Elucidation of the XX/XY Sex Determination System and Development of a Sex-Linked Molecular Marker in the Freshwater Snail Bellamya purificata
Yajun Gao, Yanhong Wen, Shaokui Yi, Yong Lin, Jinxia Peng, Xianhui Pan, Xiaoyun Zhou

TL;DR
This study identifies the XX/XY sex determination system in the freshwater snail Bellamya purificata and develops a cost-effective molecular marker to distinguish males and females.
Contribution
The study provides the first elucidation of the sex determination system and a sex-linked molecular marker in Bellamya purificata.
Findings
The sex determination system in B. purificata follows an XX/XY pattern with females being homozygous and males heterozygous.
A sex-linked InDel marker within the mis18bp1 gene was developed for cost-effective sex identification.
Candidate genes like mrc2, mis18bp1, and dmrt1 were identified as critical for sex differentiation.
Abstract
Bellamya purificata, a homotypic synonym of Sinotaia purificata, is an ecologically and economically important freshwater snail in China. Elucidation of sex determination mechanism and identification of sex-linked molecular markers are of great importance for breeding of monosex stocks. In this study, we used whole-genome resequencing data and gonadal transcriptome data to develop a sex-linked marker and elucidate the genetic architecture underlying sex determination for this species. Based on the data analysis, we obtained several important sex-associated single-nucleotide polymorphisms (SNPs), insertion–deletions (InDels) and candidate genes. The sex determination system of B. purificata follows a primarily XX/XY pattern, characterized by females being homozygous and males being heterozygous. Moreover, a sex-specific marker was successfully developed, which can identify male and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
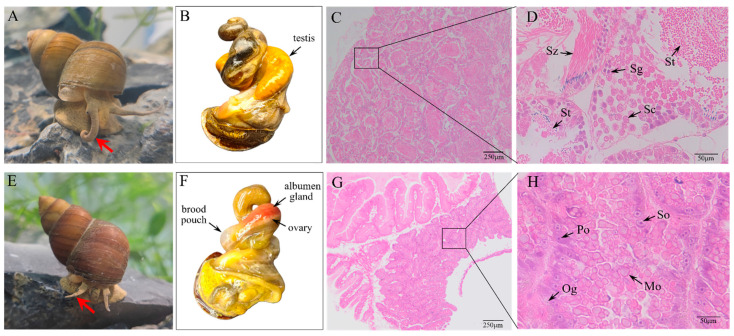 Figure 1
Figure 1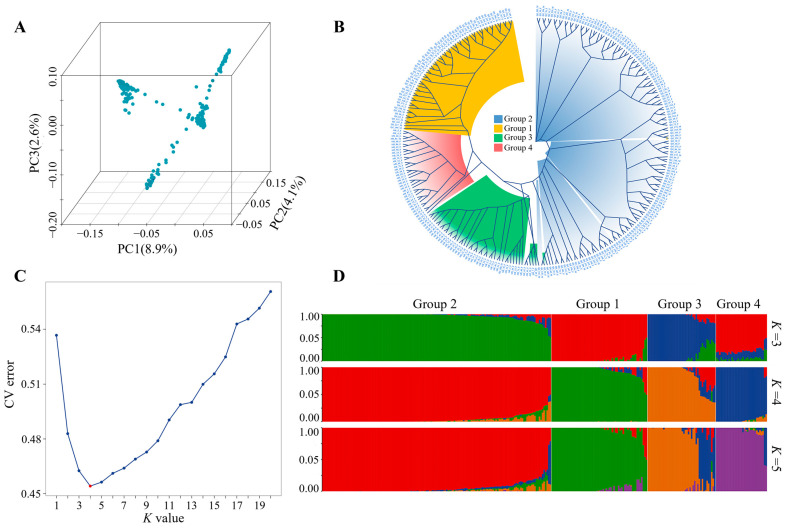 Figure 2
Figure 2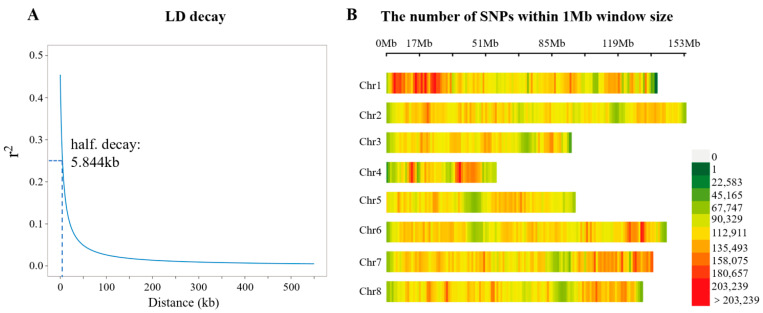 Figure 3
Figure 3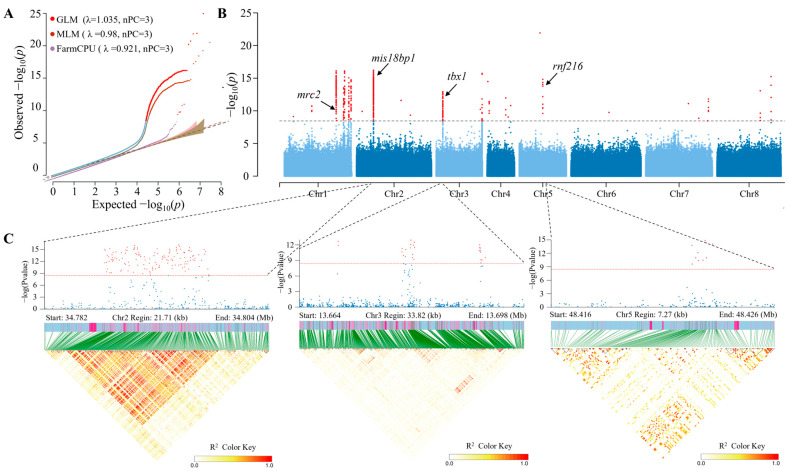 Figure 4
Figure 4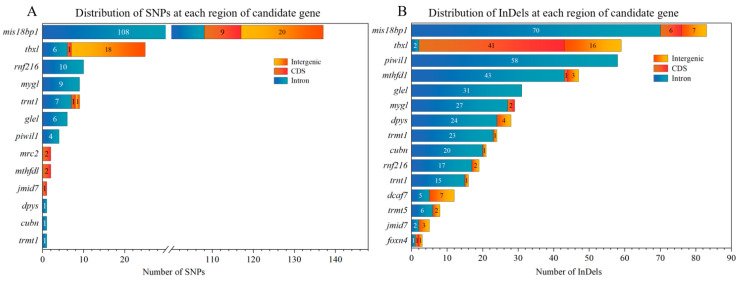 Figure 5
Figure 5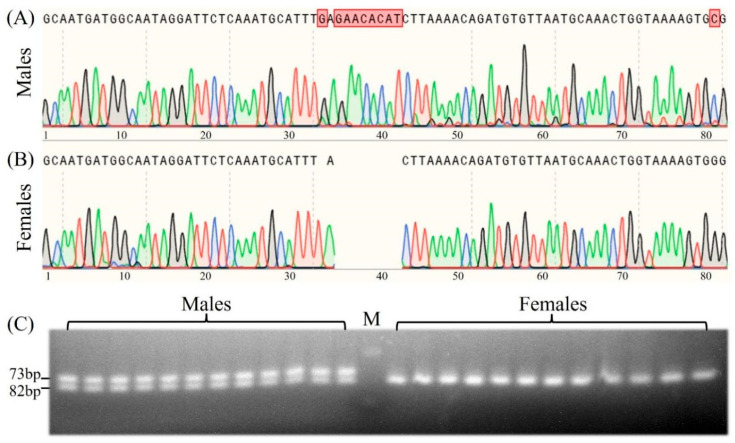 Figure 6
Figure 6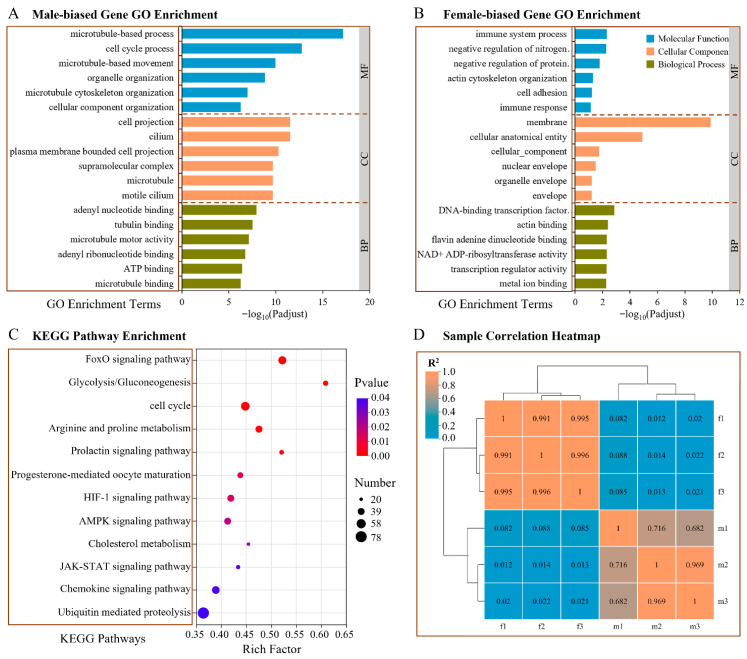 Figure 7
Figure 7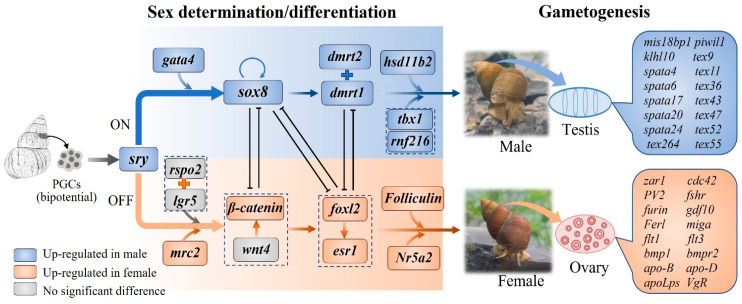 Figure 8
Figure 8- —National Key R&D Program of China
- —Guangxi Key R&D Program Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Aquatic Invertebrate Ecology and Behavior · Evolution and Genetic Dynamics
1. Introduction
The freshwater snail Bellamya purificata (Gastropoda: Viviparidae), an indigenous species in China, has a broad distribution across ponds, lakes, reservoirs, rivers, and other types of aquatic ecosystems [1]. This snail exhibits a propensity for silty substrates and sustains itself by feeding on organic detritus and algae within its habitat [2,3]. Several studies have demonstrated that freshwater snails play pivotal roles in mitigating eutrophication-driven Microcystis blooms in freshwater systems through the consumption of Microcystis pellets [4,5]. Consequently, B. purificata has been acknowledged as a crucial bioremediation organism and is extensively utilized in China’s freshwater polyculture system to optimize resource exploitation and regulate the cultivation environment [6,7,8].
In addition to its significant ecosystem functions, B. purificata is also an economically important edible snail in China. Production of “Liuzhou Luosifen”, a renowned delicacy in Liuzhou, has experienced significant growth, generating over 60 billion yuan in annual economic revenue. Bellamyinae snails serve as the exclusive ingredient for this dish, with an annual demand in the Luosifen industry reaching approximately 1.5 million tons [9]. However, the current annual production of cultured freshwater snails in China stands at a mere 94,681 tons as of 2023 [10], indicating a substantial supply–demand disparity. With the Luosifen industry’s burgeoning demand, Bellamyinae snails have become important aquaculture species in China. Regrettably, the majority of snails used for cultivation are procured from wild populations, which exhibit slow growth rates and low meat yields [9]. Therefore, the application of genomic resources to breed improved varieties is essential for bolstering productivity and economic value in snail aquaculture.
Like most aquatic animals, numerous gastropod snails exhibit sexual dimorphism, displaying differences in growth rate, maturity age, body size, and/or color pattern between the sexes [11]. For example, female freshwater snails of the genus Viviparus are larger than the males, with the former also having more globose shape, wider apertures, and body whorls [12]. Similar sex-related differences in size and shape have been observed in B. purificata, in which females are larger and have broader body whorls than males [13]. Furthermore, recent study on B. purificata has observed nutritional constituents and flavor substances differences between the sexes, with females possessing higher crude protein, glycogen, and flavor amino acids [14]. Therefore, the development of monosex stocks in B. purificata is a necessary breeding strategy that could substantially enhance production efficiency and economic benefits, especially for monosex culture [15]. To reach such aims, it is essential to thoroughly comprehend the genetic architecture underlying sex determination, as well as to identify credible sex-associated molecular markers [16]. However, as a newly emerging aquaculture species, limited genomic resources are available for this snail. In particular, the lack of a comprehensive understanding of the genetic mechanisms underlying sex determination hinders the development of efficient sex-associated molecular markers and subsequently the breeding of monosex stocks in B. purificata.
Conventional approaches for developing sex-linked markers, including Restriction Fragment Length Polymorphism (RFLP), Amplified Fragment Length Polymorphism (AFLP), Random Amplified Polymorphic DNA (RAPD), and Simple Sequence Repeat (SSR), have played a pivotal role in identifying such markers across more than 20 aquatic species [17,18]. Despite this, these traditional strategies are currently considered time-consuming, labor-extensive, and relatively inefficient when compared with state-of-the-art genomic technologies. In recent years, next-generation sequencing (NGS)-based genetic studies have offered novel perspectives on the sex determination systems in invertebrate species [19]. Particularly, genome-wide association study (GWAS)-based screening of SNP/InDel markers has become a more efficient, precise and sustainable practical tool for analyzing the genetic mechanisms underlying sex determination, with sex-associated markers and genes having been identified in various aquatic species, such as Pacific abalone (Haliotis discus hannai Ino) [20], sea urchin (Mesocentrotus nudus) [19], deep-sea mussel (Gigantidas platifrons) [21], Portuguese oyster (Magallana angulata) [16] and ivory shell (Babylonia areolate) [22]. Furthermore, RNA-Seq enables the identification and characterization of genes, along with the accurate quantification of transcript abundance [23]. Additionally, it serves as a valuable tool for deciphering gene regulation networks via the detection of differentially expressed genes (DEGs) [24]. Accumulating evidence indicates that combining GWAS and RNA-Seq can narrow the range of quantitative trait loci (QTLs) and improve the accuracy of identifying candidate genes [25]. This integrative method has been widely adopted in several aquaculture species, such as the sea urchin Mesocentrotus nudus [19] and the Japanese grenadier anchovy Coilia nasus [25], and has greatly advanced the process of molecular marker-assisted monosex breeding.
In the present study, genome-wide association studies (GWASs) and comparative transcriptome profiling were employed to identify sex-associated loci and elucidate the genetic architecture underlying sex determination in B. purificata. The results establish a fundamental framework for investigating the genetic mechanisms of sex differentiation and may facilitate the further development of monosex stocks for this species.
2. Materials and Methods
2.1. Ethics Statement
All experimental procedures related to snails were performed in strict compliance with the guidelines in the Guide for the Care and Use of Laboratory Animals of Huazhong Agricultural University. All efforts were made to minimize suffering of the snails.
2.2. Sample Collection
Five populations were sampled for this study. Among them, two populations consisting of 287 adult individuals (72♂ and 215♀) collected from the rice–crayfish coculture field of Qianjiang (Hubei Province) and Chaohu Lake (Anhui Province) were used for GWAS analysis. Nine males and nine females from the Qianjing population were selected for RNA-Seq. Three populations including those from Honghu Lake (Hubei Province) (53♂ and 97♀), rice–crayfish coculture fields of Tianmen (Hubei Province) (66♂ and 72♀), and Weishan Lake (Shandong Province) (32♂ and 65♀) were collected for sex-linked marker validation. Sampling information was listed in Table S1. Taxonomic identification of the snails was conducted based on morphological characteristics, following the descriptive criteria established by Zhang and Liu [26].
Prior to handling, snails were drained to remove excess water from the mantle cavity and then blotted dry with absorbent paper [27]. Shell width (SW) and total weight (T_W_) were measured to the nearest 0.01 mm and 0.01 g, respectively. Subsequently, the snails were deshelled and individuals were sexed. Males were identified by the presence of brownish-yellow testes and females by the presence of a red protein gland and uterus [26]. Afterwards, the abdominal foot muscle of each individual was separated and preserved in 95% ethanol and stored at −20 °C for DNA extraction. The gonads of adult B. purificata were separated and split into two parts; one was fixed with Bouin’s solution for histology to identify phenotypic sex, while the other one was immediately flash-frozen in liquid nitrogen and stored at −80 °C for subsequent RNA extraction.
For histological analysis, Bouin’s solution-fixed gonadal tissues were dehydrated in a graded ethanol series (70–100%), embedded in paraffin, and cut into 5 μm thick sections, followed by hematoxylin–eosin (H&E) staining and observation under a light microscope (4× and 40×).
2.3. DNA Extraction and Whole-Genome Resequencing
Genomic DNA of each abdominal foot muscle was isolated using the ammonium acetate method [28]. DNA quality was assessed by 1% agarose gel electrophoresis, while DNA concentration was determined using a Nanodrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Subsequently, the qualified DNA samples were sent to Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China) for DNA library construction, and sequencing was performed on the DNBSEQ-T7 platform (Complete Genomics, a subsidiary of MGI Tech, Shenzhen, China). Raw sequencing reads were subjected to preliminary quality control using fastp (v0.21.0) [29] to eliminate low-quality reads, adapter sequences, and ambiguous bases. Then clean reads were aligned to the reference genome of B. purificata (CNGBdb: CNA0142815) using BWA-MEM2 (v2.2.1) [30]. Variant detection and genotyping were performed with the Genome Analysis Toolkit (GATK, v4.4.0.0) [31]. SNPs and InDels were identified using the “HaplotypeCaller” module of GATK. Furthermore, variant filtering was conducted using GATK to eliminate low-quality variants, including those exhibiting elevated strand bias, insufficient mapping quality, aberrant read position, or substandard mapping quality rank sum scores. For SNPs, the filtering thresholds were set as follows: QD < 2.0 || FS > 60.0 || MQ < 40.0 || SOR > 3.0 || ReadPosRankSum < −8.0 || MQRankSum < −12.5. For InDels, the parameters used were QD < 2.0 || FS > 200.0 || SOR > 10.0 || ReadPosRankSum < −20.0 || MQRankSum < −12.5. Additional filtering was performed with VCFtools (v0.1.17) [32], using the following criteria: min-alleles, 2; maf, 0.05; min-meanDP, 3; and max-missing, 0.75. Functional annotation of the identified SNPs and InDels was subsequently carried out using SnpEff v4.3t [33], according to the gene annotation of the reference genome.
2.4. Population Genetic Structure and Linkage Disequilibrium Analysis
Before performing genetic association analyses, principal component analysis (PCA) was implemented using PLINK v1.9 [34] based on the filtered genotype data, while population structure was inferred using ADMIXTURE v 1.3.0 [35]. The K-value range was set from 1 to 8 during the analysis, and the optimal K-value was determined according to the minimum cross-validation error. Phylogenetic trees were constructed using the Maximum Likelihood (ML) method with IQ-TREE2 [36] and the Neighbor-Joining (NJ) approach with FastTree v2.1.11 [37], respectively. Pairwise linkage disequilibrium (LD) and the squared correlation coefficients (r^2^) between alleles at each locus were calculated with PopLDdecay v3.41 [38], and the corresponding LD decay curve was constructed.
2.5. Sex-Associated Regions and Loci Screening Using GWAS
Based on the filtered SNPs and InDels, genome-wide association studies (GWASs) between sex phenotype and genotype were conducted in rMVP v3.6.0 [39] with three models: the general linear model (GLM) [40], the mixed linear model (MLM) [41], and fixed and random model circulating probability unification (FarmCPU) [42].
2.6. Single-Marker and Haplotype Association Analysis
LDBlockShow v1.40 [43] was employed to produce locus-zoom plots and linkage disequilibrium (LD) heatmaps covering the 5.844 kb regions upstream and downstream of the lead SNP in the significant association signals, with the aim of identifying SNPs in strong LD with the lead SNP. Single-marker association analysis was subsequently conducted for SNPs located within the coding sequence (CDS) of genes harboring a high density of genetic markers with strong linkage disequilibrium. In addition, phenotypic validation of InDels in strong LD with significant SNPs was conducted to identify sex-specific fragments distinguishable by gel electrophoresis.
2.7. Development and Validation of Sex-Specific Markers
InDels longer than 8 bp were selected for further development of sex-specific markers. Based on the positions of these markers, primers were designed with NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/, accessed on 20 March 2025) using the corresponding genomic sequences. The PCR reaction mixture consisted of 12.5 μL of 2 × Taq PCR Mix, 1.0 μL of template DNA, 1.0 μL of both upstream and downstream primers (10 μM), and nuclease-free water to a final volume of 25 μL. PCR reactions were carried out in an ABI Veriti^®^ 96-well Thermal Cycler (Applied Biosystems, Foster City, CA, USA) with the following conditions: initial denaturation at 95 °C for 3 min; 35 cycles of 95 °C for 25 s, 55 °C for 25 s, and 72 °C for 30 s; and 72 °C for 5 min. The amplified PCR fragments were visualized via 3% agarose gel electrophoresis.
2.8. RNA Extraction and Transcriptome Analysis
Following sex identification, nine female and nine male gonads were selected for RNA extraction. RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), while RNA concentration was determined with a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). Qualified RNA was then used for RNA-sequencing analysis. Equal amounts of RNA from three female gonads were combined to generate three RNA pools, and the same procedure was applied to the nine male gonads. Herein, three RNA pools were prepared from both female and male gonads for RNA-Seq analysis. These samples were then sent to Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China) for cDNA library construction. Sequencing of the libraries was performed on the Illumina NovaSeq platform (Illumina, San Diego, CA, USA), generating 150 bp paired-end reads.
Raw sequencing reads were processed and filtered using Trimmomatic (v0.39) (http://www.usadellab.org/cms/?page=trimmomatic, accessed on 26 January 2025) [44] to generate clean reads. These clean reads were aligned to the B. purificata reference genome (CNGBdb: CNA0142815) using HISAT2 v2.1.0 [45]. RSEM v1.3.3 [46] was applied to count the assembled transcripts and conduct subsequent normalization for the calculation of RPKM values. DESeq2 v1.40.1 [47] was used to perform differential expression analysis, so as to screen out differentially expressed genes (DEGs) between ovarian and testicular samples with the default threshold of |log_2_ FC| > 1 and p < 0.05. Furthermore, Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were carried out for these DEGs via DAVID (https://davidbioinformatics.nih.gov/) [48].
To validate the RNA-Seq data, 18 DEGs associated with sex differentiation and gonadal development were selected for qRT-PCR experiment. Primers (Table S2) were designed using Primer Premier 6.0 (https://www.premierbiosoft.com/primerdesign/index.html, accessed on 26 January 2025) based on the gene sequences from the transcriptome data and prepared by Wuhan Tianyi Huayu Gene Technology Co., Ltd. (Wuhan, China). The qRT-PCR was performed on a CFX96TM Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Each qRT-PCR reaction system consisted of 4 μL of forward and reverse primers, 2 μL of cDNA template, 10 μL of 2 × SYBR mix (TaKaRa, Dalian, China), and 7.2 μL of ddH_2_O. Triplicate reactions were performed for each sample, with the ef1α gene serving as the internal reference. The amplification program was set as follows: 95 °C for 10 s and 40 cycles of 95 °C for 5 s, 58–60 °C for 20 s, and 72 °C for 20 s. Relative gene expression levels were quantified using the method.
2.9. Candidate Gene Identification and Functional Annotation
After obtaining the loci associated with sex-related traits, flanking regions of 5.844 kb were extracted using bedtools v2.30.0 [49] as the candidate regions. Genes located within these regions were considered potential candidate genes. Significantly differentially expressed genes (DEGs) associated with sexual differentiation identified from RNA-Seq analysis were also considered as potential candidate genes. To refine the candidate gene set, all predicted genes were compared with the RNA-Seq results, and those overlapping with the transcriptomic dataset were considered as key candidate genes.
3. Results
3.1. Morphological and Histological Appearance of Sexual Characteristics
B. purificata is a dioecious species with a unique ovoviviparous life history. However, male and female individuals can hardly be differentiated morphologically by differences in size or shell color. For individuals with well-extended tentacles, males can be identified by their tightly curved right tentacle, which is thicker and stronger than the left one and functions as the copulatory organ (Figure 1A). The female, on the other hand, has two slender, straight, equal-length tentacles (Figure 1E). After removal of the shell, the mature male can be distinguished by the yellowish testis (Figure 1B), while the female exhibits a pinkish albumen gland and a swollen brood pouch with the eggs and hatching juveniles within it appearing as irregularities on its external surface (Figure 1F). Examination of serial sections of the testis shows the spermatogonia (Sg), spermatocytes (Sc), spermatids (St) and spermatozoa (Sz) distributed within the lumen of the seminiferous tubule (Figure 1C,D), while the ovary contains oogonia (Og), primary oocytes (Po), secondary oocytes (So) and mature oocytes (Mo) (Figure 1G,H).
3.2. Genotyping and Population Structure Analyses
Whole-genome resequencing-based genotyping was performed on 215 female and 72 male B. purificata, generating a total of 3279.2 Gb of clean data. On average, each sample generated 11.4 Gb of clean data. Relative to the genome size, the average mapping rate reached 98.6%, with a mean sequencing depth of 11.6×.
Subsequently, we assessed the population structure and genetic relationships among B. purificata individuals using principal component analysis (PCA), kinship analysis, and population structure analysis in combination. Both the PCA results (Figure 2A) and the NJ-based phylogenetic tree (Figure 2B) revealed clear population stratification, with 287 individuals forming four distinct genetic clusters. During population structure analysis, the cross-validation error gradually decreased as the K value increased, with the lowest value observed at K = 4, indicating that the population could be best clustered into four subgroups (Figure 2C,D). These results indicated the presence of significant population structure within the sampled population. To control for the confounding effects of population stratification in the subsequent GWAS, the top three principal components (PC1, PC2, and PC3) with eigenvalues > 1 were incorporated as covariates in the GWAS statistical model.
3.3. Variant Calling
Following stringent quality control procedures, 14,024,434 high-quality SNPs and 2,904,311 InDels were identified across 287 individuals and subsequently used for GWAS. Linkage disequilibrium (LD) decayed rapidly within the population, reaching half of its maximum value (r^2^ = 0.239) at an average inter-SNP distance of 5.844 kb (Figure 3A), indicating abundant genetic diversity existed in the analyzed population. The reference genome applied in the present study was approximately 939.19 Mb in length. Theoretically, to guarantee the reliability of association analysis, at least 160,710 evenly distributed genetic markers (939.19 Mb/5.84 kb) would be needed. In this study, the actual number of markers employed (16,928,745) was vastly higher than this threshold. The distribution of SNPs was visualized using a SNP density plot, which revealed a relatively uniform pattern across the eight chromosomes with an average density of one SNP per 352 bp (Figure 3B).
3.4. Genome-Wide Association Studies
To identify the optimal model for population stratification correction, three distinct analytical approaches were adopted, the GLM, the MLM, and FarmCPU, all integrated with three principal components (PCs). The corresponding λ values were 1.035, 0.980, and 0.921, respectively. Generally, the optimal λ value ranges from 0.95 to 1.05, with values closer to 1 providing better control of false positives caused by population structure and kinship. Both the MLM (λ = 0.980) and GLM (λ = 1.035) models were suitable for GWAS. However, the GLM generated Manhattan plots consistent with those of the MLM while identifying more candidate SNPs. To retain an adequate number of candidate SNPs, the GLM was chosen for subsequent analysis (Figure 4A). The Manhattan plot revealed that 571 SNPs (Figure 4B, Table S3) and 1853 InDels (Table S4) reached the suggestive threshold for association with sex differentiation and gonadal development processes. Among the 571 sex-specific SNPs, approximately 40.98% were located on chromosome 1 and formed multiple significant SNP peaks. In addition, 30.65% and 20.14% of the SNPs were distributed on chromosomes 2 and 3, respectively, with one significant SNP peak for each chromosome (Figure 4B).
The genotypes of the sex-associated SNPs are summarized in Table S5. Analysis of the homozygous and heterozygous genotypes of the 163,877 SNPs from 287 individuals of 571 loci revealed that 81.01% of male genotypes were heterozygous, while 88.63% of female genotypes were homozygous, which suggests that B. purificata likely has a primarily XX/XY sex determination system.
We performed functional annotation for these sex-specific SNPs and InDels using SnpEff [33]. It revealed that the sex-associated loci corresponded to 44 candidate genes within linkage disequilibrium regions (Table S6). Furthermore, of the 571 SNPs, 32.75% (187 SNPs) were mapped to six genes, including mis18-binding protein 1 (mis18bp1) (137 SNPs), T-box transcription factor (tbx1) (25 SNPs), E3 ubiquitin-protein ligase RNF216 (rnf216) (10 SNPs), melanocyte proliferating gene 1 (myg1) (9 SNPs), piwi-like protein 1 (piwil1) (4 SNPs) and C-type mannose receptor 2 (mrc2) (2 SNPs) (Figure 5A). Similarly, 13.38% of the 1853 InDels were located at mis18bp1, tbx1, rnf216, myg1 and piwil1 (Figure 5B). LocusZoom plots illustrating the linkage disequilibrium between the lead SNP in the mis18bp1 region (Chr.2-34795421) and all genetic markers within the 5.844 kb flanking region revealed that all SNPs strongly linked to Chr.2-34795421 were concentrated near the mis18bp1 gene (Figure 4C).
3.5. Development and Detection of Sex-Linked Molecular Marker
To develop a PCR-based sex-specific molecular marker for B. purificata, sixty sexually dimorphic InDels were screened out, and primers were designed according to the genome sequence. After preliminary PCR screening, we successfully obtained an InDel located within the intronic regions of mis18bp1 gene which effectively distinguished male and female B. purificata. Using the locus-specific primer pair (F: 5′-GCAATGATGGCAATAGGATTCTCAA-3′ and R: 5′-CGCACTTTTACCAGTTTGCAT-3′), PCR amplification yielded different-length products, and subsequent Sanger sequencing of the PCR products revealed a female-specific 9 bp deletion and a sex-specific nucleotide site (Figure 6A,B). Following the refinement of electrophoresis conditions (3% agarose gel electrophoresis for 1h), distinct electrophoretic patterns were observed: two bands in males and only one band in females (Figure 6C).
Further validation showed that this locus-specific primer pair could successfully distinguish female and male samples from Honghu Lake, rice–crayfish coculture field of Tianmen, and Weishan Lake (Table S1), indicating the wide applicability of the sex-linked InDel marker. In addition, males exhibiting a heterozygous binding phenotype and females a homozygous binding phenotype at the sex-linked InDel marker suggest that the sex determination system of B. purificata is primarily male heterogametic (XX/XY).
3.6. Transcriptome Analysis of DEGs Between Testes and Ovaries
To gain a comprehensive understanding of gonadal transcriptional divergence and clarify the genetic mechanism underlying sex determination in B. purificata, we constructed and sequenced cDNA libraries from testis and ovary tissues. DEGs analysis identified a total of 7277 sex-biased genes between testes and ovaries (Figure 7A). Among these, 2996 genes were upregulated in ovaries (female-biased), while 4281 genes showed elevated expression in testes (male-biased) (Table S7). To investigate the potential roles of these DEGs in sex determination, differentiation, and gametogenesis, we performed GO and KEGG pathway enrichment analyses. GO functional enrichment analysis revealed that female-biased genes were primarily enriched in cellular component terms, including membrane and related cellular component categories. Enriched terms in the biological processes category were primarily immune system process and actin cytoskeleton organization, while the molecular functions primarily included DNA-binding transcription factor activity (Figure 7A). The male-biased genes were primarily involved in biological processes, such as microtubule-based process, cell cycle process and microtubule-based movement. The cellular components primarily included the cilium and microtubule, while the molecular functions primarily included tubulin binding and microtubule motor activity (Figure 7B). These findings imply that microtubule-associated functions play vital roles in testicular development, likely affecting biological processes including spermatogenesis and cell division. In the KEGG pathway analysis, 4014 DEGs were annotated against the KEGG database and assigned to 39 distinct pathways. The top 20 significantly enriched pathways are presented in Figure 7C. Some of the DEGs were enriched in the cell cycle, cholesterol metabolism, and FoxO signaling pathways, which are known to participate in the proliferation and function of gonadal cells, as well as regulate the biosynthesis of sex hormones. The glycolysis/gluconeogenesis signaling pathway plays a pivotal role in energy metabolism. Furthermore, the progesterone-mediated oocyte maturation (POM) and prolactin signaling pathways play crucial roles in ovarian function by modulating oocyte maturation and ovulation.
3.7. Key Genes Associated with Sex Determination, Differentiation and Gonadal Development
By analyzing the global gene expression profiles of gonads, we identified 68 candidate genes associated with multiple processes of sex determination, differentiation, and gonadal development (Table 1). Among these candidates, sixteen key genes were predominantly implicated in sex determination and differentiation. Specifically, the expression levels of sry, gata4, dmrt1, dmrt2, sox8, and hsd11b2 were significantly higher in testes than in ovaries, while the expression levels of β-catenin, esr1, Nr5a2 and foxl2 in ovary were higher than those in testis. Twenty-nine key genes were primarily associated with spermatogenesis, among which piwil1, spata4/6/17/20/24, klhl10, and tex9/11/36/43/47/49/52/55/264 exhibited significantly higher expression levels in the testis. Moreover, seven genes associated with ovarian development and six genes implicated in steroid biosynthesis and metabolism were identified in ovarian and testicular tissues. Notably, key genes known to regulate sex-determination cascades in mammals, including sry, gata4, sox8, dmrt1, and dmrt2 in males, as well as β-catenin, foxl2, esr1, and nr5a2 in females, were also detected in B. purificata. These results suggest that B. purificata and mammals share conserved genetic regulatory networks governing sex determination. The RNA-Seq data were validated by qRT-PCR analysis and the results are shown in Figure S1.
3.8. Identification of Overlapping Genes Between GWAS and RNA-Seq
In the GWAS, a candidate interval spanning 5.844 kb upstream and downstream of each significant SNP was defined. A total of 44 genes located within these intervals were extracted and functionally annotated (Table S6). These putative genes were further compared with the differentially expressed genes (DEGs) obtained from gonadal RNA-Seq analysis. Seven overlapping genes were identified between the two datasets, including mis18bp1, rnf216, tbx1, mrc2, cubn, sirt4, and dyps (Table 2). These genes were distributed across five different chromosomes and showed no obvious clustering.
4. Discussion
Over the past few years, GWAS has become recognized as a powerful tool for developing sex-specific markers, owing to its high genome coverage, capacity to identify large sex-linked fragments, and ability to discover abundant genetic markers [15]. This strategy has been widely applied to the development of sex-specific molecular markers in many aquatic species [15,16,19,20,21,22,50]. In this study, we performed a GWAS for sex-related traits in B. purificata and detected 571 SNPs and 1853 InDels that were significantly associated with sex phenotype. Among the sex-associated SNPs and InDels, one sex-linked marker located at mis18bp1 gene was successfully screened out. Diagnostic consistency reached 100% across all three validation populations, demonstrating that this sex-specific marker enables accurate and efficient sex identification in B. purificata. This result provides a powerful tool for researchers and breeders to identify the sex of B. purificata more easily and accurately.
Sex-determining genes usually display two typical characteristics: (1) the presence of sex-specific genetic variations, such as SNPs and InDels with marked divergence between males and females; (2) distinct expression patterns between testes and ovaries, indicating their functional involvement in sex determination and differentiation [16]. Such differential features have been confirmed in several well-studied sex determination and differentiation genes, including dmrt1 in freshwater snail (Cipangopaludina chinensis) [51] and Pacific oyster (Crassostrea gigas) [52], foxl2 and dmrt1l in Yesso scallop (Patinopecten yessoensis) [53], and dmy in medaka (Oryzias latipes) [54]. Genes identified through the overlap of GWAS and sex-biased expression profiles are generally considered to have higher reliability than candidates derived from a single dataset. Combining GWAS and RNA-Seq analyses, we screened seven candidate genes harboring both sex-associated SNPs/InDels and sexually dimorphic expression patterns: mis18bp1, rnf216, tbx1, mrc2, cubn, sirt4, and dyps.
Among the seven candidate genes identified, tbx1 and rnf216 appear to participate in the regulatory framework involving testicular differentiation and spermatogenesis. Tbx1 belongs to the T-box family of transcription factors and plays crucial roles during embryogenesis and organogenesis [55]. Specially, TBX1 was originally identified as a “testis-specific T-box protein”, due to its abundant and tissue-specific expression in adult testes of mice and humans [56,57]. TBX1 was also proposed to participate in the regulation of testicular differentiation in aquatic organisms. In rainbow trout (Oncorhynchus mykiss), tbx1 showed significantly higher expression in the testis during sex differentiation, with distinct expression differences between testicular and ovarian development observed soon after hatching [58]. Further studies have indicated that tbx1 regulates testis development through interaction with the retinoic acid (RA) signaling pathway in rainbow trout (Oncorhynchus mykiss) [55].
Another candidate sex-differentiation gene identified in B. purificata is rnf216, which encodes an E3 ubiquitin ligase. This gene has been reported to be indispensable for spermatogenesis and male fertility in mammals [59]. In mice, rnf216 exhibited remarkably high expression in the testis. During postnatal testicular development, rnf216 transcripts were detectable as early as 1 week postpartum, and its expression level gradually increased from 2 weeks of age through adulthood. Rnf216-deficient mice displayed reduced testis size and extensive germ cell depletion. Further evidence indicated that germ cell degeneration in these mice initiated at 2 weeks of age and peaked at 3–4 weeks, corresponding to a critical window for spermatogenesis [59]. Notably, targeted disruption of rnf216 lead to male-specific infertility, with no significant fertility defects observed in females. Ablation of rnf216 impairs meiosis prophase I: rnf216^-/-^ spermatocytes were incompletely arrested at the zygotene stage and subsequently underwent apoptosis at approximately the pachytene stage. Mechanistic investigations further demonstrated that RNF216 sustains normal meiotic progression by mediating the degradation of protein kinase A (PKA) catalytic subunits [60]. Although the function of rnf216 remains poorly characterized in aquatic organisms, emerging evidence from RNF family members supports their conserved roles in sexual regulation. For instance, rnf183 was identified as a sex-determining gene in yellow croaker (Larimichthys polyactis) [61], and rnf144a has been proposed as candidate sex-determining gene in blotched snakehead (Channa maculata) [62] and Northern snakehead (C. argus) [15].
Mrc2 (C-type mannose receptor 2) encodes an endocytic collagen receptor that participates in extracellular matrix remodeling by mediating collagen internalization and subsequent lysosomal degradation [63]. In this study, two SNPs were identified within the mrc2 genomic region, and transcriptome analysis revealed that this gene exhibited markedly higher expression in the ovary than in the testis. The Wnt/β-catenin signaling pathway is known to be an evolutionarily conserved and ancient cascade responsible for ovarian determination in diverse vertebrate groups, such as mammals, amphibians, and teleost fishes [64]. According to recent evidence, mrc2 acts as an upstream regulator of the Wnt/β-catenin pathway [65]. Based on these observations, we infer that mrc2 may play an indirect role in the sexual regulation of B. purificata.
In addition, transcriptome analysis showed that tbx1 and rnf216 exhibited significantly higher expression in the testis than in the ovary, whereas mrc2 was markedly upregulated in ovarian tissue (Table S7). Based on these results, we propose that rnf216, tbx1 and mrc2 may represent novel candidate genes involved in sex differentiation in B. purificata. Nevertheless, further experimental verifications are required to investigate their functions and regulatory mechanisms during gonadal differentiation and development in B. purificata, such as via gene-editing assays and comprehensive gene expression profiling of gonads across key sex determination and differentiation stages [62].
Mis18bp1, which encodes MIS18 bonding protein 1 (MIS18BP1), is a core subunit of the MIS18 complex (MIS18α/MIS18β/MIS18BP1). In the present study, numerous SNPs and InDels were detected within the mis18bp1 locus, and transcriptome profiling showed that this gene was significantly upregulated in the testis compared with the ovary. However, no direct evidence currently supports the involvement of mis18bp1 in sex differentiation or gonadal development. Previous studies have documented that mis18bp1 exerts essential functions in mitotic progression and kinetochore protein assembly in eukaryotes, interacts with diverse protein partners, and primarily participates in mitosis and cell cycle regulation [66]. In addition, genes closely related to mis18bp1 are predominantly implicated in biological processes such as the cell cycle, chromosome segregation, and DNA repair [66]. On this basis, we hypothesize that mis18bp1 may contribute indirectly to sexual regulation in B. purificata. Further in-depth functional studies will be necessary to elucidate the precise role of mis18bp1 during sex differentiation and gonadal development in this species.
Accumulating evidence indicates that sex determination in freshwater snails is primarily governed by genetic factors, and to date, no environmental factors have been identified that influence their sex ratio. For instance, the sex of the apple snail Pomacea canaliculata is controlled by a limited set of nuclear sex-determining genes, rather than cytoplasmic factors (such as mtDNA) [67,68,69]. The genetic basis of sex determination in viviparid snails has also been supported by analyses of family sex ratios and karyotypes [70]. Karyotype investigations suggest that certain species (e.g., Viviparus ater, V. mamillatus and V. acerosus) possess a ZW/ZZ sex-determining system [71], while an XX/XY pattern has been documented in other species, including Viviparus subpurpureus and Tulotoma angulata [72,73]. In the present study, we found that B. purificata, a gonochoric species, may primarily follow an XX/XY sex-determination pattern. This speculation is based on the segregation patterns of sex-specific SNPs and InDels. For example, of the 571 significant sex-differentiated SNP loci, females were predominantly homozygous, whereas males were mostly heterozygous. Furthermore, the sex-specific InDel marker produced only one 82 bp band in females, whereas two bands (73 and 82 bp) were present in males on agarose electrophoresis gel, which is a typical pattern for an XX/XY sex-determining system. Nevertheless, it should be noted that minor genes potentially involved in sex determination may exist on other chromosomes. The main results obtained here were consistent with previous findings in closely related species [70]. These results will be helpful for further exploration of the sex-determination mechanism of B. purificata.
Integrating the DEGs from RNA-Seq and candidate genes from GWAS, we hypothesized the possible regulatory network of sex determination, differentiation, and gonadal development in B. purificata, as shown in Figure 8. Sry may act as a key regulator in the sex-determining pathway of B. purificata, given that it functions as the master switch for testicular development in vertebrates and exhibits strict testis-specific expression. Sry directly activates the expression of sox8, which has been proven to be an alternate testis-differentiating factor of sox9 in mice [74]. The sry/sox8 suppresses ovarian-promoting genes and activates many male-specific genes, such as dmrt1 and dmrt2, leading to the differentiation of Sertoli cells [75]. In the absence of sry, the Wnt/β-catenin signaling pathway was activated by rspo2/igr5 and mrc2, which suppresses sox8 expression, allowing granulosa cell differentiation, and in turn, ovarian differentiation. In addition, foxl2 and estrogen receptor (esr1) expression are required to actively repress sox8 and dmrt1/2 expression to maintain ovarian development [76]. Following gonadal differentiation, the distinct types and expression levels of hormones secreted by the testes (e.g., hsd11b2) and ovaries (e.g., folliculin) determine the development of most secondary sexual characteristics. Our findings, together with previous reports [77,78], suggest that sex determination and differentiation in B. purificata may share more similarities with those in other vertebrates than with those of teleost fishes. The working model and novel insights obtained in this study are expected to promote further research into the sex-determining pathways in freshwater gastropods and other mollusks.
5. Conclusions
Our results indicate that B. purificata potentially follows a primarily XX/XY sex-determination system, with sry, sox8, dmrt1 and dmrt2 being critical in male sex differentiation, while β-catenin, foxl2, esr1, and nr5a2 play important roles in female sex differentiation. In addition, mis18bp1, rnf216, tbx1, and mrc2 may be important sex-associated candidate genes. However, a noticeable constraint in this study is that our transcriptomic profiles were only obtained from mature gonads, which may result in the exclusion of genes functioning during early sex determination. A sex-linked InDel marker was successfully developed, which can distinguish males and females cost-effectively. Nevertheless, population-specific variation may exist in some populations. Future investigations should prioritize early gonadal stages, broader sampling, and validation, as well as functional validation via gene editing and expression profiling. Together, these results provide a useful foundation for elucidating the genetic mechanism of sex determination and for developing monosex stocks in B. purificata.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hou J. Zhang Q. Xiong B. Zou G. Studies on toxicological effects of cadmium in Bellamya purificata J. China Agric. Univ.2009145462
- 2Cao Z. Jiang X. The influence of environmental factors on Bellamya purificate J. Shanghai Fish. Univ.19987200205
- 3Yan Y. Jin W. Wen H. Ma X. Xue T. Sun C. He J. Bing X. Estimation of genetic parameters for growth traits of Bellamya purificata in 60 days Freshw. Fish.201848108111
- 4Qu M. Qu Y. Ren W. Wang Y. Wu Q. The mechanism of controlling microcystis bloom by Bellamya aeruginosa J. Fudan Univ.201049301308
- 5Zheng Z. Lv J. Lu K. Jin C. Zhu J. Liu X. The impact of snail (Bellamya aeruginosa) bioturbation on sediment characteristics and organic carbon fluxes in a eutrophic pond Clean-Soil Air Water 20113956657110.1002/clen.201000212 · doi ↗
- 6Ma B. Wang H. Jin W. Huang B. Li Y. Li Z. Wang M. Li C. Study on the water purification effect of the algae Elodea nuttallii and snail Bellamya purificata Jiangxi Fish. Sci. Technol.2002236
- 7Chen J. Song G. Wang X. She L. Wu S. Wu M. Ding F. The efficiency of water purification by different densities of Bellamya aeruginosa and Bellamya purificata J. Anhui Agric. Sci.2012401170811709
- 8Duan X. Xie C. Lv Y. Zhang N. Zhao F. Li R. Feeding habits of Bellamya purificata and its function in water purification system of ecological ditch Fish. Mod.2013401722