Boston, MA, USA). Comparisons between two groups were conducted using a two-tailed unpaired Student’s t-test. A p value < 0.05 was considered statistically significant. Significance levels are indicated as follows: p < 0.05; * p < 0.01; ** p < 0.001; ns, not significant. Data are presented as mean ± SEM, and sample sizes are provided in the corresponding figure legends.
3. Results
3.1. Isolation and Confirmation of Primary Tenocytes
We isolated primary tenocytes from tails of WT and Scx-CreERT2;Ift88^fl/fl^ mice. After isolating primary tenocytes, we examined the tenocyte gene expression using myoblasts as a control. The isolation of myoblasts was performed as previously published [40]. We found that Scx and Tnmd genes are highly expressed in tenocytes, but not in myoblasts (Figure 1A,B). In contrast, the Myod1 and Myog genes are highly expressed in myoblasts, but not in tenocytes (Figure 1C,D). We cannot exclude the presence of minor non-tenocyte populations; however, these would be expected to be limited and unlikely to account for the observed phenotypes.
3.2. Ciliation Frequency and Cilia Length Decreased in Scx-CreERT2;Ift88fl/fl Tenocytes
We stained the isolated tenocytes for Ac-Tubulin (Green), p-SMAD 1/5 (Magenta) and nuclear localization with DAPI (Blue). We found that the p-SMAD 1/5 co-localized with Ac-Tubulin at the ciliary base (Figure 2A). The ciliation frequency of tenocytes decreased from 94% to 38% with IFT88 knock down (Figure 2B). In addition, the ciliary length decreased from 5.09 µm to 1.77 µm (Figure 2C). More cells are shown in the Supplementary Figure (Figure S1).
3.3. BMP Signaling Is Inhibited by Dysfunctional Primary Cilia
We then treated the isolated tenocytes with Cre-GFP adenovirus at an MOI of 1000. We found that about 50% of primary tenocytes cells expressed GFP (Figure 3A). We also confirmed that the Ift88 gene expression was significantly inhibited in tenocytes transfected with Cre adenovirus (Figure 3B). By Western blot analysis, we found that the IFT88 protein was reduced in the Cre-treated tenocyte cultures (Figure 3C,D). The p-SMAD 1/5 level decreased significantly in the Cre-treated tenocyte cultures (Figure 3C,E).
3.4. Chondrogenic and Osteogenic Gene Expressions Are Inhibited by Dysfunctional Primary Cilia
Primary tenocytes were isolated from Scx-CreERT2;Ift88^fl/fl^ mice and infected with Cre-GFP adenovirus at a multiplicity of infection (MOI) of 1000. Quantitative RT–qPCR analysis demonstrated that deletion of Ift88 significantly suppressed chondrogenic and osteogenic gene expression. Specifically, the mRNA levels of chondrogenic marker genes, including Sox9, Col2a1, and Acan, were markedly downregulated in Cre-treated tenocytes compared to controls (Figure 4A–C). In addition, the expression of the osteogenic marker gene Runx2 was also significantly reduced in Cre-treated tenocytes (Figure 4D), suggesting impaired chondrogenic and osteogenic differentiation capacity. Collectively, these findings indicate that Ift88 deletion disrupts primary cilia function and attenuates both chondrogenic and osteogenic gene expression in primary tenocytes treated with Cre-GFP adenovirus at a multiplicity of infection (MOI) of 1000.
3.5. Traumatic HO Is Diminished in Mice with Dysfunctional Primary Cilia
To examine the effects of dysfunctional cilia on traumatic HO formation, we crossed Scx-CreERT2 mice with Ift88^fl/fl^ mice to derive homozygous Scx-CreERT2;Ift88^fl/fl^ mice (Figure 5A). Two months after burn–tenotomy injury, we found that, compared to WT, HO volume decreased in Scx-CreERT2;Ift88^fl/fl^ mice with dysfunctional cilia (Figure 5B). The H&E staining confirmed normal endochondral bone formation at ectopic sites in tendons harvested from WT mice, whereas tendons from Scx-CreERT2;Ift88^fl/fl^ mice exhibited reduced bone formation. The Alcian blue staining suggested reduced cartilage-associated staining in the tendon region of Ift88 mutant animals. Phosphorylated SMAD 1/5 immunostaining revealed reduced p-SMAD 1/5 signal intensity in Scx-CreERT2;Ift88^fl/fl^ mice compared to WT mice (Figure 5C).
4. Discussion
Dysfunction of primary cilia causes a broad spectrum of human diseases that are collectively termed ciliopathies. For example, skeletal abnormalities, such as oral–facial malformation, short ribs/limbs, and polydactyly, are commonly associated with ciliopathies [41]. In FOP, a hallmark sign is congenital great toe malformation, but other joint malformations also occur prenatally [2]. In addition, the population and ciliation ratio of fibro/adipogenic progenitors (FAPs), one major cell types initiating HO formation in FOP [14,15], sharply increases upon muscle injury [42,43]. Recently, several studies associated primary cilia with TGF-β signaling [44] and BMP signaling in mammalian cells [45,46,47]. Also, loss of CEP128, a centriole subdistal appendage protein required for regulating ciliary signaling, leads to impaired TGF-β/BMP signaling, especially phosphorylation of Smad 2/3 in zebrafish [48].
In this study, we show that there is a strong association between BMP signaling and the integrity of primary cilia. Co-localization of Ac-Tubulin and p-SMAD 1/5 in WT tenocytes and their abrogation with IFT88 knockdown in vitro suggests an important localized structural relationship. Reduced BMP signaling after IFT88 knockdown further suggests a key structure–functional relationship. Furthermore, using homozygous Scx-CreERT2;Ift88^fl/fl^ mice, we found that traumatic HO can be mitigated in a model of dysfunctional cilia. Taken together, we hypothesize that functional primary cilia play a crucial role in mediating trauma-induced HO through the regulation of BMP signaling (Figure 6).
BMP signaling is involved in the formation of traumatic HO is supported by other evidence. For example, Activin A is expressed in response to injury in both FOP and traumatic HO models, but by different types of cells. Although wild type ACVR1 does not transduce signal when engaged by activin A, activin A is nevertheless expressed in traumatic HO. Anti-activin A does not abrogate traumatic HO, whereas antibodies that neutralize ACVR1 or ALK3-Fc (which blocks osteogenic BMPs) do partly mitigate formation of ectopic bone [49].
The reduction of HO in the Scx-CreERT2;Ift88^fl/fl^ model is robust, but not complete, suggesting that there are other signaling pathways involved in the process of traumatic HO. Indeed, traumatic HO arises through the aberrant activation of multiple osteogenic and inflammatory signaling pathways following severe tissue injury. The early inflammation activates TGF-β signaling, which is considered one of the primary drivers for osteogenic differentiation of mesenchymal progenitors in ectopic endochondral ossification [50]. Concurrently, hypoxia in the injured soft tissue stabilizes HIF-1α, which enhances chondrogenic commitment and synergizes with BMP signaling to promote endochondral ossification [39,51,52]. Additional developmental pathways—including Wnt/β-catenin, Hedgehog, and mTOR—further regulate progenitor expansion, chondrogenesis, and bone formation [36,52,53]. Traumatic HO is regulated by the PTEN/AKT signaling pathway in osteogenic and chondrogenic differentiation of tendon-derived stem cells [54]. It is plausible that primary cilia contribute to the pathological regulation of tendon-derived stem cells by interacting with PTEN/AKT signaling. Coordination among these inflammatory and osteogenic signaling networks form the molecular basis of HO pathogenesis and represent key targets for therapeutic intervention.
Primary cilia function not only as a mechanosensory hub for BMP signaling, but also as a critical regulator of cell proliferation, migration, and lineage commitment. Through their role in coordinating multiple developmental pathways—including Hedgehog, Wnt, and PDGFRα signaling—primary cilia help control the balance between progenitor cell expansion and differentiation [55]. Disruption or heightened sensitivity of ciliary signaling can therefore alter the activation state of mesenchymal progenitors and chondro-osteogenic programs, creating a permissive environment for endochondral ossification. Given these integrative roles, abnormalities in primary cilia structure or signaling may significantly influence susceptibility or progression of HO.
In summary, our findings indicate that primary cilia dysfunction is associated with reduced traumatic HO, at least partly through impaired BMP signaling. Given their central role in coordinating multiple signaling pathways, changes in primary cilia structure or function may contribute to HO susceptibility or progression and warrant further exploration as potential therapeutic targets.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Meyers C. Lisiecki J. Miller S. Levin A. Fayad L. Ding C. Sono T. Mc Carthy E. Levi B. James A.W. Heterotopic Ossification: A Comprehensive Review JBMR Plus 20193 e 1017210.1002/jbm 4.1017231044187 PMC 6478587 · doi ↗ · pubmed ↗
- 2Kaplan F.S. Xu M. Seemann P. Connor J.M. Glaser D.L. Carroll L. Delai P. Fastnacht-Urban E. Forman S.J. Gillessen-Kaesbach G. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR 1Hum. Mutat.20093037939010.1002/humu.2086819085907 PMC 2921861 · doi ↗ · pubmed ↗
- 3Pignolo R.J. Bedford-Gay C. Liljesthrom M. Durbin-Johnson B.P. Shore E.M. Rocke D.M. Kaplan F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment J. Bone Miner. Res.20163165065610.1002/jbmr.272827025942 PMC 4829946 · doi ↗ · pubmed ↗
- 4Macias-Silva M. Hoodless P.A. Tang S.J. Buchwald M. Wrana J.L. Specific activation of Smad 1 signaling pathways by the BMP 7 type I receptor, ALK 2J. Biol. Chem.1998273256282563610.1074/jbc.273.40.256289748228 · doi ↗ · pubmed ↗
- 5Yu P.B. Deng D.Y. Lai C.S. Hong C.C. Cuny G.D. Bouxsein M.L. Hong D.W. Mc Manus P.M. Katagiri T. Sachidanandan C. BMP type I receptor inhibition reduces heterotopic [corrected] ossification Nat. Med.2008141363136910.1038/nm.188819029982 PMC 2846458 · doi ↗ · pubmed ↗
- 6Fukuda T. Kohda M. Kanomata K. Nojima J. Nakamura A. Kamizono J. Noguchi Y. Iwakiri K. Kondo T. Kurose J. Constitutively activated ALK 2 and increased SMAD 1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva J. Biol. Chem.20092847149715610.1074/jbc.M 80168120018684712 PMC 2652274 · doi ↗ · pubmed ↗
- 7van Dinther M. Visser N. de Gorter D.J. Doorn J. Goumans M.J. de Boer J. ten Dijke P. ALK 2 R 206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation J. Bone Miner. Res.2010251208121510.1359/jbmr.09111019929436 · doi ↗ · pubmed ↗
- 8Shen Q. Little S.C. Xu M. Haupt J. Ast C. Katagiri T. Mundlos S. Seemann P. Kaplan F.S. Mullins M.C. The fibrodysplasia ossificans progressiva R 206H ACVR 1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization J. Clin. Investig.20091193462347210.1172/JCI 3741219855136 PMC 2769180 · doi ↗ · pubmed ↗