State-dependent promoter switching mediates integrase gene transcription in the integrative and conjugative element ICEKp1 of Klebsiella pneumoniae
Guitian Liu, Zaizhong Huang, Jiahao Guan, Cui Tai, Lilu Shi, Yanjie Chao, Ruilan Wang, Ke Wang, Hong-Yu Ou

TL;DR
This paper reveals how the ICEKp1 element in Klebsiella pneumoniae uses promoter switching to regulate integrase gene transcription, favoring integration over excision.
Contribution
The study identifies a novel state-dependent promoter switching mechanism in ICEKp1 that influences integrase gene transcription and integration preference.
Findings
Integrated ICEKp1 uses the host tRNA-asn promoter for int transcription, while excised ICEKp1 uses a 3′-end promoter.
The int 5′-UTR stem-loops act as a terminator, reducing full-length transcripts during integration.
Promoter switching is conserved in ICEclc and ICEEc1, indicating evolutionary conservation of this regulatory strategy.
Abstract
Integrative and conjugative elements (ICEs) are mobile genetic elements that widely disseminate adaptive traits among bacterial populations. Despite their prevalence, the regulatory mechanisms orchestrating ICE integration and excision remain poorly understood. Here, we investigate the transcriptional regulation of the integrase gene (int) in ICEKp1 of Klebsiella pneumoniae. Our results demonstrate that the integrated ICEKp1 exploits the adjacent host transfer RNA-asn promoter to drive int transcription, whereas the excised ICEKp1 switches to a 3′-end promoter. Remarkably, the messenger RNA stem-loops within the int 5′-UTR are organized as a potential terminator, attenuating the total amount of the full-length asn-int transcripts during ICEKp1’s integration. Ligation of the 3′ end and 5′ end of ICEKp1 maintains the activity of the 3′-end promoter for the int transcription in the excised…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
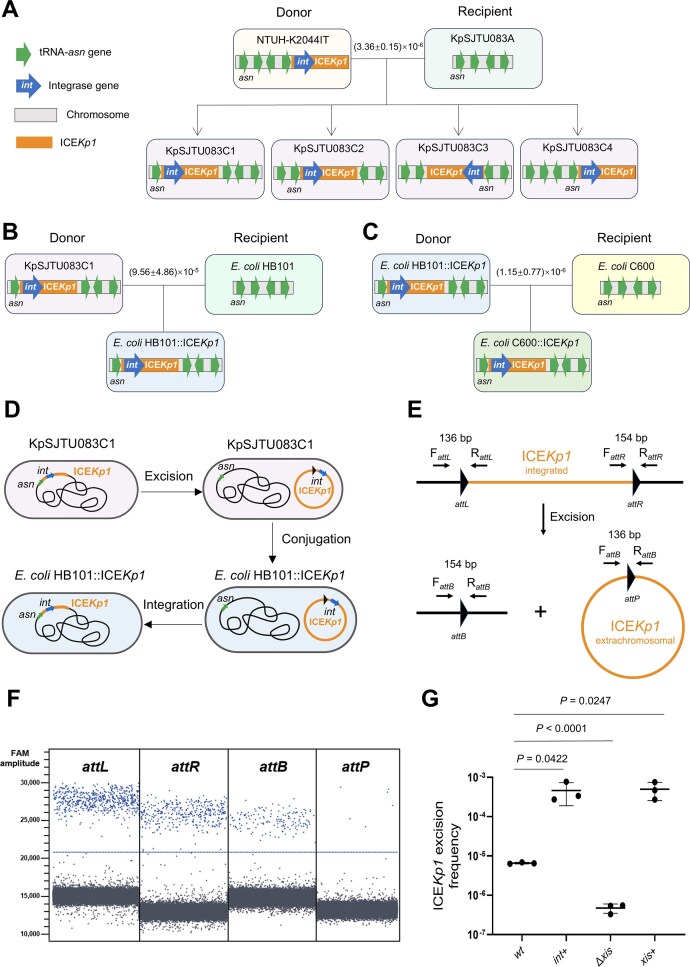 Figure 1
Figure 1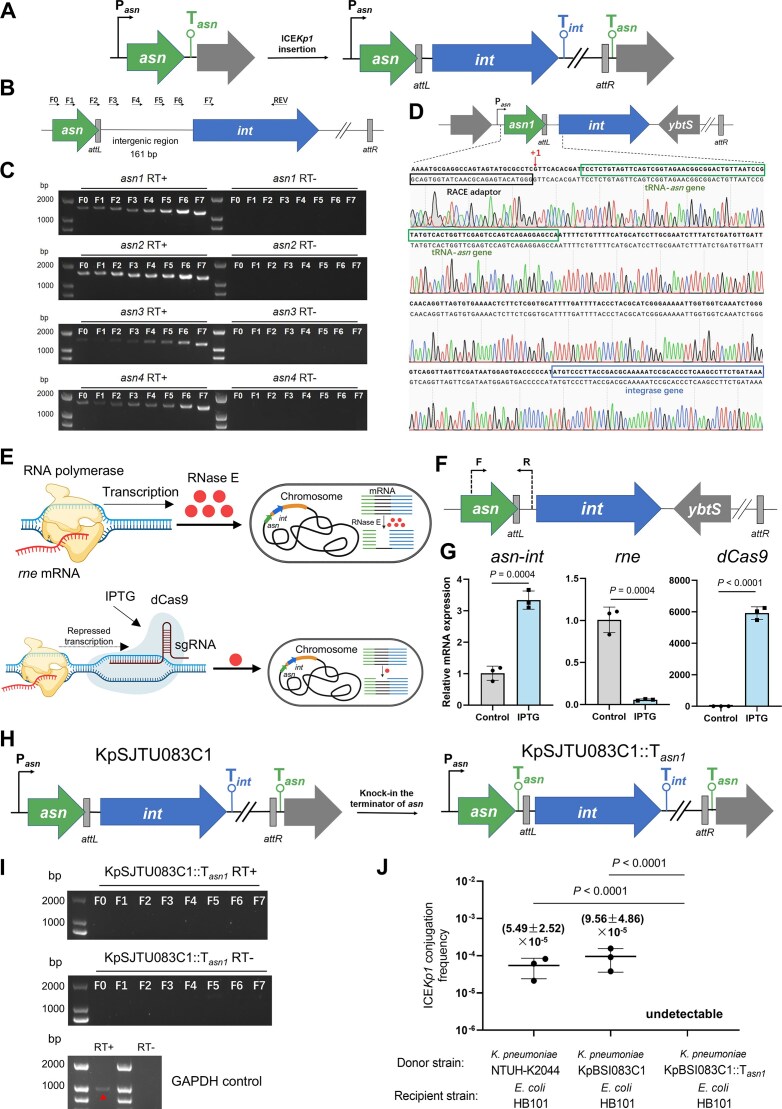 Figure 2
Figure 2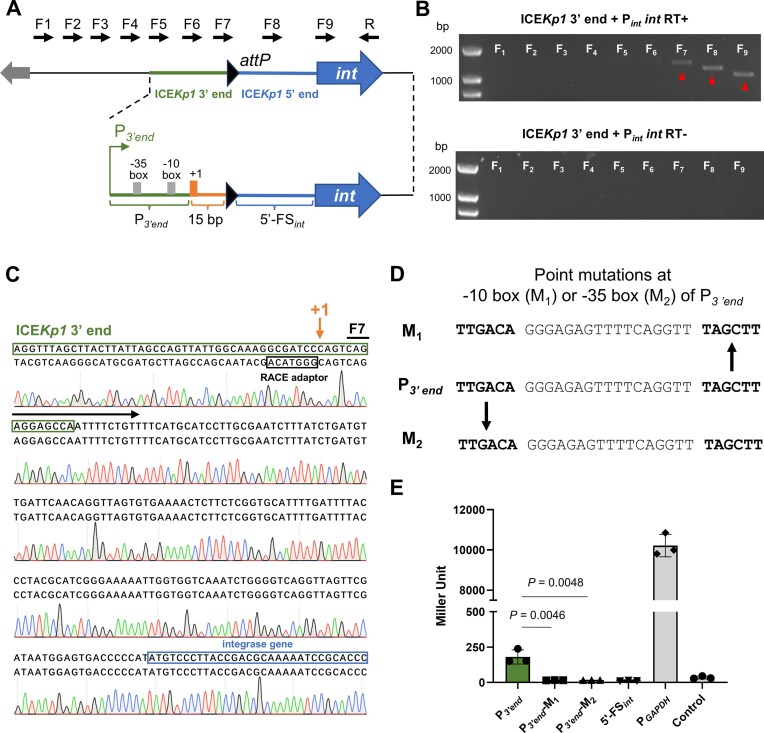 Figure 3
Figure 3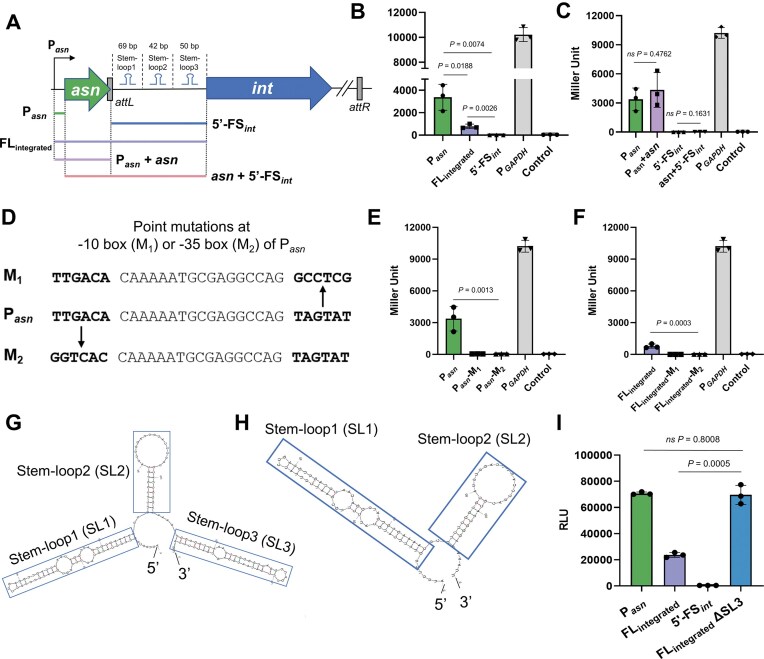 Figure 4
Figure 4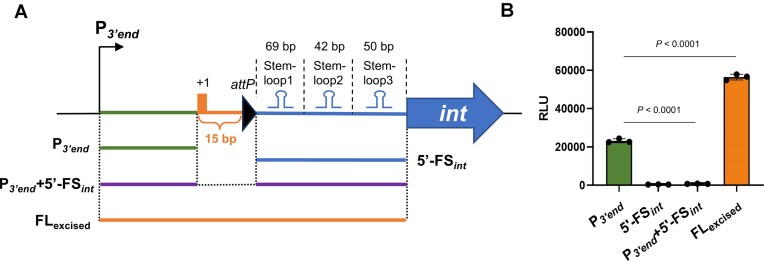 Figure 5
Figure 5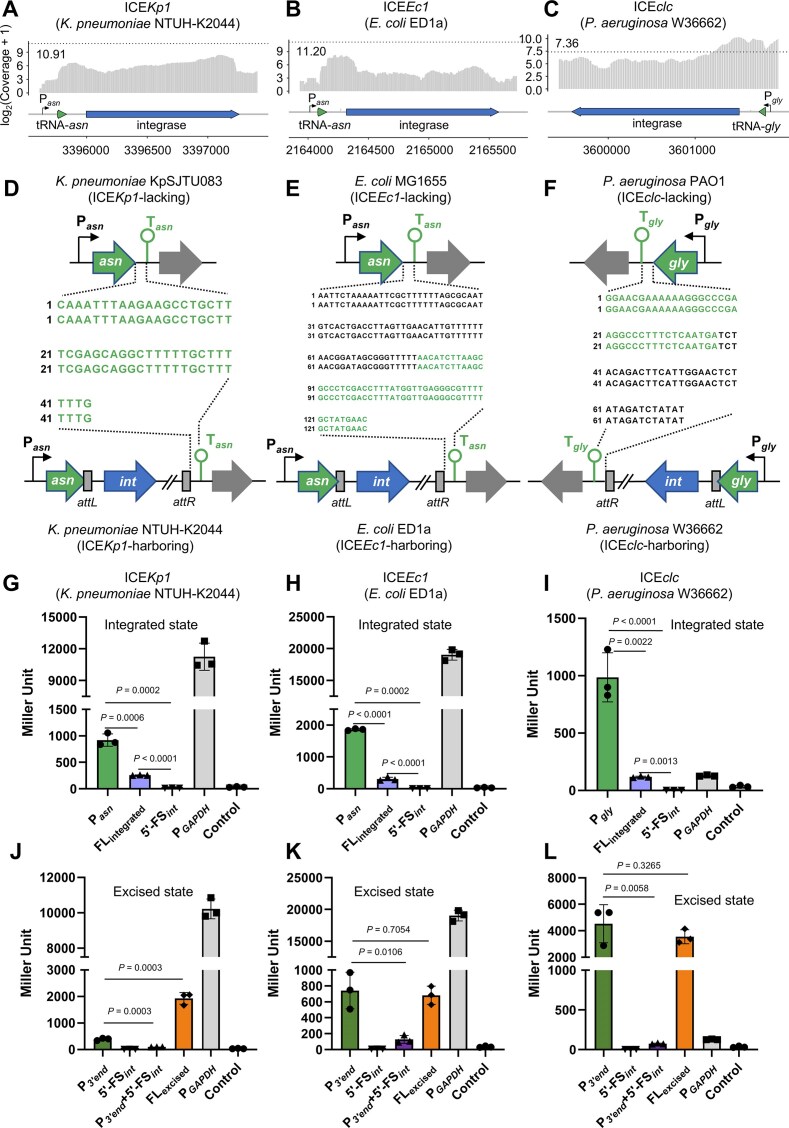 Figure 6
Figure 6- —Core Facilities and Technical Service Center at School of Life Sciences and Biotechnology
- —Shanghai Jiao Tong University10.13039/501100004921
- —National Key Research and Development Program of China10.13039/501100012166
- —National Natural Science Foundation of China10.13039/501100001809
- —Shanghai Jiao Tong University Cross-disciplinary Research Fund in Medicine and Engineering Key Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Antibiotic Resistance in Bacteria · Metalloenzymes and iron-sulfur proteins
Introduction
Integrative and conjugative elements (ICEs) are mobile genetic elements crucial for bacterial adaptation and evolution [1]. Over 2000 ICEs have been identified in completely sequenced bacterial chromosomes, with >1000 predicted in human microbiome samples [2]. These self-transferable elements often carry cargo genes conferring antibiotic resistance, virulence traits, and other adaptive functions that enhance host fitness [1, 3]. The interactions between ICEs and host bacteria, particularly regarding transfer regulation, require further investigation [4].
ICEs typically reside in an integrated state within the host chromosome [5, 6]. Upon activation, ICEs can excise, conjugate, and reintegrate [5]. However, excision and conjugation pose risks of ICE loss [5, 7]. Multiple stabilization mechanisms have been identified, including transient replication increasing copy number [8], plasmid-like partitioning systems [9], toxin–antitoxin systems [10], and inhibition of recipient restriction-modification systems [11]. The excised state is rare; for ICEclc, only 3%–5% of cells become transfer-competent [12], requiring nutrient induction [7]. Importantly, ICE excision and conjugation imposes cellular burdens; e.g., the premature integration of ICEBs1 reduces recipient viability [13].
The integration and excision of ICE is mediated by ICE-coding integrase and accessory factors (e.g., excisionase). Tight regulation maintains the low-level expression of the integrase gene (int) in integrated ICEs, involving ICE-coding specific regulators in ICEBs1 [14], ICESXT [15], and Tn916 [16]. Notably, distinct promoter modalities regulate int transcription in the integrated versus excised ICEs. For instance, in Yersinia pestis HPI, the int transcription start site (TSS) lies upstream of the integration site transfer RNA (tRNA)-asn [17], while excised ICEclc utilizes a 3′-terminal promoter [18]. Further investigation of these int transcriptional mechanisms is warranted.
The clinically significant pathogen Klebsiella pneumoniae (including multidrug-resistant and hypervirulent strains) harbors ICEKp1, a 76-kb ICE originally identified in hypervirulent strain NTUH-K2044 [19] (Supplementary Fig. S1G). It encodes important virulence factors, such as yersiniabactin and salmochelin [20]. Its 5′ region shares high similarity with the non-transmissible Y. pestis HPI (lacking a type IV secretion system). Given its low-frequency transmission [19], understanding ICEKp1’s integrase gene regulation is particularly important.
In this study, we explored the relationship between the integration tendency of ICEKp1 and the transcriptional regulation of the integrase gene. We found that the integrated ICEKp1 utilizes the adjacent host tRNA-asn promoter (Pasn) for int transcription, whereas the excised ICEKp1 switches to its’ 3′ terminal promoter (P3’end). This mechanism reinforces the integration preference of ICEKp1, a conserved feature also observed in ICEEc1 and ICEclc. These results may advance understanding of ICE regulatory strategies and their implications for bacterial genome plasticity.
Materials and methods
Strains, plasmids, and primers
All strains and plasmids used in this study are listed in Supplementary Table S1. All primers used in this study are listed in Supplementary Table S2. All deletion mutants were generated by lambda red recombination using the vector pKOBEG-Apra as previously described [21]. All strains used in this study were cultured at 37°C in lysogeny broth (LB) medium (pH = 7.2–7.4) with appropriate antibiotics.
Complete genome sequencing and assembly
The genomic DNA of K. pneumoniae KpSJTU083 and the four transconjugants that inserted ICEKp1 (KpSJTU083C1–KpSJTU083C4) was extracted and sequenced by using the combination of the 2 × 150 bp paired-end Illumina NovaSeq platform and the Oxford Nanopore MinION platform. The resulting filtered reads were then assembled by using Unicycler (v0.5.0) [22] and Flye (v2.9.1-b1781) [23]. Comparative analysis of the genome sequence was performed by using Proksee [24].
Growth curves and ICEKp1 stability determination
Growth curves of NTUH-K2044, NTUH-K2044IT, KpSJTU083, KpSJTU083A, KpSJTU083C1-C4, Escherichia coli HB101, E. coli HB101::ICEKp1, E. coli C600, and E. coli C600::ICEKp1 were determined with a Bioscreen C machine. The strains were grown in LB broth at 37°C for 24 h, and the OD_600_ was read automatically every 30 min.
The stability of ICEKp1 was determined in NTUH-K2044IT, KpSJTU083C1–C4, E. coli HB101::ICEKp1, and E. coli C600::ICEKp1 as described previously [25], with some modifications. The strains were cultured until the logarithmic growth phase, after which single colonies were selected and subjected to continuous relaxed cultivation in liquid LB medium without antibiotics for 7 days. Subculturing was performed every 24 h. Ten single colonies were selected each time and tested for ICEKp1 loss using replica plating on LB agar plates containing 200 µg/ml hygromycin, and on antibiotic-free LB agar plates. For colonies that failed to grow on the hygromycin plates (ICEKp1 negative), further confirmation was conducted via polymerase chain reaction (PCR) analysis.
Quantitative real-time PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen). Then, genomic DNA (gDNA) was removed and complementary DNA (cDNA) was produced by Evo M-MLV RT Kit with gDNA Clean for qPCR (Accurate Biology, Cat. No.AG11711). Finally, qPCR was performed using the qTOWER^3^G Touch Real-Time PCR System (Analytik Jena), and gapA was used as an internal control. The messenger RNA (mRNA) abundance level was calculated by the 2^−ΔΔCT^ method.
Knockdown the RNase E gene by using the CRISPRi system
Utilizing a CRISPR interference (CRISPRi) system under the control of a lac operator [26, 27], we knocked down the expression of the RNase E gene (rne) in KpSJTU083C1-C4 (Fig. 2E–G and Supplementary Fig. S5). One millimolar isopropyl-3-D-thiogalactopyranoside (IPTG) induces system expression to block target gene transcription via protein–RNA–DNA complex formation, achieving gene knockdown; target genes are transcribed normally without IPTG. Strains used included E. coli WM3064 [DAP auxotrophic, harboring RP4 (tra)], WM3064 derivatives with pJMP2844 (single guide RNA (sgRNA) cloning, a gift from Jason Peters; Addgene plasmid #160675; http://n2t.net/addgene:160675; RRID: Addgene_160 675) [27] or pJMP1039 (helper plasmid, a gift from Carol Gross & Jason Peters & Oren Rosenberg; Addgene plasmid #119239; http://n2t.net/addgene:119239; RRID: Addgene_119239) [26], and KpSJTU083C1-C4.
Target-specific spacers were generated by annealing (37°C for 30 min, 95°C for 3 min, slow cooling) with T4 PNK, T4 PNK buffer, Spacer-F/R, and nuclease-free water. pJMP2844 was digested with BsaI (37°C for 90 min) and ligated with diluted spacers (room temperature, 90–120 min). Ligation products were transformed into WM3064 competent cells (ice incubation, 42°C heat shock, DAP-supplemented LB recovery), followed by plating, colony sequencing, and verification.
Conjugation was performed by activating donor (pJMP2844-spacer WM3064), helper (pJMP1039 WM3064), and recipient strains, culturing to OD_600 _= 0.5, mixing equally, centrifuging, resuspending, and spotting on DAP-supplemented LB plates (30°C, overnight). Bacterial lawns were resuspended, diluted, and plated on chloramphenicol-resistant plates (no DAP). Positive transconjugants were confirmed by PCR for sgRNA insertion and preserved.
We designed specific primers for the junction sequence of asn-int (Fig. 2F) as well as argX-hisR, a previously documented co-transcript [28] used as a control (Supplementary Fig. S5A). The cleavage of RNase E was quantified by measuring the mRNA abundance of the junction sequence. After IPTG induction, the mRNA level of the rne-targeting dCas9 was increased (Fig. 2G). In contrast, the mRNA abundance of the rne was decreased. The mRNA abundance of the asn-int and argX-hisR co-transcript increased (Fig. 2G and Supplementary Fig. S5B). This result confirmed the functionality of the rne-knockdown CRISPRi system in KpSJTU083C1-C4.
β-galactosidase assay
The β-galactosidase activity was quantified based on the hydrolysis of O-nitrobenzene-β-D-galactopyranoside (ONPG). Bacteria were subcultured in LB broth to logarithmic phase, then cells were pelleted and resuspended in an equal amount of Z buffer (0.06 M Na_2_HPO_4_, 0.04 M NaH_2_PO_4_, 0.01 M KCl, 0.001 M MgSO_4_, 0.05 M β-mercaptoethanol). OD_600_ was measured blank against the Z buffer. One milliliter cell was permeabilized by adding 100 μl chloroform and 50 μl 0.1% sodium dodecyl sulfate and equilibrated for 5 min in a 28°C water bath. The reaction was started by adding 0.2 ml ONPG (4 mg/ml) at 28°C and was stopped after a sufficient yellow color had developed by adding 0.5 ml 1 M Na_2_CO_3_. The reaction time (T) was recorded precisely with a timer. Then the mixture was spined 5 min at maximum to remove debris and chloroform. OD_420_ and OD_550_ of the supernatant were measured. Units of activity were calculated with the formula as follows:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{upgreek} \usepackage{mathrsfs} \setlength{\oddsidemargin}{-69pt} \begin{document} \begin{eqnarray*} \mathrm{Miller}\ \mathrm{Units} = 1000\ \times {\mathrm{\ }}\frac{{(\mathrm{ O}{{\mathrm{ D}}_{420}} - \mathrm{ O}{{\mathrm{ D}}_{\mathrm{ 550}}}){\mathrm{\ }} \times {\mathrm{\ }}1.75}}{{T\ \times \ \mathrm{ O}{{\mathrm{ D}}_{600}}}}. \end{eqnarray*}\end{document}Luciferase assay
The luciferase assay was employed for promoter activity determination. Prepare a 6× luciferase solution with a concentration of 2.76 mg/ml as the substrate for the luciferase assay. Inoculate bacteria into a medium composed of 30 μl of substrate mixed with 150 μl of LB and culture until the logarithmic growth phase. Measure the bacteria’s OD_600_ value and luciferase activity using the Varioskan LUX plate reader (Thermo Scientific), and calculate the relative light units (RLU) with the following formula:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{upgreek} \usepackage{mathrsfs} \setlength{\oddsidemargin}{-69pt} \begin{document} \begin{eqnarray*} \mathrm{ RLU} = \frac{{{\mathrm{luciferase\ activity}}}}{{\mathrm{ OD}_{ 600}}}. \end{eqnarray*}\end{document}5′ rapid amplification of cDNA end assay
The total RNA was prepared with the RNAprotect Bacterial Reagent and RNeasy Mini Kit (Qiagen, Germany, Cat. No. 74104) following the manufacturer’s instructions. The 5′ rapid amplification of cDNA end (5′-RACE) assay was performed using SMARTer® RACE 5′/3′ Kit (Takara, Clontech) with some modifications. In the rapid amplification of cDNA ends, we substitute the SeqAmp DNA Polymerase with AdeptTect Flash HS PCR Master Mix (dye plus) (Accurate Biology, Cat. No. AG12301). The PCR program followed the manufacturer’s instructions.
Conjugation assay
The conjugation assay was performed as described previously [29], with minor modifications. Klebsiella pneumoniae NTUH-K2044IT (resistant to hygromycin) was employed as a donor strain. KpSJTU083A (KpSJTU083-pACYC184-apr, resistant to apramycin) was employed as a recipient. The donor and the recipient were cultured in LB media at 220 rpm and 37°C for 12 h. Then, the donor cells and the recipient cells were recovered by centrifugation (9000 rpm, 10 min). After that, resuspend the pellet in 1 ml phosphate buffered saline twice, then resuspend the donor cells in 10 mM MgSO_4_ (20 μl). Mix the donor cells and the recipient cells sufficiently and transfer 20 μl of the mixture on LB agar. Incubate at 37°C for 24 h. After the conjugation, LB agar with 200 µg/ml hygromycin and 50 µg/ml streptomycin or 50 µg/ml apramycin was used for the selection of transconjugants. The transconjugants were further validated by PCR. The conjugation frequency was calculated as the ratio of transconjugants to donors. All conjugation experiments were conducted in three biological replicates with three parallel tests.
Detection of the excision of ICEKp1 by droplet digital PCR
The primer pairs were designed to target the attL, attR, attB, and attP sites, with Taqman probes located at the 3′ end of the forward primers (Supplementary Table S2). The probes were labeled with a FAM fluorescent reporter group at the 5′ end and a BHQ1 quencher group at the 3′ end. KpSJTU083C1–C4 was cultured to the logarithmic growth phase, after which genomic DNA was extracted and quantified to a concentration of 16 ng/μl using NanoDrop. The DNA sample was then diluted 10 000-fold to a concentration of 1.6 × 10^−3^ ng/μl for the detection of the attL and attR sequence copy numbers by droplet digital PCR (ddPCR), while undiluted DNA samples (16 ng/μl) were used for the detection of the attB and attP sequence copy numbers. The ddPCR was performed using the Naica Crystal Digital PCR system with Sapphire chips, following the manufacturer’s instructions for the PCR program. The excision frequency of ICEKp1 was calculated with the formula as follows:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{upgreek} \usepackage{mathrsfs} \setlength{\oddsidemargin}{-69pt} \begin{document} \begin{eqnarray*} \textit{ICEKp}1\ \mathrm{excision}\ \mathrm{frequency} = {\mathrm{\ }}\frac{{2 \times \textit{attB}}}{{\textit{attL} + \textit{attR} + \left( {2 \times \textit{attB}} \right)}} \times {{10}^{ - 4}}. \end{eqnarray*}\end{document}Analysis of the transcription start site and the promoter of the excised int-ICEKp1
To identify the TSS of the excised int-ICEKp1, we firstly cloned the integrase gene, ICEKp1 5′ (161 bp) and 3′ ends (604 bp) into the high-copy number plasmid, pBluescript SK(+). Then, a 5′-RACE assay was performed to map the TSS of the excised int-ICEKp1 (5′/3′-RACE Kit, Accurate Biology, Cat. No.AG11618). Next, potential promoters of the int-ICEKp1, based on the TSS of the excised int-ICEKp1, were predicted by using BPROM [30] and SAPPHIRE [31]. Finally, the potential promoters were verified by point mutations and the β-galactosidase assay.
Statistical analysis
Student’s t-test (unpaired, equal variance, two-sided) and ordinary one-way analysis of variance were performed using the R package (https://www.r-project.org/). Statistical significance was considered when P ≤ .05. ns indicates not significant.
Results
Excision and conjugation events of ICEKp1 are infrequent in its lifecycle
To assess the dissemination potential of ICEKp1, we performed conjugation transfer assays within K. pneumoniae (intraspecies) and between K. pneumoniae and E. coli (interspecies). In the first round of the conjugation assay, we observed the transfer of ICEKp1 from the donor strain K. pneumoniae NTUH-K2044IT (derived from NTUH-K2044, knocked in the hygromycin resistance hph in ICEKp1) to the recipient strain K. pneumoniae KpSJTU083A (derived from KpSJTU083, introduced a plasmid pACYC184-apr carrying the apramycin resistance gene), occurring at a low frequency of (3.36 ± 0.15) × 10^−6^ (Fig. 1A). To detect the integration of ICEKp1 in KpSJTU083A, we performed tRIP-PCR [32] on the four tRNA-asn genetic loci (named asn1 to asn4) on the chromosome of KpSJTU083A (Supplementary Fig. S1A). All four tRNA-asn sites were found to be the integration sites for ICEKp1 (Fig. 1A and Supplementary Fig. S1B). Four types of transconjugants were obtained (KpSJTU083C1, KpSJTU083C2, KpSJTU083C3, and KpSJTU083C4). This was further verified by the whole-genome sequencing of the four transconjugants, which supported the integration of ICEKp1 into the given tRNA-asn site (Supplementary Fig. S1C and D). The analysis of growth curves and the ICEKp1 maintenance test of KpSJTU083C1-C4 (Supplementary Fig. S1E and F) indicated that the integration of ICEKp1 did not impose a heavy metabolic burden on the host strain and could stably reside in the host chromosome.
ICEKp1 excision level correlated with integrase gene expression in K. pneumoniae. Conjugation of ICEKp1 from K. pneumoniae NTUH-K2044IT to K. pneumoniae KpSJTU083A (A), K. pneumoniae KpSJTU083C1 to E. coli HB101 (B), and E. coli HB101::ICEKp1 to E. coli C600 (C). (D) Schematic map of ICEKp1 conjugation process from K. pneumoniae KpSJTU083C1 to E. coli HB101. (E) Schematic map of the excision of ICEKp1 and the primer design strategy for testing ICEKp1 excision by ddPCR. (F) The 1D droplet spots of FAM fluorescence amplitude of attL, attR, attB, and attP sites detection in KpSJTU083C1. Each point denotes a droplet, and those above the blue threshold line are classified as positive (blue points) (data of KpSJTU083C2–C4 available in Supplementary Fig. S3). (G) Correlation between ICEKp1 excision frequency and expression levels of integrase gene (int) and excisionase gene (xis).
To investigate the transfer of ICEKp1 in E. coli, a second round of the conjugation assay was conducted, utilizing KpSJTU083C1–C4 as donors and E. coli HB101 as recipient (Fig. 1B and Supplementary Fig. S2A). This demonstrated that ICEKp1 transferred from K. pneumoniae to E. coli at a low frequency approaching 10^−5^ (Fig. 1B and Supplementary Fig. S2A). In the third round of the conjugation assay, ICEKp1 subsequently transferred from E. coli HB101 to E. coli C600 at a frequency of ~10^−6^ (Fig. 1C). The assessment of growth curves and ICEKp1 maintenance tests for E. coli HB101::ICEKp1 and E. coli C600::ICEKp1 (Supplementary Fig. S2B and C) indicated that ICEKp1 was reliably inherited in non-native hosts without imposing a significant metabolic burden.
The above results demonstrated that ICEKp1 is capable of conjugative transfer both intraspecifically and interspecifically, at a low frequency. Since ICEs need to excise from the chromosome prior to conjugative transfer (Fig. 1D), we hypothesize that the low-frequency conjugative transfer of ICEKp1 may be attributed to its low excision frequency. Therefore, we detected the excision frequency of ICEKp1 in KpSJTU083C1–C4. The excision of ICEKp1 was detected by using ddPCR (Fig. 1E and F; Supplementary Fig. S3A–C), and the excision frequency was calculated based on the copy number of attB (see the “Materials and methods” section for details). The results showed that the excision frequency of ICEKp1 in KpSJTU083C1–C4 was low as (3.72 ± 0.57) × 10^−5^, (1.18 ± 0.07) × 10^−4^, (8.43 ± 1.18) × 10^−5^, and (4.16 ± 1.24) × 10^−5^, respectively, which was comparable to its conjugative transfer frequency (Fig. 1B and Supplementary Fig. S2A). ICEs typically undergo site-specific recombination under the catalysis of their encoded integrases (int) and accessory factors (e.g., excisionases, xis), thereby excising from the chromosome. Therefore, int and xis expression levels correlate strongly with ICE excision frequency. Previous studies showed that the knockout of the integrase gene of ICEKp1 abolished its excision and conjugation [19]. To further investigate the effects of int and xis on the excision frequency of ICEKp1, we separately performed int overexpression, xis knockout, and xis overexpression, followed by the detection of ICEKp1 excision frequency. The results showed that the int overexpression by itself could significantly enhance the excision frequency of ICEKp1 (Fig. 1G). In addition, overexpression of xis on its own also enhanced ICEKp1 excision frequency. Since int and xis of ICEKp1 are located on separate transcripts (Supplementary Fig. S1G), this indicates that their expression may be subject to complex regulation. Collectively, these findings suggest that the transcriptional regulation of the int restricts the excision of ICEKp1 and further impairs its conjugative transfer capability. On this basis, we subsequently investigated the transcriptional regulatory mechanism of the ICEKp1 integrase gene.
Integrated ICEKp1 displaces the terminator of the adjacent tRNA-asn gene and exploits its promoter for the integrase gene transcription
To investigate the limited transferability of ICEKp1, we focused on the transcription of its integrase gene (int-ICEKp1) located at the 5′ end of ICEKp1. This gene is positioned 161 bp downstream of ICEKp1’s insertion site, the tRNA*-asn* gene, and is transcribed in the same direction. In the ICEKp1-free strain KpSJTU083, a Rho-independent transcription terminator (Tasn) is located at the 3′ end of the empty tRNA-asn gene. However, upon ICEKp1 integration at its 3′ end (KpSJTU083C1-C4), Tasn was displaced to the downstream of the 3′ end of ICEKp1, resulting in the loss of its function to terminate transcription of tRNA-asn (Fig. 2A). Therefore, we hypothesize that in the integrated state of ICEKp1, int-ICEKp1 is co-transcribed with the upstream tRNA-asn gene in the host chromosome, resulting in the formation of a polycistronic transcript designated as asn-int.
ICEKp1 displaces the tRNA-asn terminator and exploits the tRNA-asn promoter to initiate int-ICEKp1 transcription in the integrated state. (A) Schematic map of the location change event of the tRNA-asn terminator by ICEKp1 insertion. (B) Schematic map of the asn-int polycistronic operon. F0–F7 are forward primers (from the TSS to int) used in the reverse-transcription PCR (RT-PCR). REV is the reward primer used in the RT-PCR. (C) Co-transcription of the asn-int operon in KpSJTU083C1–KpSJTU083C4 verified by RT-PCR. (D) TSS of int was identified in KpSJTU083C1 by 5′-RACE experiment. Sequence alignment was performed between the tRNA-asn1 gene (asn1) promoter (Pasn) to int (top) and the sequence identified by the 5′-RACE experiment (bottom). “+1” refers to the TSS of int (data of NTUH-K2044 and KpSJTU083C2–C4 available in Supplementary Fig. S4). (E) Schematic map of the cleavage of the asn-int co-transcript detected by CRISPRi. Intact lines (green part was the tRNA-asn region, black part was the intergenic region, and the blue part was the int region) refer to the intact asn-int transcripts. Broken lines refer to the asn-int transcript cleaved by RNase E. (F) Schematic map of the primer design for identification of the asn-int co-transcript cleavage. (G) mRNA abundance of the asn-int transcript junction sequence, rne and dCas9. The mRNA quantity was normalized against the housekeeping gene gapA. (H) Schematic map of the tRNA-asn terminator inserted in KpSJTU083C1. (I) The co-transcription of asn-int operon in KpSJTU083C1::Tasn1 verified by RT-PCR. Transcript of gapA was used as the control. (J) ICEKp1 transfer frequency determined between NTUH-K2044, KpSJTU083C1, and KpSJTU083C1::Tasn1 (donor strains) with E. coli HB101 (recipient strain).
First, we examined the TSS of the int-ICEKp1 for the integrated ICEKp1 using the reverse transcriptase (RT)-PCR and the assay of 5′-RACE assay. The RT-PCR results (Fig. 2B and C) showed that, in KpSJTU083C1–C4, int-ICEKp1 and the adjacent tRNA-asn were co-transcribed. Results of 5′-RACE further pinpointed the TSS of the int-ICEKp1 to be located 11 bp upstream of the first base pair of the tRNA-asn gene (asn1, asn2, or asn3) in KpSJTU083C1–C3, and 3 bp upstream of the first base pair of asn4 in KpSJTU083C4 and NTUH-K2044 (Fig. 2D and Supplementary Fig. S4A–D).
Next, to further verify the co-transcription of asn-int, we examined the cleavage of the asn-int co-transcript by RNase E (Fig. 2E and F). After IPTG-induced activation of the CRISPRi system targeting rne in KpSJTU083C1–C4, the significant increase in the mRNA abundance of the asn-int junction sequence revealed that the asn-int co-transcript was susceptible to cleavage by RNase E in KpSJTU083C1-C4 (Fig. 2G and Supplementary Fig. S5).
Finally, we conducted a genetic knock-in of Tasn at the 3′ end of the tRNA-asn gene in KpBSI083C1 (KpBSI083C1::Tasn1) to disrupt the formation of asn-int co-transcription (Fig. 2H). The RT-PCR results demonstrated that the tRNA-asn gene and the int-ICEKp1 could not form a co-transcript in KpBSI083C1::Tasn1 (Fig. 2I). In addition, a conjugation assay was performed to assess the potential transfer of ICEKp1 from KpBSI083C1::Tasn1 to E. coli HB101. As anticipated, no transconjugants were obtained in this assay (Fig. 2J).
Together, these results demonstrated that the integration of ICEKp1 induced the displacement of Tasn, thereby exploiting the tRNA-asn promoter to initiate the transcription of int-ICEKp1 during the integrated state of ICEKp1. It supports the hypothesis that the promoter of the tRNA-asn (Pasn) on the host chromosome initiates the transcription of the int-ICEKp1, resulting in the production of asn-int.
Excised ICEKp1 exploits the 3′ end promoter for the integrase gene transcription
In its excised state, ICEKp1 exists as an extrachromosomal entity, with the joining of its 3′ and 5′ end occurring at the attP site (Fig. 3A). Sequence alignment analyses indicate that the 3′ and 5′ ends are highly conserved among ICEKp1-like elements (Supplementary Figs S6 and S7). A putative σ^70^ promoter (P3’end) was identified in proximity to the 3′ end, and we hypothesize that this promoter may initiate the transcription of int-ICEKp1 in the 5′ end of the excised ICEKp1.
Excised ICEKp1 exploits the 3′ end promoter for the integrase gene transcription. (A) Schematic map of the primer design strategy for detection of int transcript by RT-PCR and the int promoter at the excised state, “+1” represents the TSS of the int. (B) Transcript of int was detected by RT-PCR at the ICEKp1 excised state. (C) TSS of int was identified by 5′-RACE experiment in the excised state. Sequence alignment was performed between the ICEKp1 3′ end to int (top) and the sequence identified by the 5′-RACE experiment (bottom). “+1” refers to the TSS of int. (D) Schematic map of the P3’end point mutations at the −10 box (M1) or −35 box (M2). (E) The promoter activities of the P3’end and its point mutants were determined by β-galactosidase assay.
First, the TSS of int-ICEKp1 within the excised ICEKp1 was examined through RT-PCR and 5′-RACE assay. Nine forward primers were designed to bind to various locations around the 3′ and 5′ ends, along with a reverse primer specific to the int-ICEKp1, facilitating the amplification of cDNA (Fig. 3A). Analysis through RT-PCR (Fig. 3B) and 5′-RACE (Fig. 3C) demonstrated that transcription of int-ICEKp1 initiated from the 3′ end of the excised ICEKp1. The TSS of the int-ICEKp1 was located 15 bp upstream of the first nucleotide of the 5′ end. Then, a β-galactosidase assay was conducted to evaluate the promoter activity of the P3’end and the 5′-FSint (5′-flanking sequence of the integrase gene). The assay results (Fig. 3E) indicated that the 5′-FSint region exhibited no promoter activity. In contrast, P3’end and FL_excised_ (the full-length fragment covering tRNA-asn promoter, tRNA-asn, and 5′-FSint) showed detectable promoter activity, suggesting the capacity of P3’end to drive the transcription of the integrase gene. Finally, specific mutations were introduced into the P3’end region to further investigate its promoter activity (Fig. 3D and E). Results from β-galactosidase assays analyzing mutations within the −35 and −10 boxes corroborated the hypothesis that the P3’end is crucial for int transcription (Fig. 3E).
Collectively, these findings suggest that a promoter located at the ICEKp1 3′ end, and ICEKp1 exploits this promoter to drive the transcription of the integrase gene when in the excised state.
The stem-loops within the int 5′-UTR attenuate the total amount of the full-length asn-int transcripts in the integrated ICEKp1
Within the integrated state of ICEKp1, the transcription levels of the tRNA-asn gene may lead to a corresponding increase in the transcription of int-ICEKp1, potentially resulting in the frequent excision of ICEKp1. This conflicts with the previously observed low excision and transfer frequencies of ICEKp1, as well as the documented minimal expression of the HPI-carrying integrase gene [33]. Therefore, we propose that ICEKp1 may down-regulate the activity of the tRNA-asn promoter, thereby modulating the transcription of int-ICEKp1 and facilitating its sustained integration within the host genome. To investigate this hypothesis, we conducted a β-galactosidase assay to evaluate the promoter activity in the upstream region of int-ICEKp1.
Upon the ICEKp1 integrated state, we segmented the upstream region of the int-ICEKp1 into five distinct fragments (Fig. 4A): Pasn (promoter of the tRNA-asn gene), 5′-FSint (5′ flanking sequence of int-ICEKp1), FL_integrated_ (full-length fragment; spanning from Pasn to 5′-FSint in the integrated state of ICEKp1), P_asn _+ asn (the tRNA-asn gene sequence and its promoter), and asn + 5′-FSint (the tRNA-asn gene sequence and the 5′ flanking sequence of int-ICEKp1). The results from the β-galactosidase assay (Fig. 4B) showed that 5′-FSint exhibited negligible promoter activity, while Pasn demonstrated high activity levels, indicating that the integrase gene exploits Pasn for transcription during the integrated state, whereas the 5′-FSint region is incapable of driving integrase gene transcription. Moreover, we performed point mutations on the −35 and −10 boxes of Pasn and assessed the activity of Pasn and FL_integrated_ after the mutations (Fig. 4D). The results of the β-galactosidase assay (Fig. 4E and F) showed a decrease in promoter activity for the mutation of FL_integrated_, which correlated with the decline observed in the mutation of Pasn. Similar findings were noted for KpSJTU083C2–C4 (Supplementary Fig. S8).
Stem-loops within the 5′ UTR of int attenuates the promoter activity of the full-length fragment from Pasn to int 5′-UTR in the integrated ICEKp1. (A) Schematic map of the upstream fragment of int in the ICEKp1 integrated state for promoter activity analysis. From the promoter of the tRNA-asn gene to the 5′ end of int, five groups were set: Pasn (tRNA-asn promoter), 5′-FSint (the 5′ flanking sequence of int), FLintegrated (the full-length fragment covering tRNA-asn promoter, tRNA-asn and 5′-FSint), Pasn + asn (tRNA-asn promoter and the tRNA-asn gene), and asn + 5′-FSint (the tRNA-asn gene and the 5′-FSint). (B, C) Promoter activities of Pasn, FLintegrated, 5′-FSint, Pasn + asn, and asn + 5′-FSint determined by β-galactosidase assay (data about Pasn2, Pasn3, and Pasn4 available in Supplementary Fig. S8). (D) Schematic map of the tRNA-asn1 promoter point mutations at the −10 box (M1) or −35 box (M2). The point-mutated promoter activity of the Pasn (E) and FLintegrated (F) determined by β-galactosidase assay. “−M1” and “−M2” refer to “point mutation at −10 box” and “point mutation at −35 box,” respectively. (data about asn2, asn3, and asn4 available in Supplementary Fig. S8). (G) The predicted stem-loops in the 5′ flanking region of ICEKp1. (H) The predicted mRNA secondary structure after knocking out the stem-loop3 in the 5′ flanking region of ICEKp1. (I) Under the integrated state of ICEKp1, the promoter activity of Pasn, FLintegrated, 5′-FSint, and FLintegrated ΔSL3.
In comparison to Pasn, FL_integrated_ showed comparatively reduced promoter activity, suggesting that ICEKp1 may exert a down-regulatory effect on Pasn. In addition, the promoter activities of P_asn _+ asn and asn + 5′-FSint were similar to those of Pasn and 5′-FSint, respectively (Fig. 4C), suggesting the diminished promoter activity of Pasn was not attributable to the tRNA-asn sequence. Thus, the reduced promoter activity of Pasn is likely caused by the 5′-FSint region in the int 5′-UTR. This region harbored three stem-loop (SL) structures as predicted by Mfold [34] (Fig. 4G). We hypothesized that these SL structures might exert a suppressive effect on the promoter activity of Pasn. After confirming that the knockout of individual SL structures did not induce alterations in other secondary structures within the 5′-FSint region (Fig. 4H and Supplementary Fig. S9A and B), to explore the regulatory role of the SL structures, we generated their knockouts sequentially and evaluated the consequent changes in FL_integrated_ promoter activity by using a luciferase assay (Fig. 4I and Supplementary Fig. S9C–F). The results showed that the deletion of SL3 led to a 194.2% enhancement in promoter activity compared with FL_integrated_, indicating that it exerted a significant inhibitory effect on the promoter activity of Pasn (Fig. 4I). SL1 and SL2 also exert an inhibitory effect on Pasn, albeit to a relatively weaker extent (Supplementary Fig. S9C–F). Together, these findings indicated that the step-loops (SL1–SL3) within the 5′-FSint region of the int 5′-UTR are organized as a potential terminator, which attenuates the total amount of the full-length asn-int mRNA transcripts during the integrated state of ICEKp1, thereby maintaining ICEKp1 in an integrated state.
Ligation of the 3′ end and 5′ end of ICEKp1 maintains the activity of the 3′ end promoter in the excised ICEKp1
Upon the ICEKp1 excised state, we analyzed the upstream region of the int-ICEKp1 by dividing it into four distinct fragments (Fig. 5A): P3’end (the promoter located at the 3′ end of ICEKp1), 5′-FSint (the 5′ flanking sequence of int-ICEKp1), P_3’end _+ 5′-FSint (a fusion of the P3’end sequence and the 5′ flanking sequence of int-ICEKp1 by deleting 15-bp sequence at the 3′ end of ICEKp1), and FL_excised_ (the full-length fragment). The luciferase assay results (Fig. 5B) showed that P3’end exhibited strong activity, whereas 5′-FSint displayed weak promoter activity, indicating that ICEKp1 exploits P3’end to drive the transcription of the integrase gene during the excised state. Notably, ligating P3’end (in the 3′ end of ICEKp1) to 5′-FSint (in the 5′ end of ICEKp1) to form the full-length fragment (FL_excised_) kept robust promoter activity (Fig. 5B). However, deletion of the 15-bp sequence at the 3′ end of ICEKp1 from FL_excised_ abolished promoter activity in the resulting fragment (P_3’end _+ 5′-FSint) (Fig. 5B). This suggests that the 15-bp sequence might counteract the inhibitory effect of 5′-FSint, which contains stem-loop structures (SL1–SL3) that likely function as a transcriptional terminator (Fig. 5B).
Ligation of the 3′ end and 5′ end of ICEKp1 maintains the activity of the 3′ end promoter in the excised ICEKp1. (A) Schematic map of the upstream fragment of int in the ICEKp1 excised state for promoter activity analysis. From the P3’end to the 5′ end of int, four groups were set: P3’end (the promoter located at the 3′ end of ICEKp1), 5′-FSint (the 5′ flanking sequence of int), P3’end + 5′-FSint (a fusion of the P3’end sequence and the 5′ flanking sequence of int-ICEKp1 by deleting 15-bp sequence at the 3′ end of ICEKp1), FLexcised (the full-length fragment covering P3’end, ICEKp1 3′ end from the TSS of int to attP and 5′-FSint). (B) Under the excised state of ICEKp1, the promoter activity of P3’end, 5′-FSint, P3’end + 5′-FSint, and FLexcised.
To further investigate whether the 15-bp sequence at the 3′ end of ICEKp1 can relieve terminator repression in 5′-FSint, we conducted the luciferase assay using a heterologous promoter (PgapA). As shown in Supplementary Fig. S12, 5′-FSint suppressed PgapA activity, whereas introducing the 15-bp sequence upstream of 5′-FSint restored PgapA activity, suggesting that this 15-bp sequence alleviates terminator repression.
Together, these results indicate that specific regulatory elements within the int 5′-UTR sustain P3’end activity, which may promote the reintegration of excised ICEKp1 into the host chromosome.
A similar state-dependent promoter switching mechanism for the integrase gene transcription is observed in other ICEs
We conducted further investigations to determine whether the int promoter activity alteration, observed in the integrated or excised ICEKp1, occurs in other ICEs that possess integrase genes located at their ends, adjacent to host tRNA genes.
First, we conducted an in-silico analysis examining the co-transcription of the ICE-carrying integrase gene (int) and the neighboring tRNA gene in the integration of ICE. From a total of 851 ICEs compiled in the ICEberg database [2], we selected 23 ICEs, each of which integrates at the 3′ end of the tRNA gene and possesses the int gene adjacent to the tRNA gene in the same transcriptional strand (see Supplementary Table S3). We subsequently retrieved five ICEs with RNA-seq data available in the NCBI SRA database, including the ICEKp1 of K. pneumoniae NTUH-K2044, ICEEc1 of E. coli ED1a, ICEclc of Pseudomonas aeruginosa W36662, ICEEc1 of E. coli MS14387, and ICERi1 (ICERinATCC49129) of Ralstonia insidiosa ATCC 49129 (Supplementary Table S3). Mapping the short reads to the genomic context of the tRNA gene and its adjacent integrase gene (Fig. 6A–C and Supplementary Fig. S10) indicated that these ICEs can leverage the promoters of the tRNA-asn genes to initiate transcription of the integrase genes upon the ICE integration into the host chromosome. Additionally, we analyzed the tRNA gene terminator adjacent to the int gene within these five ICE-harboring bacterial hosts (Supplementary Table S4). In contrast to ICE-containing hosts, we identified a terminator present at the 3′ end of the tRNA gene in hosts lacking ICEs (Fig. 6D–F and Supplementary Fig. S11). Upon integration of the ICE at the tRNA gene site, the terminator was displaced to the 3′ end of the ICE (Fig. 6D–F and Supplementary Fig. S11).
State-dependent promoter switching mediates int transcription in other ICE families. (A–C) “tRNA-int” co-transcription analysis through the RNA-seq data. The Y-axis represents the read coverage of the tRNA gene and int in the RNA-seq data. The X-axis represents the gene locus of the tRNA gene and int. The RNA-seq data were taken from NCBI SRA with the accession numbers: SRX10030082 for K. pneumoniae NTUH-K2044, SRX5332356 for E. coli ED1a, and SRX12812169 for Pseudomonas putida W36662. (D–F) Relocation of the tRNA gene terminator after ICE insertion. Alignment of the sequence from the 3′ end of the tRNA gene to the terminator without ICE insertion (top) against the sequence of the ICE 3′ end after ICE insertion (bottom). The sequence in green denotes that of the tRNA gene terminator. (G–I) Promoter activities of the PtRNA, FLintegrated, and 5′-FSint determined by β-galactosidase assay in the ICE integrated state. (J–L) Promoter activities of the P3’end, 5′-FSint, P3’end + 5′-FSint, and FLexcised determined by β-galactosidase assay in the ICE excised state.
Next, we performed a β-galactosidase assay to investigate the promoter activity within the upstream region of the int of the integrated ICE, with the examples of ICEEc1 of E. coli ED1a and ICEclc of P. aeruginosa W36662. The DNA fragments corresponding to the tRNA gene promoter (PtRNA), the flanking sequence of the integrase gene (5′-FSint) and the full-length (FL_integrated_) region of ICEEc1 and ICEclc were synthesized for analysis. The β-galactosidase assay performed on these various fragments of ICEEc1, ICEclc and ICEKp1 in K. pneumoniae NTUH-K2044 (served as a control) revealed that PtRNA exhibited strong activity while 5′-FSint demonstrated weak activity. The FL_integrated_ fragment exhibited medium activity in comparison (Fig. 6G–I). The 5′-FSint in the int 5′-UTRs of ICEEc1 and ICEclc are composed of multiple stem-loop structures, which are similar to those of ICEKp1, as identified by mRNA secondary structure analysis (Supplementary Fig. S13A and C). These findings suggest that the integration of ICEEc1 and ICEclc induces transcription termination downstream of the stem-loop structures, resulting in the reduction of full-length mRNA transcripts that contain both the tRNA gene and int.
Finally, we conducted a β-galactosidase assay to assess the transcriptional activity of the promoter within the upstream region of the int of the excised ICEEc1 and ICEclc. The DNA fragments corresponding to the P3’end (promoter located at the 3′ end of the ICE), 5′-FSint, P_3’end _+ 5′-FSint, and FL_excised_ (full-length fragment) of ICEEc1 and ICEclc were also synthesized for further examination. The results of the β-galactosidase assay indicated that P3’end and FL_excised_ exhibited strong activity, whereas 5′-FSint demonstrated weak promoter activity (Fig. 6J–L).
Collectively, the data suggest that ICEclc and ICEEc1 can switch the int promoter upon their integration or excision, and this modulation could facilitate their integration into bacterial host genomes.
Discussion
The ICE-encoded integrase leads to the integration and excision of ICEs via site-specific recombination [17, 35–38]. However, the factors contributing to the limited transfer frequency of ICEs, particularly in relation to integrase gene expression, remain ambiguous. In this study, we proposed a promoter switching regulation mechanism of the integrase gene transcription that occurs upon the integrated or excised state of ICEs (Graphical abstract). Our investigation concentrates on the ICEKp1 family, widespread within the hypervirulent and classical K. pneumoniae strains. Notably, ICEKp1 demonstrated the capability for self-transfer between K. pneumoniae and E. coli, occurring at a low frequency (Fig. 1). For other ICEKp families in K. pneumoniae, their 3′ and 5′ end sequences are highly conserved relative to ICEKp1 (Supplementary Figs S6 and S7), which suggests that this regulatory pattern for the integrase gene is widely prevalent across the ICEKp families. Given that most ICEs lack a replicon and cannot sustain stable existence outside the chromosome, enhanced expression of int during the excised state is advantageous for facilitating the re-integration of ICE into the donor chromosome.
The promoter for the integrase gene of non-transmissible HPI of Y. pestis was found to be located near the host tRNA-asn [17]. In this study, we also found that the integrated ICEKp1 exploits the adjacent tRNA-asn promoter (Pasn) for the int gene (Fig. 2). We further observed that the stem-loops located at the int 5-UTR might be organized as a potential terminator, diminishing the total amount of the asn-int mRNA transcripts (Fig. 4), and ensuring that the transcription of int remains at a low level, thereby maintaining the stability of ICEKp1 within the host chromosome. The molecular mechanism requires further investigation. Similar insulating gene expression is also observed in Tn916 [39], indicating that this mechanism may be present in multiple ICE families.
Following excision from the host chromosome, the left end (3′ end) and right end (5′ end) of ICE may recombine at the attP site, converting the ICE into a circular form. Within the ICEclc family, the int gene is located at the 5′ end, adjacent to the ICEclc integration site, the tRNA-gly gene. During the excised state of ICEclc, the promoter Pcir, located at the 3′ end, joins the 5′ end to initiate the int transcription [18]. Similarly, we identified a promoter (P3’end) for the integrase gene at the 3′ end of the excised ICEKp1, located upstream of the attP site (Fig. 3). Further investigation observed that the 3′ end of the excised ICEKp1 ligated to its 5′ end, generating a full-length fragment (FL_excised_) with robust promoter activity (Fig. 5). The regulatory role of the int 5′-UTR appears intricate and remains to be explored in future studies. For example, the in silico analysis of the int 5′-UTR secondary structure indicated that the 15-bp sequence at the 3′ end of ICEKp1 might be able to base-pair with the stem-loop SL3 in 5′-FSint, potentially destabilizing SL3 and relieving its inhibitory effect on the P3’end (Supplementary Fig. S13). Future studies should experimentally validate this proposed RNA–RNA interaction between the 15-bp sequence and SL3 to confirm its regulatory role.
tRNA genes are highly conserved, with their 3′ ends frequently serving as integration sites for various ICEs, such as tRNA-asn for the ICEKp family of K. pneumoniae, tRNA-gly for the ICEclc family of P. aeruginosa, and tRNA-asn for the ICEEc1 family of E. coli. Following integration into the host chromosome, the integrase gene, positioned at the 5′ end of the ICE, is located ~200 bp away from the integrated tRNA gene (Supplementary Table S3). Through comparative analyses of the tRNA gene terminators, the transcriptomic data of the ICE host strain, and the promoter activities of ICEclc and ICEEc1 (Fig. 6), we observed a similar modulation in promoter activity that influences the int transcription upon the integration or excision of ICEKp1. This observed phenomenon may extend to additional ICE families, for example, ICERi1 (ICERinATCC49129) of R. insidiosa (Supplementary Fig. S10).
The transcriptional regulation of the ICE integrase gene involves a complex network, likely mediated by multiple ICE-encoded regulators [14–16, 40]. Differential expression of these regulators between donor and recipient strains may lead to variations in integrase transcription, requiring further study. Additionally, ICE excision frequency is influenced not only by integrase regulation but also by environmental factors such as UV light and stress conditions [41, 42]. Since K. pneumoniae (the host of ICEKp1) is an opportunistic pathogen, assessing ICEKp1 excision and transfer in in vivo animal models would better reflect its real-world transmission dynamics, warranting further investigation.
In summary, we proposed that the state-dependent switching of the integrase gene promoter, promoting integration over excision of ICEKp1. This regulatory mechanism depends on ICEKp1’s state (Graphical abstract): (i) Integrated state: ICEKp1 displaces the terminator of the adjacent tRNA-asn gene and hijacks its promoter (Pasn) to drive int transcription. The mRNA stem-loop structures in the int 5′-UTR reduces the full-length asn-int transcripts. (ii) Excised state: ICEKp1 utilizes its 3′ end promoter (P3’end) for int transcription. The int 5′-UTR extends into a 15-bp region at the ICE’s 3′ end, which possesses complex mechanisms to keep P3’end activity. We propose that other ICEs carrying the integrase genes adjacent to host tRNA genes (e.g., ICEclc and ICEEc1) may employ analogous strategies. Given that the integrase gene is pivotal role in the ICE lifecycle, these findings could advance understanding of how ICEs balance stable maintenance and dissemination in bacterial hosts under stress.
Supplementary Material
gkag254_Supplemental_File
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Johnson CM, Grossman AD. Integrative and conjugative elements (IC Es): what they do and how they work. Annu Rev Genet. 2015;49:577–601. 10.1146/annurev-genet-112414-055018.26473380 PMC 5180612 · doi ↗ · pubmed ↗
- 2Wang M, Liu G, Liu M et al. IC Eberg 3.0: functional categorization and analysis of the integrative and conjugative elements in bacteria. Nucleic Acids Res. 2024;52:D 732–7. 10.1093/nar/gkad 935.37870467 PMC 10767825 · doi ↗ · pubmed ↗
- 3Botelho J, Schulenburg H. The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 2021;29:8–18. 10.1016/j.tim.2020.05.011 .32536522 · doi ↗ · pubmed ↗
- 4Gomberg AF, Grossman AD. It’s complicated: relationships between integrative and conjugative elements and their bacterial hosts. Curr Opin Microbiol. 2024;82:102556. 10.1016/j.mib.2024.102556.39423563 PMC 11625472 · doi ↗ · pubmed ↗
- 5Delavat F, Miyazaki R, Carraro N et al. The hidden life of integrative and conjugative elements. FEMS Microbiol Rev. 2017;41:512–37. 10.1093/femsre/fux 008.28369623 PMC 5812530 · doi ↗ · pubmed ↗
- 6Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Micro. 2010;8:552–63. 10.1038/nrmicro 2382.20601965 · doi ↗ · pubmed ↗
- 7Delavat F, Mitri S, Pelet S et al. Highly variable individual donor cell fates characterize robust horizontal gene transfer of an integrative and conjugative element. Proc Natl Acad Sci USA. 2016;113:E 3375–83. 10.1073/pnas.1604479113.27247406 PMC 4914192 · doi ↗ · pubmed ↗
- 8Delavat F, Moritz R, van der Meer JR. Transient replication in specialized cells favors transfer of an integrative and conjugative element. m Bio. 2019;10:e 01133–19. 10.1128/m Bio.01133-19.31186329 PMC 6561031 · doi ↗ · pubmed ↗