SoxB1-mediated chromatin remodeling promotes sensory neuron differentiation in planarians
Mallory L Cathell, Mohamad A Auwal, Sarai Alvarez Zepeda, Kelly G Ross, Ricardo M Zayas

TL;DR
This study explores how a gene called soxB1-2 helps planarian worms develop sensory neurons by changing chromatin structure.
Contribution
The study reveals soxB1-2's role in chromatin remodeling during sensory neuron differentiation in planarians.
Findings
Disrupting soxB1-2 reduces chromatin accessibility and gene expression in neural and epidermal regions.
31 candidate downstream targets, including castor and mecom, were identified as influenced by soxB1-2.
soxB1-2-responsive genes were detected in rare neural subtypes using head tissue sampling.
Abstract
Understanding how adult stem cells generate neurons is critical for advancing regenerative medicine. However, few in vivo models enable studying how stem cell fates are specified as neurons in an adult body. The planarian Schmidtea mediterranea provides a powerful system for investigating these mechanisms, owing to its abundant adult pluripotent stem cells, termed neoblasts, and its capacity to regenerate a molecularly complex nervous system. The SoxB1 family of transcription factors is broadly implicated in ectodermal lineage commitment. In planarians, the SoxB1 homolog soxB1-2 has been shown to promote neural and epidermal differentiation. However, the mechanisms by which soxB1-2 influences chromatin dynamics and transcriptional programs during adult neurogenesis remain unknown. To address this, we performed ATAC-seq and RNA-seq on neural-rich head tissues to assess how soxB1-2 RNAi…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
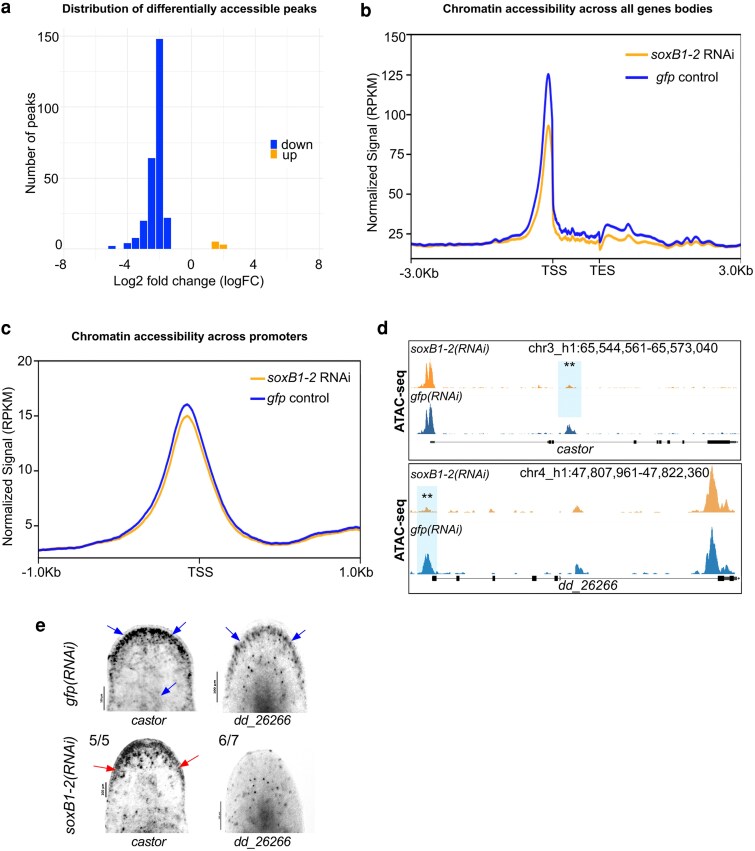 Fig. 1
Fig. 1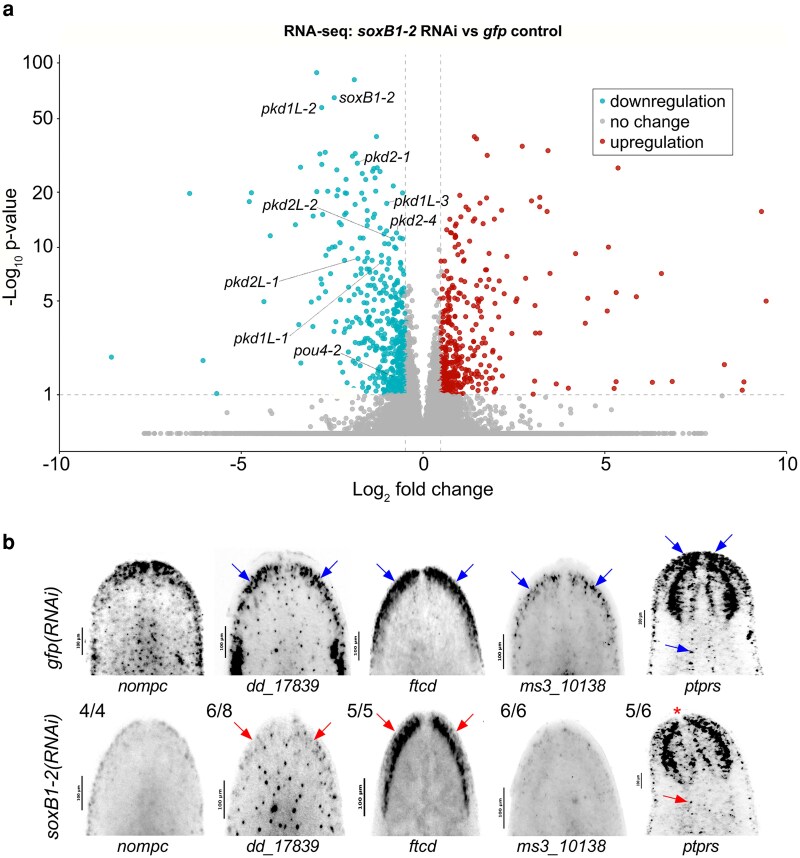 Fig. 2
Fig. 2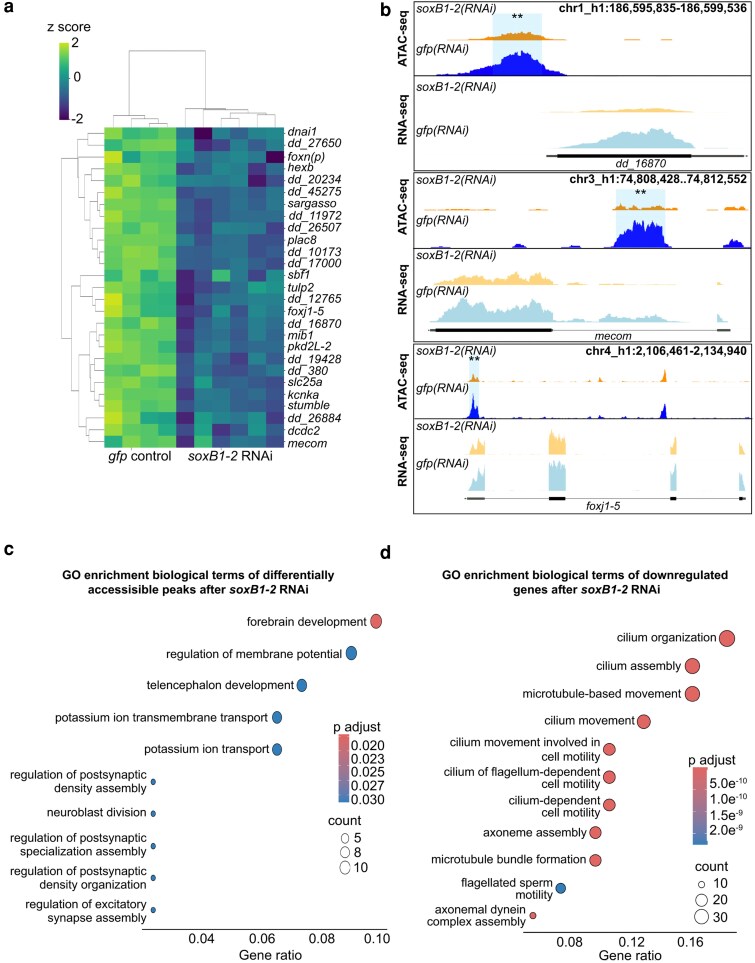 Fig. 3
Fig. 3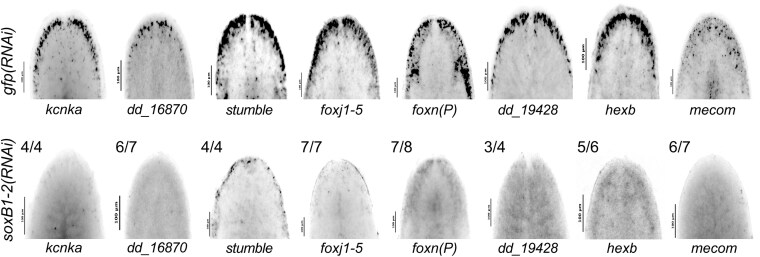 Fig. 4
Fig. 4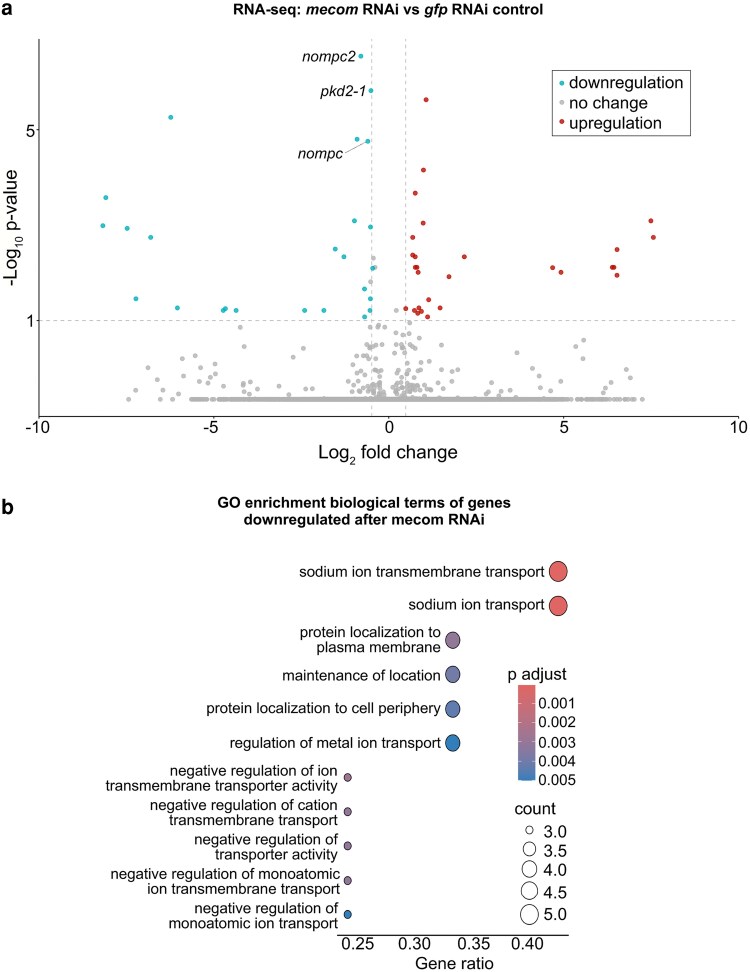 Fig. 5
Fig. 5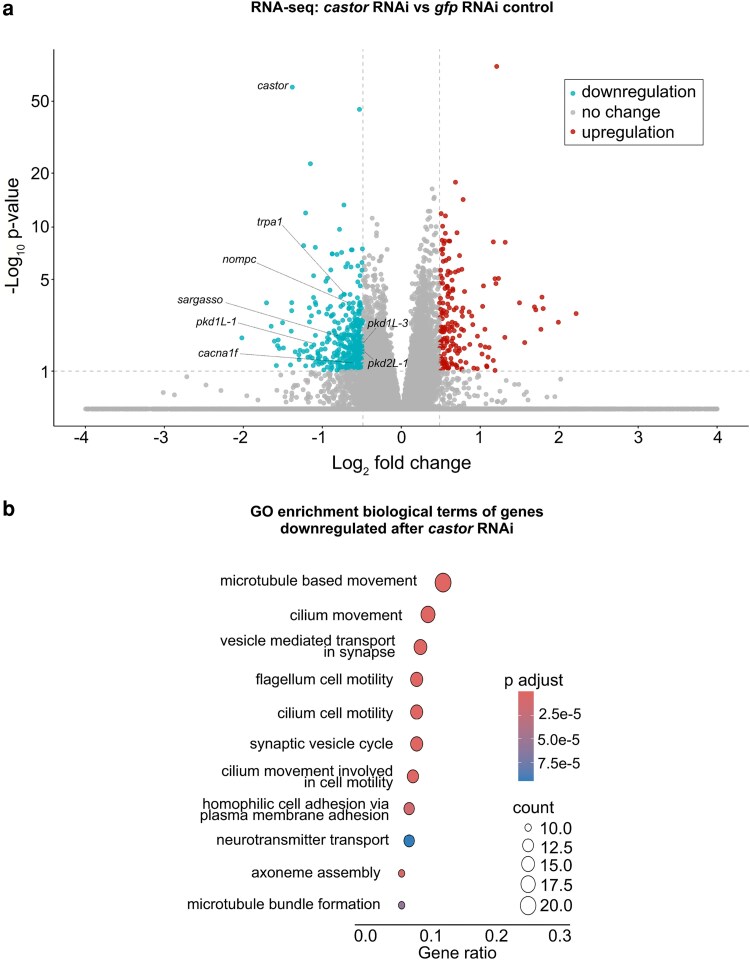 Fig. 6
Fig. 6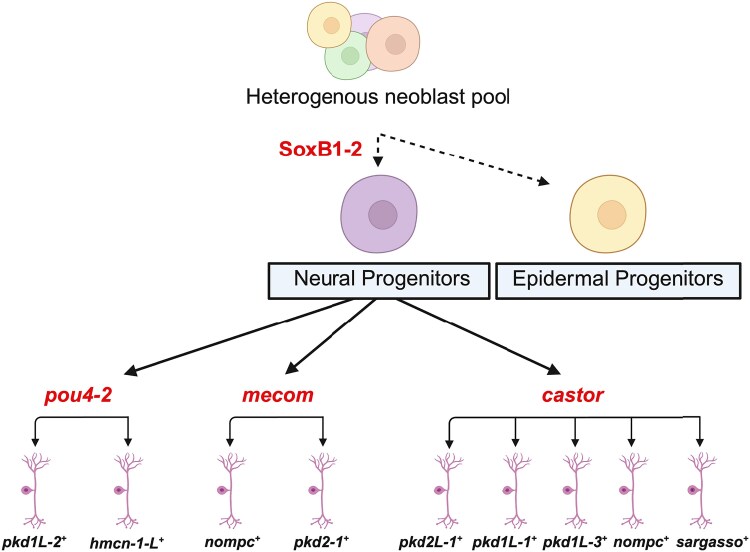 Fig. 7
Fig. 7| Gene | Transcription factor domain classification |
|---|---|
|
| Homeobox |
|
| Homeobox |
|
| Homeobox |
|
| Homeobox |
|
| Zinc finger |
|
| T-cell specific & High mobility group |
|
| Ets-1 |
|
| T-cell specific & High mobility group |
|
| SMAD |
|
| LIM & Homeobox |
- —Rees-Stealy Research Foundation
- —California Institute for Regenerative Medicine10.13039/100000900
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlanarian Biology and Electrostimulation · Developmental Biology and Gene Regulation · Pluripotent Stem Cells Research
Summary
Adult stem cells can regenerate neurons in some animals, but the mechanisms that control this process remain unclear. This study examined how the transcription factor SoxB1-2 regulates neuron formation in the regenerative flatworm Schmidtea mediterranea by combining chromatin accessibility profiling (ATAC-seq) and gene expression analysis (RNA-seq) following reduced SoxB1-2 activity. Loss of SoxB1-2 resulted in a decrease in both chromatin accessibility and expression of neural and sensory genes, including neural transcription factors such as mecom and castor. These results support a model in which SoxB1-2 promotes sensory neuron differentiation through chromatin accessibility changes at sensory neuron gene loci. Together, these findings link SoxB1-2 to chromatin accessibility and sensory neuron gene regulation during adult neurogenesis in planarians.
Introduction
The nervous system is necessary for most of the physiological and behavioral activities of metazoans. However, many adult organisms lack the ability to regenerate lost neurons de novo. Understanding the molecular programming of stem cells during adult neurogenesis may inform strategies to induce neural fates and restore damaged or lost neurons. One highly conserved class of transcription factors that has roles in neural programming during development includes the SoxB1 group proteins. Diversification of the SoxB family into SoxB1 and SoxB2 subfamilies occurred prior to vertebrate diversification, and the ancestral bilaterian had both a SoxB1 and SoxB2 gene (Zhong et al. 2011). Thus, members of the SoxB1 family are conserved across bilaterians, including SoxB1 family members Sox1, 2, and 3 in mammals, of which Sox2 is critical for maintaining stem cell potency and directing the specification of multiple neural progenitor lineages (Zhang and Cui 2014). Beyond development, SoxB1 genes are expressed in adult tissues and contribute to regenerative processes across diverse organisms (Kamachi and Kondoh 2013; Reiprich and Wegner 2015; She and Yang 2015).
SoxB1 homologs regulate neurogenesis across various neural lineages by modulating transcription factor networks that drive terminal fate decisions (Miyagi et al. 2009). They are thought to function as pioneer transcription factors, capable of binding to closed chromatin and altering accessibility, thus facilitating the binding of downstream factors and initiating broad regulatory cascades (Stevanovic et al. 2021). Despite their well-characterized roles in development, the genomic activity and function of SoxB1 genes influencing cell fate specification and differentiation during adult neural regeneration remain poorly understood.
Integrating epigenomic and transcriptomic approaches offers a path to dissect how SoxB1 genes orchestrate adult stem cell-driven neurogenesis. This approach enables identification of SoxB1 regions targeted for chromatin alteration and links the changes in chromatin dynamics to gene regulation, offering mechanistic insight into SoxB1-driven cell fate specification. One such strategy involves comparing chromatin accessibility and gene expression profiles in systems where SoxB1 activity is perturbed to controls. Characterization of these target loci can illuminate how chromatin-level regulation underlies SoxB1-mediated neurogenesis.
The planarian Schmidtea mediterranea presents an ideal model for investigating neural regeneration. Schmidtea mediterranea harbors a large population of pluripotent stem cells, enabling complete tissue regeneration, including a molecularly complex nervous system (Cebrià 2007; Rink 2013, 2018; Ross et al. 2017). Previous work has shown that inhibition of the planarian SoxB1 gene, soxB1-2, results in severe phenotypes, including seizure-like behaviors and loss of mechanosensation (Ross et al. 2018). Co-expression analyses, conducted via in situ hybridization with cell-type-specific markers, along with pseudotime analysis of single-cell RNA sequencing data, suggested that soxB1-2^+^ stem cells constitute ectodermal progenitors destined for epidermal or neuronal fates (Ross et al. 2018).
Functional screens from our lab have identified soxB1-2 downstream targets, including the BRN3/POU4 homolog pou4-2, which plays a role in specifying a subset of mechanosensory neurons (McCubbin et al. 2025). Knockdown of soxB1-2 leads to depletion of the pou4-2^+^ mechanosensory population, suggesting a positive regulatory relationship (McCubbin et al. 2025). Additionally, RNAi of pou4-2 results in loss of the sensory neuron terminal gene pkd1L-2, a phenotype also observed following soxB1-2 disruption (Ross et al. 2018; McCubbin et al. 2025). However, the mechanisms by which soxB1-2 regulates sensory neural fate trajectories remain unclear. One hypothesis is that soxB1-2 acts as a pioneer transcription factor in planarians, binding to silent chromatin and modifying the chromatin landscape at target loci to permit binding of additional transcription factors that direct the differentiation of terminal sensory and epidermal lineages.
In this study, we tested the hypothesis that soxB1-2 primes chromatin for the acquisition of sensory and epidermal progenitor fates through the integration of epigenetic and transcriptomic approaches focused on isolated planarian head tissues to enrich for neuronal cell populations. We assessed changes in chromatin accessibility (via ATAC-seq) and gene expression (via RNA-seq) following soxB1-2 RNAi knockdown to identify direct and functional targets. Our findings revealed that loss of soxB1-2 leads to reduced chromatin accessibility at genomic regions associated with sensory neuronal and epidermal genes, some of which encode transcription factors. Among these, we identified mecom and castor transcription factor genes, which we found are involved in the regulation of mechanosensory signaling genes. Together, these results provide mechanistic evidence that soxB1-2 functions as a pioneer factor in priming chromatin for the emergence of sensory neuronal and epidermal progenitor fates.
Materials and methods
Animal husbandry
Asexual S. mediterranea (clonal strain CIW4) planarians were kept in 9-cup Ziploc reusable containers in 1× Montjuïc salts (1.6 mM NaCl, 1.0 mM CaCl_2_, 1.0 mM MgSO_4_, 0.1 mM MgCl_2_, 0.1 mM KCl, 1.2 mM NaHCO_3_) dissolved in ultrapure filtered water at 20 °C. Animals were fed beef calf liver weekly, as previously described (Merryman et al. 2018). Animals were starved for 1 week prior to all experiments.
RNA interference
Double-stranded RNA (dsRNA) was expressed in HT115 Escherichia coli containing cDNA inserts in either pJC.53.2 (Collins et al. 2010) or pPR-T4P (Liu et al. 2013) as described previously (Ross et al. 2018). Worms measuring 3–4 mm (for in situ hybridization) or 5–7 mm (for ATAC-seq and RNA-seq) were fed dsRNA, mixed 1:3 with a paste composed of 200 μL ultrapure water, 500 μL pureed calf liver, and 40 μL food dye. Animals were cleaned with 1× Montjuïc salt exchanges after each feeding and again the following day. Animals were fed dsRNA twice per week, for a total of five to ten feedings depending on the target gene. soxB1-2 RNAi animals received five feedings, mecom RNAi animals received six feedings, and castor RNAi animals received 10 feedings*. Green fluorescent protein* (gfp) dsRNA was used as the negative control. For soxB1-2 RNAi experiments, animals were amputated anterior to the pharynx 3 days after the fifth feeding for RNA-seq and ATAC-seq or fixed for in situ hybridization 7 days after the fifth feeding. For mecom and castor RNAi experiments, whole worms (3–4 mm) were fixed for in situ hybridization or processed for RNA-seq 7 days after the sixth feeding or tenth feeding, respectively.
ATAC-seq assay and analysis
Two independent replicates of both soxB1-2 RNAi and gfp control groups (30 heads per replicate) were placed directly into ATAC-seq lysis buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl_2_, 0.1% Digitonin, 0.1% Tween-20, 0.1% NP-40, in water) and pipetted up and down until homogeneous. The slurry was run through a 50-micron filter, and the ATAC-seq assay was performed as previously described (Corces et al. 2017) with the Illumina tagment DNA enzyme and buffer kit (cat# 20034197). Samples were purified using Zymo DNA Clean and Concentrator-5 Kit (cat# D4014) and stored at −20 °C until sequencing. Samples were quality-checked on an Agilent 2100 Bioanalyzer and sequenced to a read depth of 38 million paired-end reads on the Illumina NovaSeq 6000 at the University of California, San Diego IGM Genomics Center, Inc. (San Diego, CA).
ATAC-seq libraries were assessed for quality using FastQC (v0.11.9) (Andrews 2010), and fragment size distributions were evaluated with ATACseqQC (v1.26.0) (Ou et al. 2018). Adapter trimming and merging of forward and reverse reads were performed using NGmerge (v0.3) (Gaspar 2018). Trimmed reads were aligned to both the S3h1 and S2F2 S. mediterranea genome assemblies using Bowtie2 (v2.4.4) (Langdon 2015). Samtools (v1.2.0) software was used to remove mitochondrial and improperly paired reads and to generate BAM files (Li et al. 2009). To control for differences in sequencing depth across ATAC-seq libraries, reads were down-sampled prior to peak calling and footprinting to equalize the number of properly paired, uniquely mapped reads between conditions. Subsampling was performed using samtools (v1.17) with the -s flag, which performs Bernoulli sampling on alignments. For each replicate, BAM files were name-sorted and subsampled using a unique random seed and fraction to achieve matched read counts across samples. Picard (v2.26.4) was used to remove PCR duplicates. Deduplicated BAM files were used for peak calling with Genrich (BioGrids) using parameters -j -v -p 0.01 -a 150. Peaks were called for individual and pooled replicates. Genrich-generated BedGraph files were converted to BroadPeak format and processed as previously described (Reske et al. 2020). Consensus peaks were mapped to gene annotation models for both genomes using BEDTools (v2.31.1) (Quinlan and Hall 2010). RPKM-normalized coverage tracks (bigWig) and profile plots were generated using deepTools (v3.5.1) (Ramirez et al. 2014). Differential accessibility analysis between gfp controls and soxB1-2 RNAi samples was performed using csaw (v1.36.0) (Lun and Smyth 2016), applying an FDR cutoff ≤ 0.05 as described previously (Reske et al. 2020). Gene Ontology enrichment was performed using enrichGO from clusterProfiler (v4.12.6) (Yu et al. 2012). The resulting datasets were compared to the human proteome using BLASTX (cut-off e-value < 1e^−3^). Human UniProt IDs (UniProt Consortium 2015) were used as input for annotation and overrepresentation analysis. Statistical analyses were carried out in R (v4.4.0) (R Core Team 2024), and figures were generated using ggplot2 (Wickham 2009).
Motif enrichment analysis
Differentially accessible (DA) regions were first identified independently using csaw and edgeR (Bioconductor v3.19), based on a unified peak set generated across all ATAC-seq samples. Peaks with FDR ≤ 0.05 were considered significantly differentially accessible. Peaks with positive log₂FC values were classified as more accessible in soxB1-2(RNAi) samples, while peaks with negative log₂FC values were classified as more accessible in controls. Significant DA peaks were separated by direction and resized to a fixed width of 350 bp centered on the peak midpoint using GenomicRanges to standardize regions for footprinting. Footprint scores were computed across the unified peak set for each biological replicate using TOBIAS FootprintScores (Bentsen et al. 2020). Condition-level footprint signal tracks were generated by averaging replicate footprint bigWig files using bigwigCompare (deepTools 3.5.6; –operation mean) to produce a single representative footprint track for control and soxB1-2 RNAi samples.
Differential TF binding analysis was performed using TOBIAS BINDetect (Bentsen et al. 2020), supplying the averaged footprint tracks, the unified resized peak set, and the S2F2 S. mediterranea genome. A curated set of transcription factor motifs derived from JASPAR (Castro-Mondragon et al. 2022) was used as input motifs. BINDetect was run with default settings to identify motif sites and infer differential motif occupancy between conditions (Quinlan and Hall 2010; Ramirez et al. 2014; Bentsen et al. 2020).
Gene identification and cloning
The soxB1-2 cDNA used in all experiments was previously cloned into pJC.53.2 (Collins et al. 2010; Ross et al. 2018) in our lab. All other gene sequences were identified through PlanMine (Rozanski et al. 2019) using the dd_Smed_v6 transcriptome and cloned from S. mediterranea cDNA into pPR-T4P (Liu et al. 2013) using ligation-independent cloning and gene-specific primers as described in Adler and Alvarado (2018) (all primer sequences and gene identifiers listed in Supplementary Table 6). In the text, dd_Smed_v6 transcripts are referred to as dd_transcript no. for brevity.
Wholemount in situ hybridization
Antisense probes were generated from DNA templates amplified from pJC.53.2 (Collins et al. 2010; Ross et al. 2018) or pPR-T4P (Liu et al. 2013), incorporating digoxigenin-labeled UTPs, as previously described (Pearson et al. 2009). Samples were incubated in 5% N-acetyl cysteine (NAC) in 1× PBS for five minutes to euthanize the animals, then fixed in 4% formaldehyde in 1× PBS with 0.1% Triton X-100 for 25 min. Animals were processed for in situ hybridization as described previously (King and Newmark 2013), except for mecom, a low-abundance transcript, for which hybridization and heated washes were performed at 58 °C.
RNA-seq assay and analysis
For soxB1-2 RNAi experiments, head fragments from four independent gfp control and six independent soxB1-2 RNAi replicates were homogenized in TRIzol (Invitrogen), and RNA was extracted and purified according to the manufacturer's protocol. For mecom and castor RNAi experiments, the same procedure was applied to whole worms, using five independent gfp controls and five mecom or castor RNAi replicates. RNA was treated with the TURBO DNA-free Kit and column-purified using the Zymoclean Miniprep Kit (cat. no. 11-330). RNA libraries were sequenced on an Illumina NovaSeq 6000 platform to a depth of 24 million reads per sample (12 million paired-end reads) at MedGenome, Inc. (Foster City, CA). Reads were pseudo-aligned to the S2F2 transcriptome using kallisto (Bray et al. 2016), and differential gene expression analysis was performed using Bioconductor (Huber et al. 2015) and DESeq2 (Love et al. 2014) with an FDR (P-adj) cut-off value ≤ 0.1 and fold-change threshold of 1.4. Gene Ontology enrichment was conducted using enrichGO from clusterProfiler (v4.12.6) (Yu et al. 2012). Gene sets were compared to the human proteome using BLASTX (e-value cut-off < 1e^−3^). Resulting UniProt human gene IDs were used for annotation and GO overrepresentation analysis. Statistical analyses were carried out in R (v4.4.0) (R Core Team 2024), and figures were generated using ggplot2 (Wickham 2009).
Results
Neural genes exhibit reduced chromatin accessibility following soxB1-2 RNAi
To investigate how soxB1-2 activity influences chromatin accessibility, we performed ATAC-seq (Buenrostro et al. 2015) on head tissues (amputated in the “neck” region anterior to the pharynx and posterior to the auricles; see Supplementary Fig. 1) from soxB1-2(RNAi) and gfp(RNAi) (control) animals to enrich for neural cell populations. The optimal RNAi feeding regimen and timepoint for obtaining soxB1-2 knockdown samples were determined based on prior studies conducted in our laboratory (Ross et al. 2018). Sequencing data were aligned to the S. mediterranea genome assembly (S3h1) (Ivankovic et al. 2024), and peaks were mapped to the nearest annotated gene model (see Materials and Methods). Peak calling identified 27,560 reproducible accessible chromatin regions in control animals and 26,679 regions in soxB1-2(RNAi) animals with 66 and 64% overlap between biological replicates, respectively (P ≤ 0.01, minimum peak length of 150 bp; see fragment size distributions from each ATAC-seq replicate in Supplementary Fig. 2a). Replicate accessibility profiles were highly correlated (Pearson r = 0.96 for both conditions; Supplementary Fig. 2b, c and Supplementary Table 1), confirming strong reproducibility across samples.
Subsequent differential accessibility analysis using csaw (FDR ≤ 0.05; see Materials and Methods) identified 264 significantly differentially accessible (DA) peaks. The majority of these (257 peaks) were present in controls but significantly reduced in soxB1-2(RNAi) animals (down). In comparison, only seven peaks were significantly increased in soxB1-2(RNAi) animals relative to controls (up) (Supplementary Table 1). This overall reduction in accessible peaks in the soxB1-2(RNAi) animals was also observed as a cumulative decrease in signal across all gene bodies and promoter regions in the soxB1-2 RNAi groups (Fig. 1a–d). Thirteen genes associated with transcription factor binding were identified within DA chromatin regions, such as castor (Fig. 1d, e), which has been linked to neurodevelopmental processes in both invertebrate and vertebrate models (Isshiki et al. 2001). Most of the peaks that were shared between the gfp and soxB1-2 RNAi groups were located in distal intergenic regions (54.7 and 57.54%, respectively; Supplementary Fig. 3). Promoter regions accounted for 22% of peaks in controls and 21.25% in soxB1-2 RNAi samples, while internal intronic regions contained 17.53 and 16.23%, respectively. Only a small fraction mapped to internal exonic regions, 3% in controls and 2.56% in soxB1-2 RNAi. In contrast, DA peaks showed a markedly different distribution: only 25.52% were in distal intergenic regions, while 36.39% mapped to internal intronic regions and 9.96% to internal exonic regions (Supplementary Fig. 3).
soxB1-2 RNAi reduces chromatin accessibility in S. mediterranea. a) Histogram of log2 fold changes (log2FC) in differentially accessible ATAC-seq peaks between gfp and soxB1-2 RNAi groups, with 264 peaks exhibiting decreased accessibility (blue) and seven peaks showing increased accessibility (orange) (FDR ≤ 0.05). Sample size: N = 2 for both control and soxB1-2 RNAi groups. b) Profile plot of global ATAC-seq signal across gene bodies in gfp control (blue) and soxB1-2 RNAi (orange) groups. TSS, transcription start site; TES, transcription end site. c) Profile plot of global ATAC-seq signal across promoter regions in gfp control (blue) and soxB1-2 RNAi (orange) groups. TSS, transcription start site. d) Genome tracks showing reduced chromatin accessibility at the mecom and dd_26266 locus. e) In situ hybridization of genes enriched in neural and ciliated neural cell populations that display reduced chromatin accessibility post-soxB1-2 RNAi. Blue arrows indicate regions of in situ signal loss following soxB1-2 RNAi, with specific anatomical areas marked using red arrows. Sample size: For controls, N ≥ 3 worms were analyzed, and all samples showed consistent expression patterns. For RNAi-treated animals, the number of biological replicates (N) is indicated at the top left of each treatment group.
Distinguishing direct chromatin-opening activity of SoxB1-2 from indirect effects mediated through downstream regulators is an important consideration. To evaluate this, we performed motif enrichment analysis following Neiro et al. (2022) using differentially accessible regions between control and soxB1-2(RNAi) animals. Sox-family binding motifs, including SoxB1-like sites, were significantly enriched in regions that remain accessible in control animals relative to soxB1-2(RNAi) (Supplementary Fig. 4). This supports a model in which SoxB1-2 contributes to establishing or maintaining chromatin accessibility at these loci, consistent with pioneer-like activity as described for other SoxB1 factors (Neiro et al. 2022).
To validate ATAC-seq results and determine whether genes that lose chromatin accessibility are downstream targets of soxB1-2, we selected two genes, castor and dd_26266, for in situ hybridization following soxB1-2 RNAi. These genes are expressed in the same neural or ciliated neural clusters as soxB1-2 (Fincher et al. 2018). In situ analysis revealed reduced expression of castor and dd_26266 in the head margin (Fig. 1e), a region enriched in sensory neurons (Okamoto et al. 2005; Wang et al. 2016; Reho et al. 2022). Additionally, castor exhibited further expression loss in the dorsal epidermis and dorsal ciliated stripe (an organ associated with water flow sensing in planarians) (Hyman 1951; Farnesi and Tei 1980; Ross et al. 2018) (Fig. 1e). The loss of chromatin accessibility and gene expression in sensory neuron-rich regions supports a key role of soxB1-2 in regulating downstream genes with potential roles in neuronal specification or terminal differentiation, including other transcription factors.
soxB1-2 RNAi results in loss of sensory and neural gene expression
To examine the relationship between gene regulation and chromatin accessibility in the soxB1-2 RNAi amputation scheme, we repeated soxB1-2 RNAi experiments and performed RNA-seq on RNA extracted from animal heads (Supplementary Fig. 1). RNA-seq analysis identified 533 downregulated genes meeting our significance cut-offs (fold-change ≥ 1.4, P-adj ≤ 0.1) (Fig. 2a, Supplementary Table 2). Among these, we observed a significant reduction in genes previously shown to be downstream targets of soxB1-2 that are involved in neural and mechanosensory function. These included pou4-2, pkd1L-1, pkd1L-2, pkd1L-3, pkd2L-1, pkd2L-2, pkd2-1, and pkd2-4 (Fig. 2a) (Ross et al. 2024; McCubbin et al. 2025).
Genes identified by ATAC-seq are regulated by soxB1-2. a) Volcano plot of differentially expressed genes in gfp control vs soxB1-2 RNAi groups, with downregulated (blue) and upregulated (red) genes (FC ≥ 1.4, P-adj ≤ 0.1). Sample size: N = 4 and N = 6 for control and soxB1-2 RNAi groups, respectively. b) In situ hybridization of putative sensory neural genes identified by RNA-seq following soxB1-2 RNAi. Blue arrows indicate regions of in situ signal loss following soxB1-2 RNAi. Red arrows mark anatomical regions with undetectable signal; red asterisk denotes loss of expression in the tip of the head. Sample size: For controls, N ≥ 3 worms were analyzed, and all samples showed consistent expression patterns. For RNAi-treated animals, the number of biological replicates (N) is indicated at the top left of each treatment group.
To validate the differential expression results, we isolated all genes that were significantly differentially expressed between gfp controls and soxB1-2 RNAi-treated animals and generated a heatmap (Supplementary Fig. 5). This visualization confirmed consistent expression patterns across replicates, with upregulated genes in the control group showing corresponding downregulation in the RNAi group, and vice versa, indicating robust sample clustering for this gene set. To further investigate these expression changes and validate patterns observed in our dataset, we selected no mechanoreceptor protein channel c (nompc), dd_17839, formimidoyltransferase cyclodeaminase (ftcd), ms310138, and receptor-type tyrosine-protein phosphatase S (ptprs) as representative genes downregulated in the soxB1-2(RNAi) group. These genes were co-expressed with soxB1-2 specifically within neural and ciliated neural populations in published single-cell RNA-seq datasets (Wurtzel et al. 2015) (Supplementary Fig. 6). To assess whether they were potential downstream targets of soxB1-2, we performed in situ hybridization on gfp and soxB1-2 RNAi animals. Consistent with the RNA-seq results, these genes exhibited reduced expression following soxB1-2 RNAi, particularly in sensory neuron-enriched regions concentrated in the head rim and dorsal ciliated stripe (Fig. 2b).
The observed transcriptional changes following soxB1-2 RNAi prompted us to examine how these patterns compared to those in previous RNA-seq datasets. Given that our prior studies were conducted on RNA extracted from whole worms (Ross et al. 2018), we sought to determine whether broader tissue sampling may have masked gene expression changes in rare neural populations in the whole-worm datasets. Comparing this head fragment dataset to previous whole-worm soxB1-2 RNAi RNA-seq results identified 134 shared downregulated genes (Supplementary Table 2). Gene Ontology (GO) enrichment analysis indicated that shared genes are primarily associated with “ion homeostasis”, “transmembrane transport regulation”, and “cell communication”, with key processes including “negative regulation of ion and cation transport”, “sodium ion transmembrane transport”, and “response to metal ions” (Supplementary Fig. 7a). Additionally, the presence of electrical coupling-related terms suggests a role in direct cell-to-cell communication, particularly in excitable tissues such as neurons and muscle.
In contrast, 396 genes were uniquely identified in the RNA-seq dataset from head tissue compared to our previously published day-14 whole worm dataset (Supplementary Table 2). GO analysis of these genes revealed enrichment in “ciliary and flagellar dependent cell motility”, “movement”, “axoneme assembly”, and “microtubule bundle formation” (Supplementary Fig. 7b). These features are fundamental not only for locomotion and fluid movement in the ventral epidermis but also for sensory-neural functions given the presence of cilia in sensory neurons (Rompolas et al. 2013; Azimzadeh and Basquin 2016).
To explore whether loss of soxB1-2 causes changes in gene expression that could be correlated with neoblast progeny adopting alternative differentiation fates, we also performed GO enrichment analysis on genes upregulated in soxB1-2(RNAi) animals. This upregulated gene set was enriched for metabolic processes, xenobiotic detoxification, potassium ion homeostasis, and endocytic pathways (Supplementary Fig. 7c). These categories are not strongly associated with known alternative lineage programs or fate switches. However, because these data were generated from bulk head tissue, the upregulated signatures could also reflect shifts in the relative abundance of specific cell types following loss of soxB1-2-expressing populations. Thus, we find no evidence from this analysis for a clear transcriptional shift toward alternative lineage programs. Consistent with a primary effect on neural gene expression programs, RNA-seq of head tissue revealed additional downregulated genes enriched for ciliated sensory neuron function that were not detected in whole-worm RNA-seq, suggesting a more specialized role for soxB1-2 in these populations. We therefore next examined the genome-wide overlap between RNA-seq and ATAC-seq datasets to further define the regulatory effects of soxB1-2 loss.
Integrating ATAC-seq and RNA-seq identifies candidate downstream targets of soxB1-2 in neural and ciliated neural cells
To uncover potential downstream targets of soxB1-2, we integrated our head region ATAC-seq and RNA-seq datasets. This combined approach allowed us to identify genes that not only exhibit changes in chromatin accessibility but also corresponding changes in transcriptional output. While we found consistent signals across both assays in some genes, others were detected only by one method, underscoring the complementary nature of chromatin and transcriptomic profiling.
While differences between ATAC-seq and RNA-seq datasets are expected due to the distinct regulatory layers they capture, their integration revealed a set of 31 genes exhibiting both decreased chromatin accessibility and transcriptional downregulation (Fig. 3a), highlighting them as strong candidates for direct downstream targets of soxB1-2. We assessed enrichment of the 31 candidate genes in neural and ciliated neural cell clusters and examined their co-expression with soxB1-2 using single-cell RNA-seq datasets. Single-cell databases indicated most of the 31 genes were either enriched in the same neural and ciliated neural clusters where soxB1-2 is also enriched or co-expressed with soxB1-2 within these populations (Wurtzel et al. 2015; Fincher et al. 2018). Exceptions included dd_380, enriched in epidermal clusters, and dd_8471 and dd_8567, which lacked specific cluster enrichment and exhibited broad co-expression across all cell types (Supplementary Table 3).
ATAC-seq and RNA-seq reveal coordinated loss of chromatin accessibility and downregulation of neural genes. a) Heatmap of differentially expressed genes identified through RNA-seq (FC ≥ 1.4, P-adj ≤ 0.1) in gfp control vs soxB1-2 RNAi groups. Z-scores indicate upregulation (2), no change (0), and downregulation (−2). Labeled genes are putative neural genes identified from both ATAC-seq and RNA-seq experiments. b) Genome tracks showing reduced chromatin accessibility and gene downregulation following soxB1-2 RNAi at putative neural gene loci (foxn(p), foxj1-5, and stumble) in S. mediterranea. c) GO term enrichment analysis of genes associated with reduced chromatin accessibility following soxB1-2 knockdown. d) GO term enrichment analysis of downregulated genes in RNA-Seq experiments following soxB1-2 knockdown.
Among the genes identified, we were particularly interested in transcription factors, as these are likely to act as intermediate regulators within the broader soxB1-2 gene regulatory network. By integrating ATAC-seq and RNA-seq, we could prioritize transcription factors that were either losing accessibility and downregulated at the chromatin and transcriptional levels or uniquely detected through chromatin accessibility changes, suggesting potential regulatory priming. This allowed us to expand the architecture of the soxB1-2 pathway by highlighting candidate nodes acting downstream of soxB1-2.
Notably, transcription factors, including castor, were exclusively detected through ATAC-seq assays (Fig. 1d); they were not identified as differentially expressed in RNA-seq following soxB1-2 RNAi treatment (Table 1), potentially due to low transcript abundance or subtle expression changes. However, some transcription factors, such as mecom and foxj1-5, exhibited significant decreases in both chromatin accessibility and expression in soxB1-2(RNAi) animals (Fig. 3b). The mecom gene is a neural transcription factor that is expressed in the same neural and ciliated neural clusters as soxB1-2 (Wurtzel et al. 2015; Fincher et al. 2018). ATAC-seq and RNA-seq peaks at the mecom locus in both soxB1-2(RNAi) and control animals are shown in Fig. 3b, demonstrating a significant reduction in chromatin accessibility following RNAi of soxB1-2.
GO analysis revealed distinct enrichment patterns between the ATAC-seq and the RNA-seq datasets (Fig. 3c, d). ATAC-seq enrichment was associated with direct neural functions, with “forebrain development” as the top term, alongside pathways such as “potassium ion transport”, which is critical for neural processes like action potential generation and neurotransmitter release (Meir et al. 1999; Wang et al. 2023). Additionally, ATAC-seq revealed enrichment for “synaptic development and assembly” (Fig. 3c). In contrast, RNA-seq data GO enrichment predominantly involved cilium-related processes, including movement, assembly, and development (Fig. 3d), consistent with our previous findings on gene expression associated with ciliated neurons and the epidermis (Ross et al. 2018). The enrichment of cilium-related terms aligns with the observed loss of chromatin accessibility and gene downregulation following soxB1-2 RNAi in genes that have sensory or mechanosensory-related functions. Additionally, this gene enrichment was reflected in spatial expression data, as in situ hybridization of these potential soxB1-2 target genes showed a decrease in expression in body regions strongly associated with ciliated sensory neurons (Figs. 1e, 2b, and 4 and Supplementary Table 3).
In situ hybridization validation of neural genes identified in ATAC-seq and RNA-seq. a) In situ hybridization of neural genes expressed in the head rim, dorsal ciliated stripe, and periphery in gfp control (top) and soxB1-2 RNAi (bottom) animals. Sample size: For controls, N ≥ 3 worms were analyzed, and all samples showed consistent expression patterns. For RNAi-treated animals, the number of biological replicates (N) is indicated at the top left of each treatment group.
In addition to neural gene regulation, we observed overlapping chromatin accessibility loss and downregulation of multiple genes enriched in epidermal progenitor and late epidermal lineages, as well as genes shared by both neural and epidermal populations. These include candidates potentially involved in epidermal fate determination, such as dd_12765, foxj1-5, dd_13134, and dd_380 (Supplementary Table 3) (Vij et al. 2012; Fincher et al. 2018; Pascual-Carreras et al. 2021). The presence of both neural and epidermal signatures suggests that soxB1-2 plays a broader role in ectodermal lineage identity. However, because this study primarily focuses on its function in neuronal lineages, our downstream analyses were concentrated on genes enriched in neural populations.
To validate that genes showing overlap in both datasets are regulated by soxB1-2, we cloned 14 of the 31 candidate targets and assessed their expression using in situ hybridization in both control and soxB1-2 RNAi animals. All tested genes were expressed in the head margin and other regions enriched for sensory neurons, and all showed reduced or absent expression following soxB1-2 knockdown (Fig. 4, Supplementary Fig. 8). These results support the model that soxB1-2 activity regulates these genes and provide strong functional validation of our integrated RNA-seq and ATAC-seq approach for identifying soxB1-2 targets in neural and ciliated neural populations. Furthermore, the validated genes include several with known or predicted roles in neural development or function (Fig. 3c). For example, kcnka, a potassium channel homolog, was previously shown to be downregulated following coe RNAi, a neural transcription factor involved in nervous system development in S. mediterranea (Cowles et al. 2014).
Distinct and overlapping roles of mecom and castor in mechanosensory signaling, ion transport, and ciliary regulation
Based on the significant loss of chromatin accessibility at the mecom and castor loci and their downregulation following soxB1-2 RNAi, we investigated these genes as candidate downstream transcription factor targets of soxB1-2 activity. While only mecom was significantly reduced in the soxB1-2(RNAi) RNA-seq dataset, we observed a reduction in expression for both genes in sensory regions by in situ hybridization (Figs. 1e and 4). Combined with their known roles in neural and sensory development in other model systems, these observations prompted us to define the regulatory targets of mecom and castor to better understand their contribution to the soxB1-2-dependent transcriptional network. To determine the optimal RNAi timepoints for transcriptomic analysis, we performed a time-course experiment across ten RNAi feedings and monitored mecom and castor expression levels. mecom transcript depletion was consistently observed by RNAi day 24 (Supplementary Fig. 9a, b). castor transcript loss occurred by day 37 (Supplementary Fig. 10a, b). RNA-seq analysis revealed that knockdown of mecom and castor resulted in the significant downregulation of 25 and 391 genes, respectively (Supplementary Tables 4 and 5). The relatively small number of mecom-dependent genes detected is consistent with its low baseline expression in a small number of cells (Fig. 4). Among the downregulated genes, nompc was the only mechanosensory receptor reduced in both mecom and castor RNAi samples (Figs. 5 and 6; Supplementary Figs. 9 and 10). pkd2-1, another known soxB1-2 target involved in mechanosensation, was downregulated exclusively in the mecom dataset (Fig. 5a and Supplementary Fig. 9c). In contrast, castor knockdown led to downregulation of additional mechanosensory and ion channel-related genes, including transient receptor potential ankyrin 1 (trpa1) and pkd family members pkd1L-1, pkd1L-3, and pkd2L-1 (Fig. 6a and Supplementary Fig. 10c), which were all previously implicated in soxB1-2-dependent mechanosensory regulation (Ross et al. 2024). In addition to mechanosensory and ion channel-related genes, castor RNAi resulted in the downregulation of numerous genes associated with ciliary structure and function, including dystonin (dst) and multiple dynein component homologs. These findings suggest that while mecom and castor regulate distinct gene sets, both contribute to a mechanosensory and ion channel gene expression network downstream of soxB1-2.
mecom RNAi disrupts mechanosensory and ion channel-like gene expression. a) Volcano plot of differentially expressed genes in gfp control vs mecom RNAi groups, with downregulated (blue) and upregulated (red) genes (FC ≥ 1.4, P-adj ≤ 0.1). Sample size: N = 5 for control and mecom RNAi groups. b) GO term enrichment analysis of downregulated genes following mecom knockdown.
castor RNAi disrupts genes involved in synapse and ciliary function. a) Volcano plot of differentially expressed genes in gfp control vs castor RNAi groups, with downregulated (blue) and upregulated (red) genes (FC ≥ 1.4, P-adj ≤ 0.1). Sample size: N = 5 for control and castor RNAi groups. b) GO term enrichment analysis of downregulated genes following castor knockdown.
GO enrichment analysis of downregulated genes in mecom RNAi revealed significant enrichment for “ion transporter activity”, consistent with the functional roles of several affected genes. Some of these genes encode TRP channels, which detect mechanical stimuli and mediate ion flux across membranes, including pkd2-1 and nompc. Additionally, genes involved in ion channel regulation, such as cav3, which encodes a homolog to human caveolin 3, were also downregulated (Supplementary Table 4), establishing a potential role for mecom in planarians in mechanosensory signaling and ion transport regulation. In contrast, although multiple mechanosensory and ion channel-related genes were downregulated in the castor RNA-seq dataset following RNAi, enriched GO terms were primarily associated with ciliary functions, including “cilium movement”, “microtubule bundle formation”, “axoneme assembly”, and “microtubule-based movement” (Fig. 6b). Given the role of cilia in planarian sensory neurons, this enrichment is consistent with castor's potential role in sensory regulation. GO terms related to the detection of mechanical stimuli were also enriched, supporting the observed downregulation of mechanosensory genes, such as nompc and members of the PKD gene family, in planarians. Additionally, GO terms enriched among castor-regulated genes overlapped with those identified in the soxB1-2 RNAi dataset (Figs. 3d and 6b). Together, these findings suggest that castor plays a key role in regulating genes involved in mechanosensation, ion transport, and ciliary function, positioning it as a critical component of the soxB1-2 downstream regulatory network in planarian mechanosensory biology.
Discussion
Evidence for a transcriptional activator role of soxB1-2 in chromatin remodeling during neural and epidermal specification
Planarians provide a powerful in vivo model for investigating adult neurogenesis and lineage specification. Leveraging this system, we have begun to dissect the role of soxB1-2 in directing adult stem cell fate toward ectodermal lineages, with a particular focus on neural differentiation. Although prior studies established that soxB1-2 promotes ectodermal identity in planarians (Ross et al. 2018), the molecular mechanisms underlying this process had not been explored. In both vertebrate and invertebrate systems, SoxB1 family members are known to play critical roles in maintaining pluripotency, sustaining progenitor identity, and promoting ectodermal development (Buescher et al. 2002; Meulemans and Bronner-Fraser 2007; Kurtova et al. 2024). However, except for mammalian SOX2 homologs, few studies have addressed the mechanisms by which SoxB1 factors regulate these processes (Iwafuchi-Doi and Zaret 2014; Soufi et al. 2015). Here, we show that loss of soxB1-2 leads to a marked reduction in chromatin accessibility at numerous neuronal and epidermal loci, some of which encode transcription factors. These findings suggest that soxB1-2 may function as a transcriptional activator, initiating chromatin remodeling to prime progenitor cells for neural and epidermal differentiation.
Although our findings indicate that soxB1-2 modulates chromatin accessibility and regulates downstream transcription factors including mecom, foxj1-5, and castor, we have not yet provided direct biochemical evidence of SoxB1-2 binding to closed chromatin. However, motif enrichment analysis revealed that regions losing accessibility upon soxB1-2 knockdown are significantly enriched for Sox-family motifs, particularly those resembling the SoxB1 subclass, consistent with a model in which SoxB1-2 directly establishes or maintains chromatin accessibility at its target loci. To definitively test this pioneer-like mechanism, future work will require generation of a SoxB1-2–specific antibody to enable chromatin profiling approaches such as CUT&RUN or ChIP–seq. Integrating these assays with ATAC-seq will allow us to distinguish direct vs indirect SoxB1-2 regulatory events and determine whether SoxB1-2 engages inaccessible chromatin in vivo.
Despite the expanding use of transcriptomic tools in planarians, genome-scale chromatin accessibility datasets remain scarce. Applying ATAC-seq in this system not only enables mechanistic investigation of soxB1-2 function but also broadens the epigenomic resources available for this emerging regenerative model. By generating high-quality chromatin accessibility profiles from control and soxB1-2(RNAi) animals, our study establishes a foundational dataset for investigating adult neural fate specification and offers a framework for integrating chromatin and transcriptional regulation in planarian stem cell biology.
Mechanistic insights into soxB1-2 function via ATAC-seq and RNA-seq integration post-RNAi
ATAC-seq has enhanced our understanding of the chromatin landscape in planarian neoblasts and throughout regeneration (Pascual-Carreras et al. 2023; Poulet et al. 2023; Ivankovic et al. 2024). When combined with RNAi-mediated perturbations, this approach has enabled detailed investigation of the epigenomic effects resulting from disruption of key regulators, including the stem cell marker smedwi-2, the chromatin remodeler BPTF (a core subunit of the Nucleosome Remodeling Factor, NuRF), and BAF complex components such as brg1 and smarcc2 (Li et al. 2021; Wiggans et al. 2023; Verma et al. 2025).
More broadly, the integration of ATAC-seq with RNA-seq has become a widely adopted strategy for linking chromatin accessibility to gene expression, with applications spanning neural development, cancer biology, and chromatin remodeling dynamics (Xu et al. 2023; Bai et al. 2024). Building on this, recent studies have begun applying this integrative strategy in planarians following RNAi (Bai et al. 2024). In our work, combining ATAC-seq and RNA-seq enabled a mechanistic analysis of soxB1-2-driven lineage specification and the regulation of its direct downstream targets. Although both assays were performed on head fragments, the number of biological replicates differed (ATAC-seq: N = 2 per condition; RNA-seq: N = 4 to 6 per condition), and we interpret cross-assay comparisons in light of these differences in statistical power. Nevertheless, the concordant loss of accessibility and gene expression at key neural regulators across assays supports a robust requirement for soxB1-2 in neural lineage specification. We identified 31 genes that exhibited both differential chromatin accessibility and altered transcript levels following soxB1-2 knockdown. These genes were either enriched in or co-expressed with soxB1-2 across multiple cell types, including ciliated neural cells, general neural populations, early epidermal progenitors, and neoblasts (Supplementary Fig. 6 and Supplementary Table 3). Among these, ciliated neural cells showed the highest levels of enrichment and co-expression (Supplementary Table 3). These patterns support a model in which soxB1-2 activity regulates ectodermal progenitor states through coordinated control of chromatin accessibility and gene expression (Fig. 7).
Hypothetical model of SoxB1-2-regulated neuronal cell fates uncovered by integrating epigenomic and transcriptomic approaches. Previous work showed that soxB1-2 is first expressed in a piwi-1+ progenitor population that acquires epidermal or neuronal cell fates that terminally differentiate into ciliated sensory neurons or epidermis (Ross et al. 2018). Subsequent work has focused on understanding the mechanisms downstream of SoxB1-2 involved in specifying sensory neuron subclasses. McCubbin et al. (2025) found that the Pou4 homolog, pou4-2, regulates the differentiation of ciliated sensory neurons in the ciliated stripes. In this study, we show that mecom and castor also function in the soxB1-2 regulatory network by regulating neuronal gene expression. Created in https://BioRender.com.
Furthermore, in situ hybridization of select candidate targets (Fig. 4, Supplementary Fig. 8) confirmed reduced or ablated expression of the putative target genes following soxB1-2 RNAi, supporting their status as downstream, transcriptionally regulated genes. Interestingly, changes in chromatin accessibility following soxB1-2 RNAi did not always correspond to changes in gene expression; some loci showed decreased accessibility without changes in transcript levels, while others exhibited altered expression despite stable accessibility. Independent validation of several of these loci (Figs. 1e and 2b) supports a model in which soxB1-2 initiates chromatin remodeling that may precede or occur independently of transcriptional activation. This model aligns with findings from Xenopus spinal cord regeneration, where chromatin changes have been shown to prime cells for future gene expression (Kakebeen et al. 2020). These findings highlight the power of combining ATAC-seq and RNA-seq to reveal regulatory mechanisms that underlie lineage specification and transcriptional control during planarian tissue homeostasis or regeneration.
Enhanced resolution of neural regulation via targeted analysis of head tissue
Similar to previous studies (Nakazawa et al. 2003; Roberts-Galbraith et al. 2016; Wang et al. 2016), using head tissue enriched for neural and ciliated neural cell populations improved our ability to detect expression changes in neurons. This spatially targeted sampling revealed gene expression changes in sensory neurons undetectable in previous whole-worm RNA-seq datasets (Ross et al. 2018), particularly within rare or spatially restricted cell types. For example, mecom exhibited significantly reduced chromatin accessibility and transcript levels following soxB1-2 RNAi in the head tissue dataset. These changes were not observed in the whole-body RNA-seq dataset. In both our whole-body and head tissue RNA-seq datasets and head tissue ATAC-seq datasets, the samples represent heterogeneous cell populations, not isolated neural or soxB1-2^+^ cells. This heterogeneity inherently introduces noise and likely obscures additional SoxB1-2 targets or changes in rare cell populations, even in our neural-enriched head tissue. Notably, we did not detect changes in chromatin accessibility or transcript levels in sensory populations marked by pou4-2, despite research in our lab showing strong downregulation following soxB1-2 RNAi (McCubbin et al. 2025). This may reflect indirect regulation or a dilution effect due to the limited expression of pou4-2 and the reduced sensitivity of bulk assays to capture changes in rare cell types. Future studies employing single-cell RNA-seq or FACS-based enrichment of soxB1-2^+^ populations, pending the development of a validated antibody, will be critical for resolving these questions.
mecom and castor are downstream of soxB1-2 and are putative mechanosensory signaling, synapse, and ion transport regulators
Chromatin accessibility at regulatory regions near mecom and castor was markedly reduced following soxB1-2 RNAi. Consistent with this, in situ hybridization showed a loss of mecom expression in cells and a substantial reduction in castor expression. These findings indicate that mecom and castor act downstream of soxB1-2 and suggest that the corresponding neural cell populations are very likely not produced in the absence of soxB1-2, although further functional or cell-type-specific analyses will be needed to formally confirm their absence.
As cilia mediate mechanical sensing in planarians, these findings suggest that mecom and castor regulate mechanosensory populations downstream of soxB1-2, which is consistent with known roles of these genes in other organisms. For example, Mecom promotes supporting cell proliferation and sensory hair cell differentiation in the mammalian cochlea, and Castor specifies early-born central complex neurons involved in sensory integration in Drosophila (Plath and Barron 2015; Chen et al. 2022; Dillon and Doe 2024). In the context of sensory neuron development, mecom overexpression has been shown to promote supporting cell proliferation and hair cell differentiation by modulating Tgf-β, Notch, and Wnt signaling pathways (Chen et al. 2022). The identification of soxB1-2 downstream targets exhibiting both chromatin accessibility loss and transcriptional downregulation highlights its regulatory role in neural and ciliated neural populations, further supporting the involvement of genes like mecom in these developmental processes. Additional analyses will be required to delineate the precise cellular relationships among these factors.
Although transcript knockdown was confirmed by in situ hybridization prior to RNA-seq (Supplementary Figs. 9b and 10b), RNAi targeting mecom did not result in significant downregulation of mecom transcript levels in the mecom RNA-seq data. This likely reflects mecom's low baseline expression or its spatially restricted enrichment at the head tip in both control and RNAi-treated animals. Because mecom is an uncharacterized gene in planarians, RNA-seq was performed on whole worms, which likely diluted transcripts originating from the head tip region. Future studies using spatially enriched sampling, such as head dissections, may improve detection sensitivity. Alternatively, the lack of statistical significance may reflect the use of stringent analytical thresholds.
In contrast, following soxB1-2 RNAi, some downstream transcription factors exhibited clear reductions in both chromatin accessibility and transcript levels, whereas others showed loss of chromatin accessibility without meeting RNA-seq significance cutoffs. For example, castor did not meet RNA-seq significance cutoffs in the soxB1-2 RNA-seq dataset, despite clear accessibility changes at its locus. Such discrepancies between ATAC-seq and RNA-seq are not uncommon and may reflect regulatory complexity, temporal separation between chromatin remodeling and transcriptional output, or transcript stability that allows mRNA to persist despite reduced chromatin accessibility. Despite these caveats, the combined ATAC-seq and RNA-seq data support mecom and castor as components of a conserved regulatory program linking soxB1-2 to sensory neuron differentiation (summarized in Fig. 7). Future work will be required to define the precise cellular contexts and regulatory mechanisms through which these factors function.
Conclusion
Our study demonstrates that soxB1-2 plays a critical role in lineage specification in S. mediterranea by modulating chromatin accessibility and gene expression. Integration of ATAC-seq and RNA-seq revealed that soxB1-2 knockdown reduces chromatin accessibility at neural and epidermal loci, supporting the idea that soxB1-2 functions as a pioneer-like transcription factor that primes chromatin for differentiation. We identified mecom and castor as downstream targets implicated in mechanosensory signaling, ion transport, and synapse formation, thereby expanding the regulatory network governed by soxB1-2. In addition to its mechanistic insights, this work establishes one of the few chromatin accessibility datasets in planarians, offering a foundational resource for the regeneration community and a platform for future hypothesis generation. Beyond planarian biology, these findings contribute to a broader understanding of adult neurogenesis and chromatin remodeling in regeneration-competent systems. The regulatory principles uncovered here provide a conceptual framework that could inform stem cell reprogramming strategies in less regenerative organisms, including humans, where restoring neurogenic capacity remains a critical therapeutic challenge.
Supplementary Material
iyag002_Supplementary_Data
iyag002_Peer_Review_History
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adler CE, Alvarado AS. 2018. Systemic RNA interference in planarians by feeding of ds RNA containing bacteria. Methods Mol Biol. 1774:445–454. 10.1007/978-1-4939-7802-1_17.29916170 · doi ↗ · pubmed ↗
- 2Andrews S . 2010. Fast QC: a quality control tool for high throughput sequence data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 3Azimzadeh J, Basquin C. 2016. Basal bodies across eukaryotes series: basal bodies in the freshwater planarian Schmidtea mediterranea. Cilia. 5:15. 10.1186/s 13630-016-0037-1.26998257 PMC 4799839 · doi ↗ · pubmed ↗
- 4Bai Y et al 2024. Integrative analysis based on ATAC-seq and RNA-seq reveals a novel oncogene PRPF 3 in hepatocellular carcinoma. Clin Epigenetics. 16:154. 10.1186/s 13148-024-01769-w.39501301 PMC 11539654 · doi ↗ · pubmed ↗
- 5Bentsen M et al 2020. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun. 11:4267. 10.1038/s 41467-020-18035-1.32848148 PMC 7449963 · doi ↗ · pubmed ↗
- 6Bio Grids . Genrich: Detecting sites of genomic enrichment. Git Hub repository. Available at: https://github.com/jsh 58/Genrich.
- 7Bray NL, Pimentel H, Melsted P, Patcher L. 2016. Near-optimal probabilistic RNA-Seq quantification. Nat Biotechnol. 34:525–527. 10.1038/nbt.3519.27043002 · doi ↗ · pubmed ↗
- 8Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 109:21.29.1–21.29.9. 10.1002/0471142727.mb 2129 s 109.PMC 437498625559105 · doi ↗ · pubmed ↗