Pulmonary Sclerosing Hemangioma Presenting As Perihilar Lung Nodule in a Patient With Papillary Thyroid Carcinoma: A Case Report and Review of the Literature
Hunter Stecko, Marissa Guo, Aaron Guo, William MacDonald, Desmond D'Souza

TL;DR
This case report describes a rare lung tumor in a patient with thyroid cancer and a family history of cancer.
Contribution
The novelty lies in presenting a challenging diagnostic case of pulmonary sclerosing pneumocytoma in a patient with multiple cancer risks.
Findings
The patient had a perihilar lung nodule diagnosed as pulmonary sclerosing pneumocytoma after surgery.
The case highlights the difficulty in diagnosing this rare tumor due to its mimicry of other diseases.
A review of the literature on clinical presentation and management is provided.
Abstract
Pulmonary sclerosing pneumocytoma is a rare, typically asymptomatic, tumor with potential for malignant transformation. Diagnosis is frequently difficult due to its rarity and proclivity to mimic other pathologies. Here, we describe a particularly challenging case of a patient with a personal and familial history of other malignancies who presented with an incidental lung nodule, found to be pulmonary sclerosing pneumocytoma on pathologic examination after an anatomic lung resection. We conclude with a review of the natural history, clinical and histopathological presentation, and management options for this rare entity.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
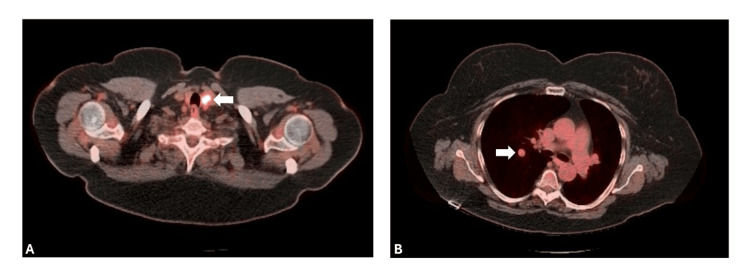 Figure 1
Figure 1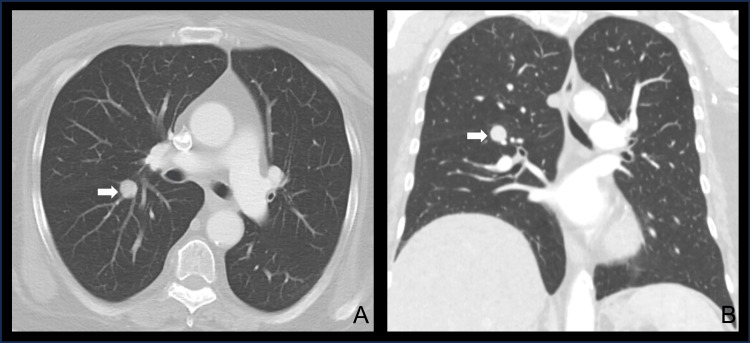 Figure 2
Figure 2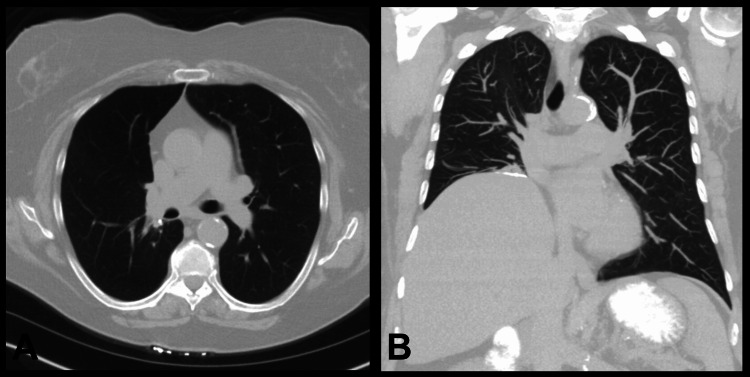 Figure 3
Figure 3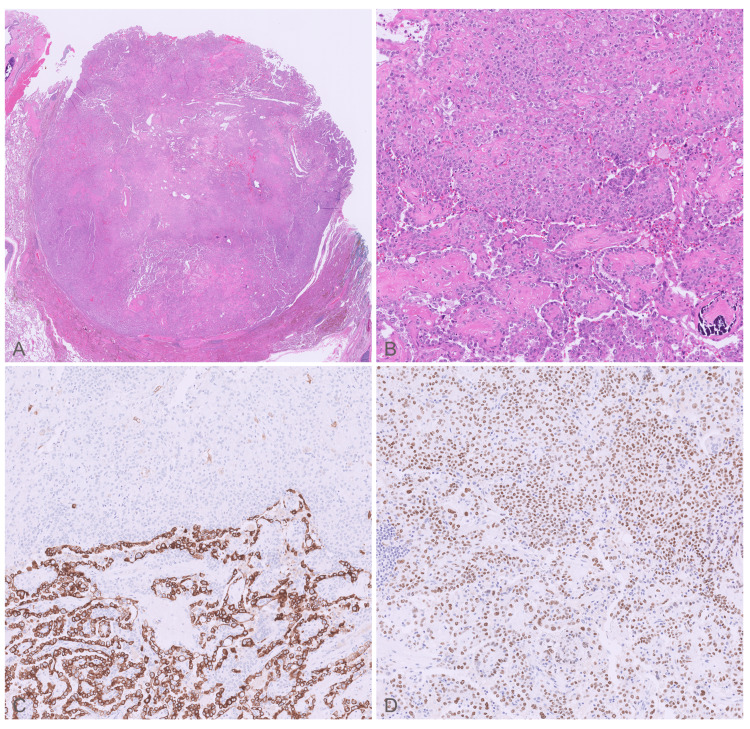 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMedical Imaging and Pathology Studies · Interstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Pleural and Pulmonary Diseases
Introduction
Pulmonary sclerosing pneumocytoma (PSP), previously called pulmonary sclerosing hemangioma, is a rare benign tumor with uncertain histogenesis, although studies have suggested that these lesions arise from primitive respiratory epithelium [1]. This proposed epithelial origin is further supported by the consistent expression of thyroid transcription factor-1 (TTF-1) and epithelial membrane antigen (EMA) in both surface and round cell populations, as well as recurrent activating AKT1 mutations, implicating dysregulation of the PI3K/AKT/mTOR pathway in PSP. PSP has a strong female predominance and is commonly diagnosed in the fifth decade [2]. While there have been reports of PSPs causing symptoms, such as cough, hemoptysis, chest pain, and fever, the majority of patients are asymptomatic, and the discovery of PSP is often incidental. On computed tomography (CT) scan, PSP generally appears as a singular ovoid lesion with smooth borders. While PSP has a propensity to be located peripherally, other presentations, including endobronchial involvement, have been described [3].
Pathologically, PSP comprised two cell types: cuboidal cells resembling bronchiolar epithelium and type II alveolar pneumocytes lining the surfaces of papillary and tubular structures, and interstitial round cells with relatively consistent morphology [1,4]. Immunohistochemistry typically shows positive staining for TTF-1 and EMA [1,4]. Alterations to mTOR-related pathway genes, particularly AKT1, are frequently implicated in the pathogenesis of PSP [5], though mutations in BRAF and IDH1 have also been reported [6,7].
The clinical presentation of PSPs has been found to mimic carcinoid tumors [8]. Carcinoid tumors are frequently diagnosed with nuclear imaging using somatostatin receptor (SSTR)-targeted radiotracers, such as (86)Ga-DOTATATE and (86)Ga-DOTANOC [9]. However, PSP may also show avidity on these scans, possibly due to the expression of SSTRs by respiratory epithelial tissues [10]. The rate at which this occurs is currently unreported in the literature due to the rare nature of PSP. To add further complexity, some lesions have been demonstrated to have concurrent carcinoid tumors with PSP [11]. Additional case descriptions are needed to inform clinicians on how to approach the diagnosis and management of this rare tumor. Differentiating PSP from carcinoid tumor is particularly important given the difference in need for surveillance and approach to resection in patients with PSP vs. carcinoid tumor. For this reason, we herein present the case of a patient with diagnostically complex PSP.
Case presentation
A 65-year-old woman with a remote smoking history presented with an incidentally identified lung mass while undergoing work-up for a thyroid nodule. Her medical history otherwise included hypertension, hyperlipidemia, type 2 diabetes, and obstructive sleep apnea. Her family history was significant for multiple instances of breast cancer among female relatives, ovarian cancer, and thyroid cancer. Positron emission tomography (PET)/CT scan demonstrated 18F-fluorodeoxyglucose (FDG) uptake by the thyroid lesion and adjacent lymph nodes, suggestive of malignancy (Figure 1A). In addition, it showed a well-circumscribed right-sided perihilar lung nodule with a standardized uptake value (SUV) of 3.5 (Figure 1B). She underwent total thyroidectomy with left neck dissection, and the final pathological diagnosis was consistent with locally advanced papillary thyroid carcinoma with lymph node metastases. Notably, post-procedural radioactive iodine uptake studies showed no uptake in the previously identified lung mass.
Positron emission tomography/computed tomography imaging of the neck and chest(A) A 1.2 cm left thyroid mass demonstrating high 18F-fluorodeoxyglucose (FDG) avidity (SUV 9) (white arrow). (B) Incidental finding on the same diagnostic scan demonstrating a 1.2 cm right perihilar lung nodule with mild FDG avidity (SUV 3.5) (white arrow).
Following her recovery, she was referred to thoracic surgery for further evaluation. A CT chest scan demonstrated a round, noncalcified nodule measuring 1.6 cm by 1.5 cm in the right lower lobe (RLL) (Figure 2). Endobronchial biopsy of the RLL nodule and mediastinal lymph nodes via navigational bronchoscopy was non-diagnostic. Subsequent CT-guided percutaneous biopsy of the pulmonary lesion revealed atypical cells; however, the yield was insufficient for a conclusive diagnosis. A SSTR-targeted PET/CT scan was also obtained to evaluate for a neuroendocrine tumor, which demonstrated a stable RLL nodule with mild tracer avidity. Pulmonary function testing was within normal limits.
Pre-operative computed tomography scan of the chestA computed tomography scan demonstrating an axial (A) and coronal (B) view of a round, perihilar mass located within the patient’s right lower lobe (white arrow).
Ultimately, the patient was recommended for robotic-assisted right lower lobectomy given concern for carcinoid tumor. Dissection of thoracic lymph nodes from stations 2, 4, 7, 9, 10, and 11 was performed during the operation. The patient tolerated the procedure well and had an uncomplicated post-operative course. Given her extensive family history of cancer, she eventually underwent somatic genomic testing, which revealed no pathogenic variants, including any within MLH1, MSH2, MSH6, PMS2, EPCAM, BRCA1, or BRCA2.
Her inpatient course was uncomplicated, and she was discharged on post-operative day 2 in good condition. She was readmitted to an outside hospital on post-operative day 5 due to shortness of breath. Residual hydropneumothorax was visualized on a spiral CT scan obtained to rule out pulmonary embolism as a cause of her dyspnea. She was discharged on home oxygen, which she discontinued within a week following her discharge on post-operative day 6.
On one-month follow-up, she was recovering well, and her surgical incisions were well-healed. She likewise was doing well at the six-month follow-up. Her six-month follow-up CT scan demonstrated normal post-operative changes without disease recurrence or lymphadenopathy (Figure 3).
Six-month post-operative computed tomography scan of the chestA computed tomography scan demonstrating an axial (A) and coronal (B) view of the patient’s lungs following resection of the right lower lobe. Normal post-operative changes are demonstrated and no recurrence of the tumor is visualized.
Histopathological examination
Gross examination showed a 1.7 cm ovoid, bulging mass with tan-white cut surfaces near the superior hilum of the RLL. No necrotic or hemorrhagic foci were present. Histopathologically, the tumor was well-circumscribed with papillary and solid architectural patterns. The surfaces of the papillae are lined by bland cuboidal cells with minimal atypia. The underlying stroma has variable collagen deposition and cellularity composed of uniform, large, round cells with distinct cell borders and abundant cytoplasm. The solid pattern consists of round cells arranged in sheets. Immunohistochemistry demonstrated diffuse TTF-1 and EMA expression in both the surface epithelial and stromal round cell populations, whereas cytokeratin expression (AE1/AE3, CK7, and CAM5.2) was restricted to the surface cells, a biphasic immunophenotypic pattern that helps distinguish PSP from primary pulmonary adenocarcinoma (Figure 4). All seven lymph nodes sampled were benign. Genomic analysis identified a 13-amino acid AKT1 internal tandem duplication, further supporting the diagnosis of PSP. Immunostains were negative for thyroglobulin, PAX8, CK5/6, p40, CK20, and no genetic mutations were detected in ALK, BRAF, EGFR, ERBB2, KEAP1, KRAS, MAP2K1, MET, NFE2L2, or PIK3CA.
Pathologic features of the surgical specimen(A) Light microscopic examination showed a well-circumscribed tumor with variable cellularity and collagen deposition, H&E, original magnification ×2. (B) The tumor comprised two distinct cell populations, including round cells arranged in sheets (top of the panel) and surface cells lining papillary structures (bottom of the panel); original magnification ×20. The surface cells are immunoreactive with (C) cytokeratin 7 (CK7) and (D) thyroid transcription factor-1 (TTF-1), while the round cells are immunoreactive only with TTF-1; immunohistochemistry, original magnification ×20.
Discussion
Most PSPs appear as solitary, incidental pulmonary nodules with nonspecific findings [1], engendering a wide differential diagnosis. Here, we presented a case in which a concomitant diagnosis of thyroid cancer added further complexity to the evaluation of a patient’s pulmonary mass, highlighting the challenge of diagnosing PSPs prior to surgical resection. A previous study investigating the diagnostic accuracy of CT imaging for PSP found that up to 21% of cases were interpreted as primary lung cancer, while 16% were interpreted as a metastasis in patients with an underlying malignancy [12]. While the majority of PSPs appear hypometabolic (SUV <2.5) on FDG-PET [12], at least one other report has described a case of bilateral PSPs with high FDG uptake [13]. In our case, the primary consideration was a low-grade malignancy, such as a carcinoid tumor, given the lesion’s mild avidity on both FDG-PET and SSTR-PET.
Due to its limitations, tissue diagnosis of PSP obtained from small biopsies may lead to overdiagnosis as adenocarcinoma. PSPs are characterized by a biphasic cell population (surface and round cells) organized into four typical architectural patterns: papillary, sclerotic, solid, and hemorrhagic [1]. As a result, the reliability of specimens obtained by fine needle aspiration, core needle biopsy, or intraoperative excision for frozen section is relatively low due to the tumor’s histologic heterogeneity. PSP also shows overlap in immunoprofile with pulmonary adenocarcinoma, both of which express TTF1. Therefore, the use of wider immunostain panels, with particular attention given to their expression in tumor cell subpopulations, is preferred. However, this may or may not be possible in a setting of cytology or small biopsy samples. Nevertheless, preoperative differentiation of PSPs, the majority of which are benign and slow growing, from more aggressive neoplasms can be critical in determining the best therapeutic option for the management of these lesions.
Overall, given their rarity, a standardized approach to treating PSPs has not been established. According to the World Health Organization’s classification of lung tumors, PSPs are categorized as benign adenomas [14], and some authors have advocated for a conservative strategy of observation [15]. A retrospective study by Kim et al. found that all-cause mortality did not differ between patients with PSP and those without, irrespective of whether PSP patients underwent surgery [16]. On the other hand, there have been several instances reported of PSPs with lymph node or extrapulmonary metastases [17], suggesting that a subset of these lesions may have malignant potential. Other studies have described PSPs with rapid or unchecked growth [18], as well as malignant transformation [19]. Moreover, combined lesions have been found comprising both PSP and carcinoid tumor [11]. Such presentations provide support for early surgical resection of PSPs. This may be accomplished via local resection, such as wedge resection and tumor enucleation, segmentectomy, or lobectomy. For patients who are unable to tolerate an operation, stereotactic body radiotherapy or radiofrequency ablation represent non-surgical options [17]. In the case presented above, the decision was made to proceed with right lower lobectomy given the uncertain diagnosis and central location of the pulmonary nodule. It is also worth noting the possibility that local resection may carry a higher risk of recurrence [20]. Thus, though lung-sparing resection (i.e., wedge resection or segmentectomy) could be considered, and perhaps would be in a more well-defined disease process, such as non-small cell lung carcinoma, the lack of evidence supporting sublobar resection in PSP caused us to opt for a more extensive resection to minimize recurrence risk.
Conclusions
PSP presents a challenging diagnostic dilemma due to its nonspecific radiological findings and histological heterogeneity. Difficulty differentiating PSP from malignant lesions, particularly in patients with a history of cancer, can lead to ambiguity regarding surgical management. PSP should remain in the differential diagnosis of well-circumscribed perihilar nodules with mild FDG avidity on PET imaging in patients with known malignancy, and definitive classification may require surgical resection in cases with inconclusive biopsy results. Additional case descriptions are needed to guide future management of these rare tumors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A clinicopathologic study of 100 cases of pulmonary sclerosing hemangioma with immunohistochemical studies: TTF-1 is expressed in both round and surface cells, suggesting an origin from primitive respiratory epithelium Am J Surg Pathol Devouassoux-Shisheboran M Hayashi T Linnoila RI Koss MN Travis WD 9069162420001089581310.1097/00000478-200007000-00002 · doi ↗ · pubmed ↗
- 2Pulmonary sclerosing pneumocytoma - a comprehensive exploration of clinical, radiological, and surgical dimensions IJISRT Pathak D Sun X 76677192024
- 3Sclerosing pneumocytoma: case report of a rare endobronchial presentation Medicine (Baltimore) Khanna A Alshabani K Mukhopadhyay S Lam L Ghosh S 098201910.1097/MD.0000000000015038 PMC 648589730985653 · doi ↗ · pubmed ↗
- 4Pulmonary sclerosing pneumocytoma containing spindle cells with sarcomatoid features: a case report with literature review Diagn Pathol Liang J Du Q Ye X Huang W 161820233675985710.1186/s 13000-023-01296-x PMC 9912602 · doi ↗ · pubmed ↗
- 5Molecular genetic landscape of sclerosing pneumocytomas Am J Clin Pathol Boland JM Lee HE Barr Fritcher EG 39740415520213314559010.1093/ajcp/aqaa 136 · doi ↗ · pubmed ↗
- 6Coexistence of glandular papilloma and sclerosing pneumocytoma in the bronchiole Pathol Int Kitawaki Y Fujishima F Taniuchi S 4254306820182969674310.1111/pin.12677 · doi ↗ · pubmed ↗
- 7Unexpected diagnosis of multiple sclerosing pneumocytomas in a patient with chondrosarcoma of the hand BMJ Case Rep Van Laer SL Vandendriessche A Somville J Van Schil PE 514202110.1136/bcr-2020-238375 PMC 783989633495182 · doi ↗ · pubmed ↗
- 8Pulmonary sclerosing pneumocytoma masquerading as a carcinoid lung tumor Chest Shah V Patton B Lazzaro R Husta B 6341542018