Gut Microbiota of Captive and Wild Siberian Cranes and Links to Soil in Poyang Lake Wetlands
Zheng Lai, Liting Xiao, Huilin Yang, Wenjing Yang, Qinghui You, Chaosheng Zhang, Minfei Jian

TL;DR
This study compares gut microbiota in wild and captive Siberian cranes and finds links to soil microbes in their wetland habitat.
Contribution
The study reveals distinct gut microbiota patterns in captive vs. wild cranes and identifies microbial similarities with their surrounding soil.
Findings
Captive cranes have richer and more even gut microbiota than wild cranes.
Soil and gut microbiota show partial overlap, with Firmicutes dominating gut communities.
Wild cranes and soil samples have higher levels of Escherichia-Shigella compared to captive cranes.
Abstract
The gut microbiota of captive and wild Siberian cranes (Leucogeranus leucogeranus) and their associations with soil microbiota in the Poyang Lake wetlands were analyzed and compared in this study. Captive cranes had richer and more even gut microbiota communities than their wild counterparts. While gut and soil microbiota were distinct overall, the gut communities of captive and wild cranes showed some similarity. Firmicutes was the dominant gut microbiota group. Notably, captive cranes had higher levels of certain microbiota like Ligilactobacillus, while wild cranes and soil samples showed higher levels of Escherichia-Shigella. Crucially, the analysis revealed compositional similarities between soil and gut samples. This knowledge can help inform better management and conservation strategies for the Siberian cranes. Gut microbiota are integral to host health and ecological adaptation,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1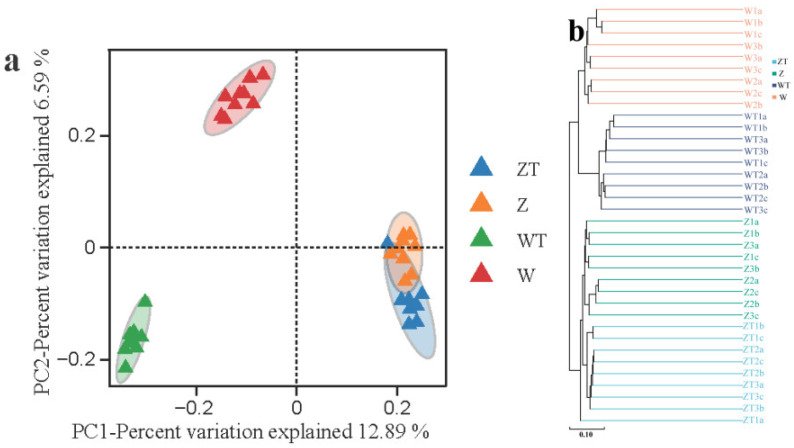 Figure 2
Figure 2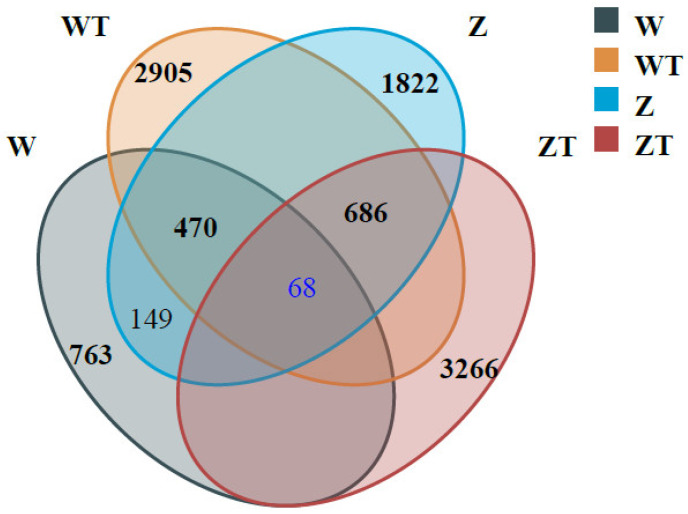 Figure 3
Figure 3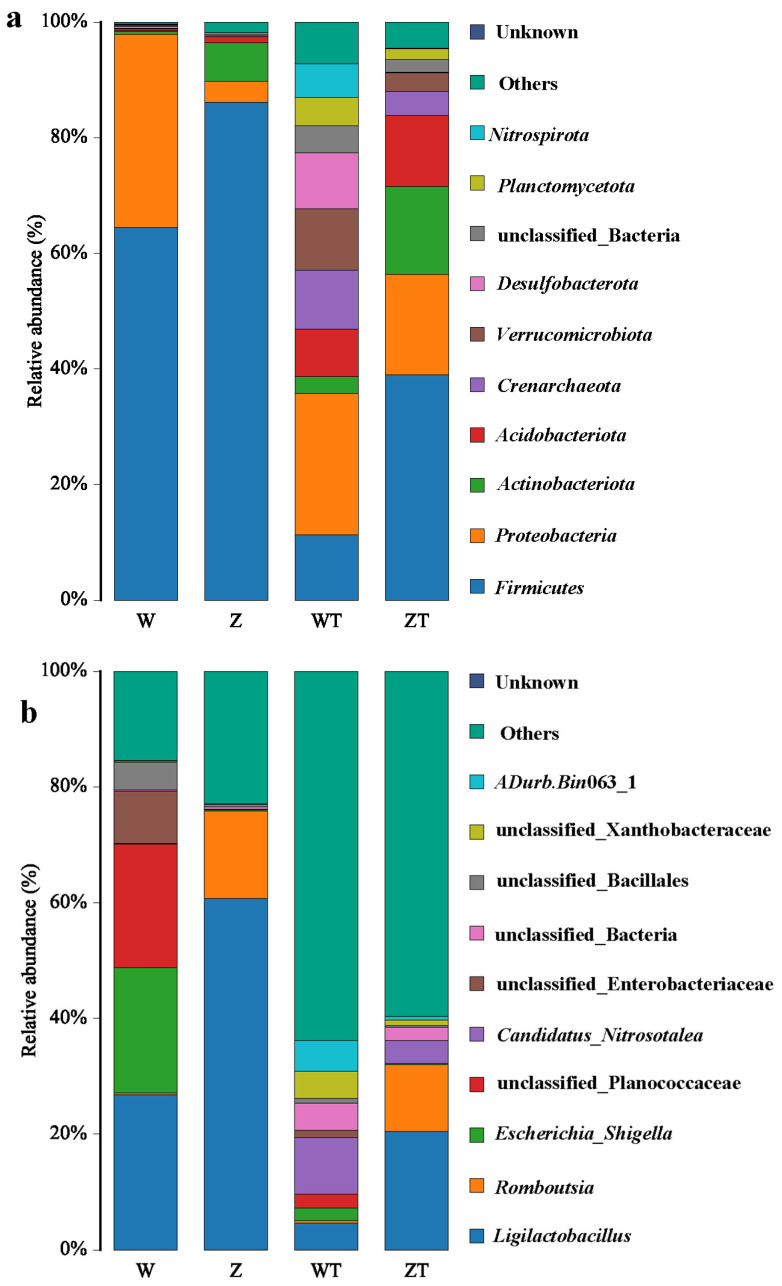 Figure 4
Figure 4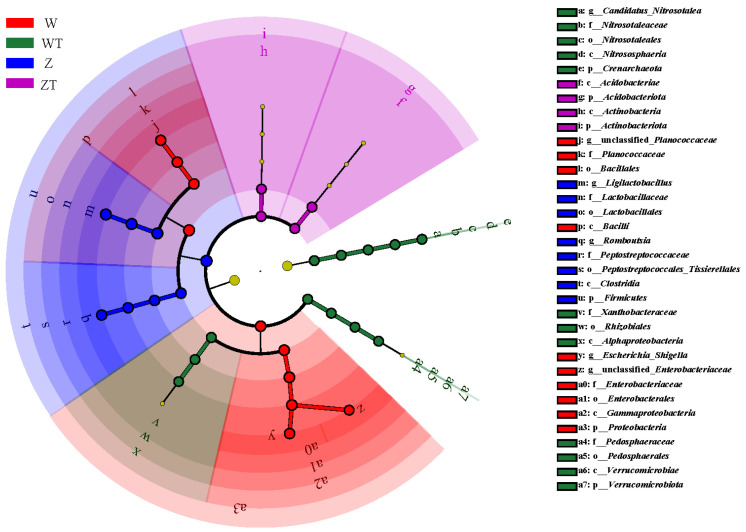 Figure 5
Figure 5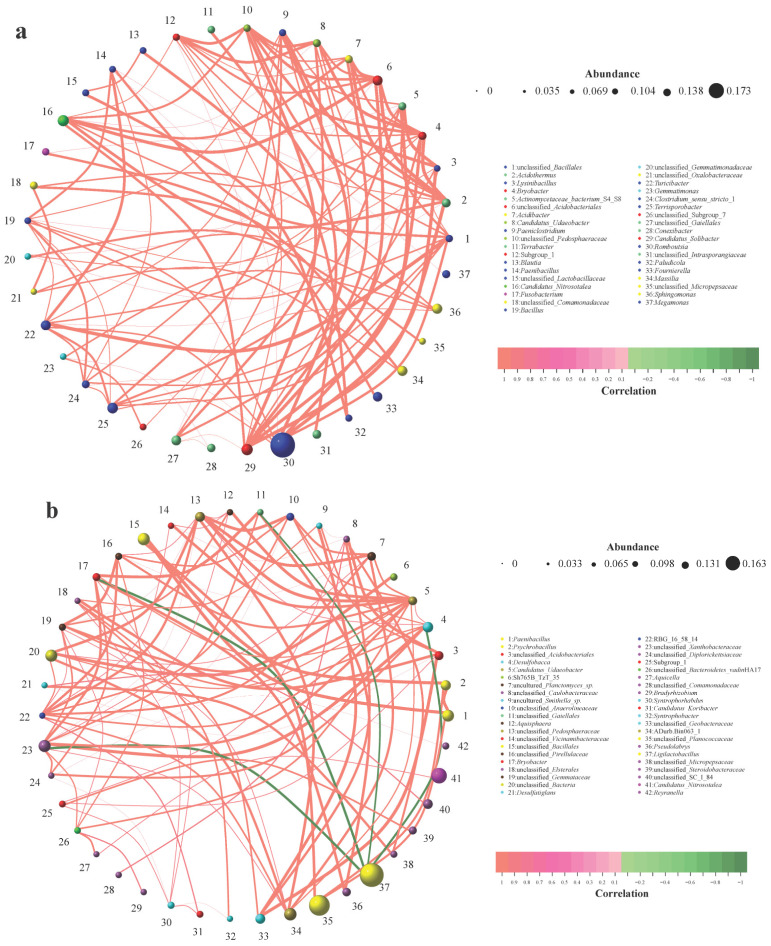 Figure 6
Figure 6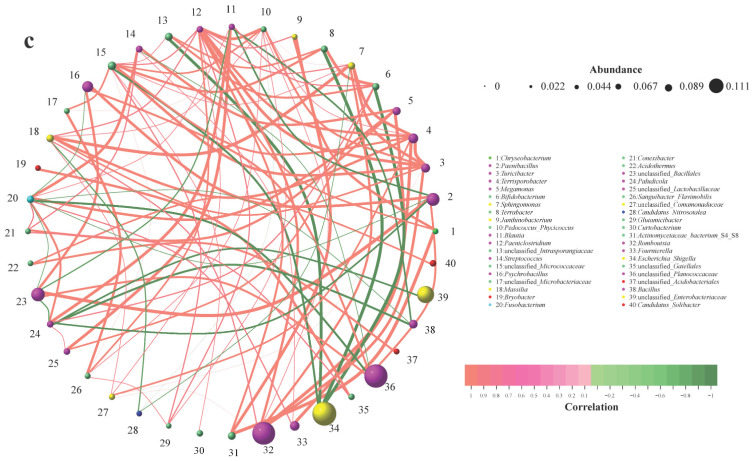 Figure 7
Figure 7- —National Natural Science Foundation of China
- —Jiangxi Provincial Natural Science Foundation Key Project
- —Jiangxi Provincial Natural Science Foundation
- —China Scholarship Council
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Aquaculture disease management and microbiota · Pharmaceutical and Antibiotic Environmental Impacts
1. Introduction
Gut microbiota play a vital role in host health and physiology, contributing to digestion, metabolism, immune homeostasis, and overall fitness [1,2]. Their composition and diversity are shaped by intrinsic (genetics, immunity, age) and extrinsic factors (diet, habitat, environmental microbiota) [3,4,5,6]. Understanding these microbial communities is crucial for assessing ecological adaptations and conservation strategies for endangered species [3]. Captive breeding is essential for species conservation but imposes significant environmental shifts that can alter gut microbiota [7,8]. Factors such as controlled diets, reduced microbial exposure, antibiotic use, and human interactions may lead to lower microbial diversity, increased opportunistic pathogens, and functional impairments in digestion and immunity [4,9]. While some studies suggest captivity may enhance immune status, its overall impact remains debated [10]. Thus, research on gut microbiota can improve survival of animals in different living environments [11]. Evaluating gut microbiota differences between captive and wild individuals is critical for optimizing conservation management.
The Siberian crane was classified as the critically endangered (CR) crane species by the International Union for Conservation of Nature (IUCN) and faces serious threats from habitat degradation and climate change [12]. Currently, the global population of Siberian cranes is estimated to be approximately 3500–4000 individuals. Among these, Poyang Lake wetlands serve as the most critical wintering habitat, supporting over 98% of the global population [13]. With Poyang Lake wetlands as its key wintering ground, conservation efforts have focused on captive breeding and reintroduction. However, microbiota shifts in captivity may influence health and adaptability, potentially affecting reintroduction success. Despite studies on Siberian crane diet, behavior, and gut microbiota composition [14,15,16,17], the role of environmental microbiota, particularly soil microbes, remains unclear.
As a vast microbial reservoir, soil plays a fundamental role in shaping gut microbiota through feeding, foraging, and excretion [18,19,20]. Microbial exchange between soil and gut communities can influence host digestion, metabolism, and immunity [21,22], yet this interaction remains understudied in avian conservation.
Our previous research [17] primarily characterized the compositional structure of the gut microbiota in wintering Siberian cranes. Building upon this foundation, the present study shifts the focus from a descriptive analysis to the exploratory investigation of the associations between the gut microbiota and the soil microbiota of the habitat. This study aims to: (1) characterize gut microbiota differences between captive and wild Siberian cranes for first time, (2) analyze soil microbiota composition in their respective habitats, and (3) explore gut–soil microbial associations to assess environmental influences on gut microbiota. This exploratory study aims to investigate whether differences exist in gut microbiota composition between captive and wild Siberian cranes and whether habitat conditions are associated with soil microbial community composition. Using 16S rRNA high-throughput sequencing, this study describes the gut and soil microbial communities associated with captive and wild Siberian cranes, offering a reference for future rewilding and conservation research.
To ensure the validity of comparisons between captive and wild populations, the sampling sites were carefully selected to minimize confounding environmental background interference. The sampling sites are located within the same climatic zone and share comparable geographical features. This geographical proximity and climatic consistency help to ensure that any observed differences in gut and soil microbiota between captive and wild cranes can be more reliably attributed to captivity-related factors rather than to large-scale climatic or geographical variations.
2. Materials and Methods
2.1. Study Area and Sample Collection
Poyang Lake (28°11′–29°51′ N, 115°49′–116°46′ E), located at the confluence of the middle and lower reaches of the Yangtze River, in northern Jiangxi Province, is the largest freshwater lake in China. The region benefits from abundant sunlight, with a multi-year average temperature ranging from 16.5 to 17.8 °C. It experiences hot, rainy summers and cold, dry winters, with an annual average precipitation of 1450–1550 mm. The well-developed hydrological network and rich food resources provide an optimal habitat for wintering waterfowl, particularly migratory species [13]. This study was conducted at two strategically selected sampling sites in proximity to Poyang Lake (Figure 1): the Siberian Crane Conservation Area (SCS) within the state-owned Nanchang Wuxing Reclamation Farm and the Poyang Lake National Wetland Park (NWP). The SCS (28°43′–28°48′ N, 116°11′–116°19′ E) is located in the eastern suburbs of Nanchang, adjacent to the eastern margin of Poyang Lake. This area serves as a critical conservation zone for the endangered Siberian Crane, providing a semi-natural habitat that mimics the ecological conditions of their natural wintering grounds. The NWP (28°56′–29°13′ N, 116°23′–116°44′ E), situated in Poyang County along the eastern shore of Poyang Lake, is a nationally designated wetland park that supports a diverse avian community. This site is particularly significant for its role in harboring multiple nationally protected bird species, including the Oriental White Stork (Ciconia boyciana), Tundra Swan (Cygnus columbianus), Hooded Crane (Grus monacha), and Red-breasted Goose (Branta ruficollis).
Both study sites are separated from the main body of Poyang Lake by a levee system, yet they remain in close proximity to the natural wintering habitats of Siberian cranes. These locations share similar climatic and geographical characteristics, including comparable temperature regimes, precipitation patterns, and hydrological dynamics, which facilitate comparative analyses of microbial communities across captive and wild crane populations as well as their interactions with the soil microbial community in the wetland ecosystem.
This study design enables a comprehensive investigation of the microbial ecology in both managed conservation areas and natural wetland habitats, providing valuable insights into the ecological relationships between captive and wild Siberian Cranes and their environment.
Wild Siberian cranes overwinter in Poyang Lake (28°22′–29°45′ N, 115°47′–116°45′ E) from late October to late March (mean residence: 150 ± 7 days). During this time, individuals inhabiting artificial wetlands, such as rice paddies or lotus fields, remain until local food resources are exhausted. Wild Siberian cranes are primarily fed tubers such as lotus roots or rice.
Captive Siberian cranes were housed together in a single outdoor enclosure measuring approximately 100 m^2^. The enclosure floor consisted of simulated wetland soil, constructed from a mixture of local soil, sand, and organic matter, designed to mimic the natural wetland substrate. The enclosure included a shallow artificial pond (approximately 1 m^2^, depth 20 cm) that provided access to water for wading and drinking. Water in the artificial pond was refreshed every 48 h using clean groundwater to maintain hygiene. No additional bedding materials were provided. The cranes were fed a standardized diet consisting of corn and carrots, which were provided three times daily (morning, noon, and afternoon). Food was placed on a designated raised feeding platform to minimize direct contact with soil and reduce potential contamination of food with environmental microbes. This consistent feeding schedule and diet composition were maintained throughout the study period to ensure uniformity. No antibiotics or other medications were administered to any of the captive or wild cranes during the sampling period, based on records provided by the breeding center and field observations. Routine but limited human contact occurs during daily care activities such as feeding and cleaning.
Feces and soil samples were collected from Siberian crane habitats at two locations: SCS and NWP, following their overwintering patterns. Sampling at SCS was conducted in 15 March 2022, while at NWP, it took place in 25 March 2022. At SCS, feeding hotspots were identified using binoculars, and three major crane flocks were selected for sampling. To minimize cross-species contamination, sampling was restricted to areas where no other bird species were present within a 50 m radius during feeding. Immediately after crane departure, feces and soil samples were collected from feeding sites, identified based on crane footprints and feeding pits. Three biological replicates of feces samples were collected from each of the three wild crane flocks, with three adjacent soil samples obtained within a 5 m radius per feces deposit. This yielded a total of 18 paired samples (9 feces + 9 soil). Due to the challenges of non-invasive sampling of wild endangered cranes, the fecal samples represent population-level snapshots from each flock, and we could not control for individual identity, sex, or age. Feces samples were designated as the W group (biological triplicates: W1a–c, W2a–c, W3a–c; n = 9), and their associated soil samples as the WT group (Paired triplicates: WT1a–c, WT2a–c, WT3a–c; n = 9). For captive Siberian cranes at the National Wetland Park (NWP), an identical paired sampling protocol was implemented to maintain methodological consistency. From three separate enclosures (biological replicates), triplicate feces samples were collected per enclosure and immediately matched with adjacent enclosure soil samples (≤5 m radius, 0–10 cm depth). Feces samples from captive cranes were labeled as the Z group (Z1a–c, Z2a–c, Z3a–c; n = 9 biologically independent samples), while corresponding enclosure soil samples were categorized as the ZT group (ZT1a–c, ZT2a–c, ZT3a–c; n = 9 spatially paired controls).
To ensure sample integrity and prevent contamination, fresh feces and soil samples were collected using sterilized gloves and forceps. Any material that had come into contact with the ground was removed before transferring the samples into 10 mL sterile EP tubes.
The collection process was conducted with minimal disturbance to the crane populations. feces aliquots were flash-frozen in liquid nitrogen within 15 min of collection, soils were sieved (2 mm mesh) and stored at −80 °C until DNA extraction. All procedures complied with IUCN guidelines for research on endangered species (Approval No.: 20220315-001)
2.2. DNA Extraction, Amplification and High-Throughput Sequencing
Genomic DNA was extracted from feces samples (n = 18) using the feces Microbial DNA Kit (D2700, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) with bead-beating lysis (0.1 mm zirconia beads, 6 m/s for 45 s) and from soil cores (n = 18) with the Foji Soil DNA Kit (DE-05514, Chengdu Foregene Biotechnology Co., Ltd., Chengdu, China) incorporating humic acid removal steps. The extraction procedures were strictly followed according to the manufacturer’s instructions for each kit. DNA integrity was verified by 1% agarose gel electrophoresis and stored at −20 °C for subsequent use.
The V4 hypervariable regions of bacterial 16S rRNA genes were amplified using barcoded primers 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACNVGGGTWTCTAAT-3′) with Phusion High-Fidelity DNA Polymerase ((Thermo Fisher Scientific Inc., Ltd., Waltham, MA, USA)) [23]. PCR conditions included: 95 °C for 4 min (initial denaturation); 32 cycles of: 95 °C/30 s, 55 °C/30 s (annealing), 72 °C/30 s; Final extension: 72 °C/10 min. Negative controls (n = 3 per batch) were processed alongside samples. Paired-end sequencing was performed on Illumina MiSeq PE250 platform at Biomarker Technologies (Beijing, China, CAP-accredited lab.).
2.3. Bioinformatics Processing and Ecological Statistical Analysis
Raw data quality control: Raw sequencing reads were quality-filtered using Trimmomatic-v0.33 (SLIDINGWINDOW:4:20, MINLEN:100). Adapter sequences were removed using cutadapt-v1.91.Amplicon Sequence Variant (ASV) Analysis: The high-resolution ASV analysis was performed to characterize microbial community composition with single-nucleotide precision. Cleaned sequencing reads were processed using the DADA2 pipeline (v1.16.0) implemented in QIIME2 (v2020.6) to generate exact biological sequence variants (ASVs) [24].Diversity Analysis: Alpha diversity indices (including observed ASVs, Chao1, ACE, Shannon, and Simpson) and species accumulation curves were calculated using R software (version 3.1.1) with the picante package (version 1.8.2) (Table 1). Beta diversity was assessed using weighted UniFrac distances to account for phylogenetic relatedness among microbial communities. Principal coordinate analysis (PCoA) and hierarchical clustering (UPGMA) were performed based on this distance matrix with significance testing via PERMANOVA (999 permutations) [25] to visualize community structure (Figure 2).Taxonomic Annotation and Biomarker Analysis: Taxonomic classification of amplicon sequences was performed using the SILVA database (version 138) with a confidence threshold of 85% [26]. Sequences were assigned to taxonomic ranks using the naïve Bayesian classifier implemented in QIIME2 [25]. To minimize potential misclassifications, only sequences with a confidence score above the predefined threshold were retained for downstream analyses. The community composition of each sample was statistically analyzed at various levels (phylum, class, order, family, genus, species). QIIME2 was used to generate abundance tables at different classification levels, and R was used to visualize the community structure of each sample at various taxonomic levels. Furthermore, biomarker taxa were identified using LEfSe (Kruskal–Wallis test, LDA score > 2, and FDR-adjusted p-values (using the Benjamini–Hochberg procedure) for multiple testing correction) [27].Cross-Domain Correlation and Network Analysis: Inter-domain microbial correlations were assessed using SparCC methods (Sparse Correlations for Compositional Data) with thresholds of 100 bootstrap iterations, 95% confidence intervals, |r| > 0.6 and p < 0.01. Network visualization was performed in Gephi, (v0.9.2) employing the Fruchterman-Reingold force-directed layout [28].
Raw reads with an average length of 276 bp were obtained after denoising by Illumina MiSeq sequencing. The total number of raw reads, clean reads, and the mean length of the reads were compared in all the 36 samples. The number of sequences varied from 25,391 to 170,149, and a total of 18,399 ASVs with 99% sequence similarity threshold. Detailed sequencing statistics for each individual sample are provided in Supplementary Table S1.
2.4. Nutritional Composition Data
Nutritional composition data for dietary items were obtained from the following sources: corn and carrot nutrient profiles from the USDA FoodData Central database (https://fdc.nal.usda.gov, accessed on 26 February 2026); lotus root and rice nutrient profiles from the China Food Composition Tables [29].
3. Results
3.1. Distinct Microbial Diversity, Composition, and Host-Environment Overlap Between Gut and Soil Microbiota
High-resolution alpha diversity metrics revealed systematic variations in microbial complexity between soil and gut compartments (p < 0.05, Table 1), with pronounced effects of captivity status. Soil microbiota exhibited consistently higher Ace, Chao, Simpson, and Shannon indices compared to the gut microbiota of Siberian cranes, indicating greater microbial richness and diversity in the soil environment. Among all groups, the WT (wild soil) group demonstrated the highest values for these indices, suggesting that the soil microbiota in the habitat of wild Siberian cranes harbors the most complex and diverse microbial communities.
In contrast, the gut microbiota of wild and captive Siberian cranes displayed distinct diversity patterns. Wild Siberian cranes exhibited significantly lower gut microbial richness and diversity compared to their captive counterparts, as reflected in reduced Ace, Chao, Simpson, and Shannon indices. However, an opposite trend was observed in the soil microbiota associated with their habitats, where soil from wild crane habitats showed higher diversity than that from captive environments. These findings suggest that captivity may influence gut microbial diversity while simultaneously reducing the complexity of the surrounding soil microbial communities.
PCoA based on weighted UniFrac distances demonstrated a clear separation between gut and soil microbiota across the four groups (R^2^ = 0.23, p = 0.001, PERMANOVA), with Axis 1 accounting for 12.89% of the variance (Figure 2a). Notably, the W group and their corresponding WT group exhibited the most pronounced separation, highlighting the greatest disparity in microbial community structures between these two groups.
Furthermore, hierarchical clustering analysis based on Bray–Curtis dissimilarity further supported this distinction, delineating four well-defined branches corresponding to the four groups (Figure 2b). All samples within the same group clustered together, forming distinct branches. The dendrogram showed that groups W and WT formed distinct major clusters from groups Z and ZT, reflecting the similarity between host gut and sympatric soil microbiota. Z group and W group formed two distinct subclusters, with a relatively large inter-cluster distance, indicating substantial divergence in gut microbial composition between captive and wild populations.
High-resolution ASV analysis demonstrated distinct microbial niche specialization across host and environmental compartments (Figure 3). Only 68 ASVs (0.7% of total detected) were shared among all four groups (gut: W/Z; soil: WT/ZT).
W group shared 470 ASVs (35.8% of their gut microbiota) with WT group. In contrast, Z group shared a higher number of ASVs with ZT group (686 ASVs), though this represented a lower proportion (26.8%) of their gut community due to their greater overall gut microbial richness. Soil compartments (WT/ZT) accounted for 70.5% of the total unique ASVs, with ZT group displaying the highest niche specialization (37.3% unique ASVs).
3.2. Phylum and Genus-Level Divergence in Gut and Soil Microbiota of Captive and Wild Siberian Cranes
Taxonomic profiling at the phylum level revealed a distinct host–environment dichotomy in microbial community composition (Figure 4a). In the gut samples (groups W and Z) and ZT group, the dominant phyla were Firmicutes, Proteobacteria, and Actinobacteriota, with relative abundances ranging from 38.97% to 86.08%, 3.68% to 33.34%, and 0.56% to 15.14%, respectively. In contrast, WT group was dominated by Proteobacteria (24.48%), Firmicutes (11.32%), and Verrucomicrobiota (10.64%).
Notably, the relative abundance of Firmicutes was substantially higher in both gut groups (W: 64.51%; Z: 86.08%) than in the soil groups (WT: 11.32%; ZT: 38.97%). Conversely, Acidobacteriota exhibited the opposite trend, being more prevalent in soils (WT: 8.24%; ZT: 12.33%) than in gut samples (W: 0.24%; Z: 1.07%) (p < 0.001).
Comparing the two gut groups, W group showed a significantly higher relative abundance of Proteobacteria (W: 33.34%; Z: 3.68%) but significantly lower abundances of Firmicutes (W: 64.51%; Z: 86.08%), Actinobacteriota (W: 0.56%; Z: 6.64%), and Acidobacteriota (W: 0.24%; Z: 1.07%) relative to Z group.
At the genus level, distinct microbial signatures distinguished captive from wild cranes (Figure 4b). The most abundant genera across all samples were Ligilactobacillus, Romboutsia, and Escherichia–Shigella. However, their distributions varied markedly among groups. Specifically, Ligilactobacillus was substantially enriched in Z group (60.71%) compared to W group (26.85%), WT group (4.62%), and ZT group (20.42%) (p < 0.001 for all). Similarly, Romboutsia showed higher abundance in Z group (15.15%) than in the other groups (W: 0.28%; WT: 0.48%; ZT: 11.59%; p < 0.001). In contrast, Escherichia–Shigella was significantly more abundant in W group (21.59%) than in Z group (0.22%) and both soil groups (WT: 2.12%; ZT: 0.17%) (p < 0.001).
In the soil groups (WT and ZT), a large proportion of sequences could not be assigned to known genera, with unidentified genera accounting for 63.83% and 59.63% of the relative abundance in WT and ZT, respectively, reflecting the high proportion of uncharacterized microbial diversity in environmental samples.
3.3. Differential Microbial Abundance in Gut and Soil Microbiota Revealed by LEfSe Analysis
LEfSe analysis (LDA score > 2, p < 0.01) revealed 34 phylogenetically conserved biomarker taxa across five taxonomic levels, distinguishing microbial communities in the gut and soil environments (Figure 5). At the phylum level, Firmicutes predominated in the gut microbiota, while Proteobacteria was the dominant phylum in the soil microbiota, highlighting ecosystem-specific microbial composition differences.
Comparative analysis of the gut microbiota of wild and captive Siberian cranes demonstrated significant variation in their microbiota profiles. W group exhibited 10 unique biomarkers, while Z group showed 8. The gut microbiota of wild cranes was characterized by higher relative abundances of Bacilli and Escherichia-Shigella, whereas captive cranes were enriched in Ligilactobacillus and Romboutsia.
In the soil, WT group was characterized by Candidatus Nitrosotalea, while ZT group showed enrichment in Actinobacteriota.
3.4. Network-Based Insights into Gut and Soil Microbiota Interactions in Siberian Cranes
From the microbial network relationship graph between captive Siberian crane and the soil of their habitat, it is observable that all the bacterial genera exhibit positive correlations (Figure 6a). Among them, the genera that have the most intimate interaction with other genera are unclassified_Bacillales, Acidothermus, unclassified_Acidobacteriales, unclassified_Pedosphaeraceae, Subgroup_1, Bacillus, Turicibacter, Candidatus_Solibacter and Romboutsia. These genera are concurrently significantly and positively correlated with 9 other genera (p < 0.01).
From the microbial network relationship graph between wild Siberian crane and the soil in the same area, it can be noted that all other genera are positively correlated, except for Ligilactobacillus and its associated genera, which present negative correlations (Figure 6b). Among them, the genus with the most intimate interaction with other genera is Candidatus_Udaeobacter, which is significantly and positively correlated with 13 other genera (p < 0.01). Additionally, unclassified_Xanthobacteraceae also has a close interaction with other genera and is significantly correlated with 12 other genera (p < 0.01). Furthermore, unclassified_Pedosphaeraceae14 is significantly correlated with 11 other genera (p < 0.01).
From the microbial network relationship graph between captive and wild Siberian crane, it can be discerned that there are both positive and negative correlations among the bacterial genera (Figure 6c). Among them, the genera with the most intimate interaction with other genera are Paeniclostridium and unclassified_Micrococcaceae. Paeniclostridium was significantly and positively correlated with 12 other genera (p < 0.01), while unclassified_Micrococcaceae was significantly and positively correlated with 11 other genera (p < 0.01) and significantly negatively correlated with one genus (p < 0.01). Moreover, Escherichia_Shigella is solely significantly and negatively correlated with other genera (p < 0.01).
4. Discussion
The significantly higher microbial diversity and richness observed in the soil of Siberian crane habitats compared to their intestinal microbiota underscore the distinct environmental conditions that govern microbial community assembly. The soil environment, characterized by aerobic and weakly alkaline conditions, contrasts sharply with the anaerobic and weakly acidic intestinal environment of the cranes [21]. These stark physicochemical differences impose strong selective pressures on microbial colonization, leading to distinct community structures in each habitat. Oxygen availability, pH gradients, and nutrient composition likely drive these ecological divergences, shaping microbial interactions and functional adaptations.
A key finding of this study is the higher gut microbial richness and diversity in captive Siberian cranes relative to their wild counterparts. The finding is consistent with patterns observed in red-crowned cranes (Grus japonensis) [30] and common cranes (Grus grus), suggesting that similar ecological changes associated with captivity may influence gut microbiota across crane taxa. However, given the limited sample size in each study, further research with larger sample sizes is needed to confirm this pattern. The finding aligns with the Dietary Filtering Hypothesis [31,32]. Standardized captive diets may relax niche competition, supporting greater microbial richness compared to specialized wild foraging. Captive cranes were fed corn (~64% starch, ~74% carbohydrates) and carrots (~4.7% sugars, ~2.8% fiber) three times daily. This combination provides diverse carbohydrate substrates (resistant starch, cellulose, hemicellulose, pectins), creating multiple metabolic niches that support broader microbial taxa [33]. Consistent food availability may also reduce competitive exclusion [32]. In contrast, wild cranes forage on lotus roots (moderate starch: ~11%; fiber: ~4.9%) and rice stubble. While rice grains are starch-dense (~77%), the cellulose-rich rice straw (35–40% cellulose) dominates their intake, presenting a structurally complex but compositionally narrower substrate. Cellulose digestion requires specialized cellulolytic bacteria [34], potentially constraining diversity by favoring fiber-degrading specialists. Seasonal and spatial heterogeneity of wild resources may further filter microbial communities [35].
The degree of similarity between the gut microbiota of Siberian cranes and their habitat soil underscores potential microbial exchange. Specifically, 26.77% of total gut ASVs in captive cranes and 35.80% in wild cranes overlapped with their respective soil microbiota. In contrast, only 16.08% of gut ASVs in adult black-billed gulls (Larus saundersi) were shared with soil [36], indicating stronger microbial associations between Siberian cranes and their habitat. This disparity may be attributed to foraging behavior—Siberian cranes feed by excavating plant roots and stems in shallow waters, increasing direct soil contact, whereas black-billed gulls primarily consume aquatic prey, limiting soil interaction. These findings align with the habitat filtering hypothesis, which posits that environmental constraints shape microbial community assembly [37]. The minimal ASV overlap (149) between wild and captive cranes suggests strong host-environment physiological filters, such as gut pH, bile acids, and immune responses, which regulate microbial colonization and create a barrier effect [38]. However, the greater ASV sharing between captive cranes and their habitat soil (Z-ZT = 686) compared to wild cranes and their soil (W-WT = 470) suggests that captivity may weaken microbial transmission barriers. This could result from altered diets and reduced exposure to diverse environmental microbial sources, leading to a captivity gradient [39]. The high number of unique ASVs in captive soil (ZT = 3266) likely reflects anthropogenic selection pressures, including fertilizer use and reduced plant diversity, which reshape soil microbial composition. These changes may indirectly influence gut microbiota diversity in captive birds by modifying environmental microbial pools [40].
Our findings contribute to the growing evidence that avian gut microbiota assembly is shaped by factors across multiple spatial and temporal scales. While our study highlights broad contrasts between captive and wild environments, recent research emphasizes that fine-scale variation within populations can be equally substantial. For instance, a study showed that microhabitat heterogeneity structures microbiota in European pied flycatchers at a local scale [41]. Similarly, studies on finches have demonstrated strong host-specific signatures in finches even under controlled conditions, underscoring host identity as a key variable [42], while others have revealed that seasonal effects interact with environmental and individual-level factors rather than acting alone [43]. Most directly relevant, foraging environment shapes gut microbiota in cranes within a single wetland system [44], corroborating our observation that local habitat conditions, such as soil microbial communities, may contribute to microbial assembly. Collectively, these studies suggest that the patterns we observed arise from a complex interplay between captivity, seasonal migration, and fine-scale habitat variation, rather than any single factor in isolation. This interpretation, however, remains speculative and requires direct experimental testing.
This study identified Firmicutes, Proteobacteria, and Actinobacteriota as the dominant bacterial phyla in the intestinal microbiota of both captive and wild Siberian cranes, consistent with findings from wintering Siberian cranes in Poyang Lake [17]. Among these, Firmicutes was the most prevalent, which aligns with studies on the gut microbiota of other crane species, such as the white-naped crane (Grus vipio) [45] and the red-crowned crane (Grus japonensis) [46]. Firmicutes play a crucial role in the breakdown of complex carbohydrates, fatty acids, and polysaccharides in the gut, facilitating host energy metabolism [34]. Notably, the relative abundance of Firmicutes in wild Siberian cranes (W group) was 86.08%, significantly higher than in captive Siberian cranes (Z group) at 64.51%. This difference may be attributed to the higher sugar content in the natural diet of wild cranes, which could promote Firmicutes proliferation. Additionally, captive conditions may lead to a relaxation of environmental microbial filters, reducing the number of discriminatory taxa by approximately 67% compared to wild-type soil microbiota. This reduction could weaken microbial transmission barriers and homogenize gut microbial niches in captivity [47].
At the genus level, Ligilactobacillus and Romboutsia exhibited the highest relative abundances in captive Siberian cranes, whereas Escherichia-Shigella was most abundant in wild individuals. LEfSe analysis further confirmed that Ligilactobacillus, and Romboutsia were significantly enriched in captive Siberian cranes. Ligilactobacillus is known to ferment sugars and produce lactic acid [48], its enrichment in captive cranes may reflect dietary starch and sugar content, though functional consequences for host health require direct investigation. Similarly, Romboutsia plays a key role in host well-being, with its higher abundance potentially enhancing survival [49]. In contrast, Escherichia-Shigella was more abundant in wild cranes. While some members of this genus can be pathogenic, this pattern may reflect environmental exposure rather than disease risk [50].
We acknowledge several limitations. The small sample size of cranes (n = 3) limits statistical power and generalizability. The cross-sectional design captures only a snapshot, failing to reflect temporal dynamics. 16S rRNA sequencing provides taxonomic profiles without functional verification, and uncontrolled host variables (age, sex) may confound comparisons. LEfSe can be unstable in small or sparse datasets and may inflate Type I error rates [51]. Future research should employ metagenomic/metabolomic analyses to elucidate microbial functions, longitudinal monitoring across overwintering periods to capture dynamic changes, and larger-scale individual-level sampling to account for host heterogeneity. Addressing these limitations will deepen our understanding of captivity-induced microbiota shifts and their implications for crane conservation.
5. Conclusions
This study provides a comparative analysis of the gut microbiota of captive and wild Siberian cranes and their associations with soil microbial communities in the Poyang Lake Wetlands. By integrating compositional profiling with co-occurrence network analysis, we observed distinct microbial community structures between captive and wild individuals, as well as between gut and soil environments. Notably, we observed a distinct microbial network structure in wild Siberian cranes, in contrast to the associations observed in captivity. A key finding is the significant negative correlation between the gen and the gut microbiota of wild cranes, which is an interaction absent in captive environments. Additionally, the pattern of exclusively positive correlations between captive cranes and their soil microbiota differs from the assumption that captivity universally disrupts host-microbiota interactions. Instead, our results suggest that captivity may be associated with changes in microbial interactions and gut microbiota functions. However, due to the limited sample size, these findings should be interpreted as preliminary and require further validation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Liu H. Liao C. Wu L. Tang J. Chen J. Lei C. Zheng L. Zhang C. Liu Y. Xavier J. Ecological dynamics of the gut microbiome in response to dietary fiber ISME J.2022162040205510.1038/s 41396-022-01253-435597888 PMC 9296629 · doi ↗ · pubmed ↗
- 2Lindsay E.C. Metcalfe N.B. Llewellyn M.S. The potential role of the gut microbiota in shaping host energetics and metabolic rate J. Anim. Ecol.2020892415242610.1111/1365-2656.1332732858775 · doi ↗ · pubmed ↗
- 3Sun F. Chen J. Liu K. Tang M. Yang Y. The avian gut microbiota: Diversity, influencing factors, and future directions Front. Microbiol.20221393427210.3389/fmicb.2022.93427235992664 PMC 9389168 · doi ↗ · pubmed ↗
- 4Li C. Liu Y. Gong M. Zheng C. Zhang C. Li H. Wen W. Wang Y. Liu G. Diet-induced microbiome shifts of sympatric overwintering birds Appl. Microbiol. Biotechnol.20211055993600510.1007/s 00253-021-11448-y 34272578 · doi ↗ · pubmed ↗
- 5Li L. Ye J. Yu M. Jiang J. Guo X. Yu W. Rong K. Dynamic changes in the avian gut microbiome in response to diverse lifestyles Appl. Microbiol. Biotechnol.202516733134410.1111/ibi.13388 · doi ↗
- 6Hill S.C. François S. ThézéJ. Smith A.L. Simmonds P. Perrins C.M. van der Hoek L. Pybus O.G. Impact of host age on viral and bacterial communities in a waterbird population ISME J.20231721522610.1038/s 41396-022-01334-436319706 PMC 9860062 · doi ↗ · pubmed ↗
- 7Eliades S.J. Brown J.C. Colston T.J. Fisher R.N. Niukula J.B. Gray K. Siler C.D. Gut microbial ecology of the critically endangered Fijian crested iguana (Brachylophus vitiensis): Effects of captivity status and host reintroduction on endogenous microbiomes Ecol. Evol.2021114731474310.1002/ece 3.737333976843 PMC 8093715 · doi ↗ · pubmed ↗
- 8Tang S. Li Y. Huang C. Yan S. Li Y. Chen Z. Wu Z. Comparison of gut microbiota diversity between captive and wild tokay gecko (Gekko gecko)Front. Microbiol.20221389792310.3389/fmicb.2022.89792335783386 PMC 9248866 · doi ↗ · pubmed ↗