CRISPR-Cas–associated SCCmec Variants in Methicillin-resistant Staphylococcus aureus Evade Rapid Diagnostic Detection
Magdalena Podkowik, Alice Tillman, Courtney Takats, Heloise Carion, Gregory Putzel, Julian McWilliams, Benjamin See, Guiqing Wang, Sigridh Munoz-Gomez, Caitlin Otto, Karl Drlica, Luciano Marraffini, Alejandro Pironti, Sarah Hochman, Christopher Kerantzas, Bo Shopsin

TL;DR
Some methicillin-resistant Staphylococcus aureus clones evade rapid diagnostic tests due to a CRISPR-Cas linked SCCmec variant, risking misdiagnosis and improper treatment.
Contribution
The study reveals a novel CRISPR-Cas–associated SCCmec variant that causes false-negative results in rapid MRSA diagnostics.
Findings
A CRISPR-Cas–linked SCCmec variant was identified in 64 MRSA clones from 45 patients.
The variant evades detection by Xpert and BCID2 assays, leading to false-negative results.
The variant is associated with mecA instability and is prevalent in healthcare settings.
Abstract
Rapid molecular assays guiding treatment of methicillin-resistant Staphylococcus aureus detect SCCmec (Xpert) or the SCCmec–orfX junction (BCID2). Sequence variation in this region can disrupt primer binding, yielding false-negative results. Investigation of a missed bloodstream infection linked escape to a CRISPR-Cas–associated SCCmec variant, leading to identification of 64 variants from 45 patients—2% of 2432 screened. Misdiagnosis was restricted to clonal complex 5, a hospital-associated lineage; 11 of 40 SCCmec/junctions evaded detection by BCID2 or Xpert. Variants had mecA instability and circulated in healthcare settings. Our findings reveal a unique escape mechanism and underscore a threat to diagnostic accuracy. This study identifies methicillin-resistant Staphylococcus aureus clones carrying a CRISPR-Cas–linked SCCmec variant that escapes detection by rapid, point-of-care…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
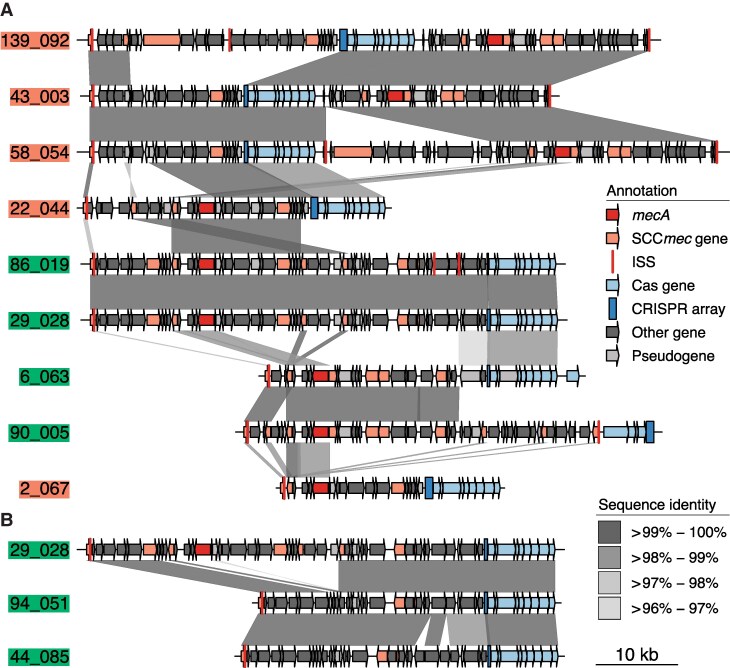 Figure 1
Figure 1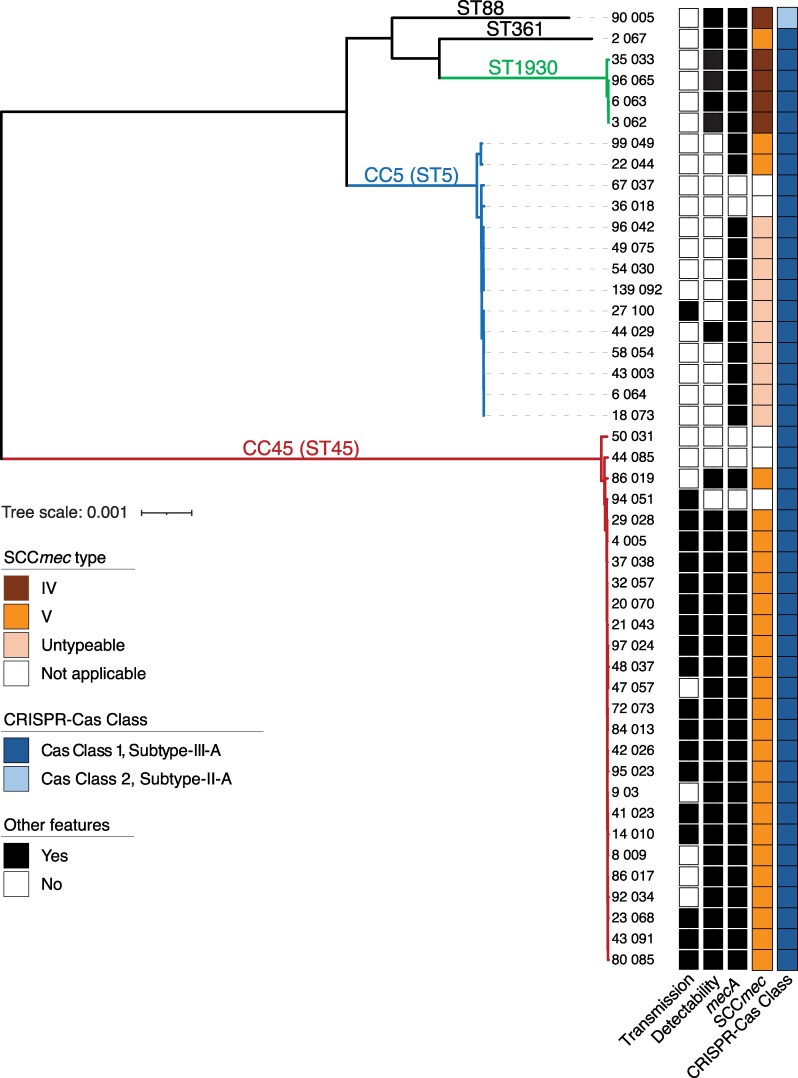 Figure 2
Figure 2- —National Institutes of Health10.13039/100000002
- —CDC10.13039/100031003
- —NYULH Antimicrobial-Resistant Pathogens Program
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCRISPR and Genetic Engineering · Antimicrobial Resistance in Staphylococcus · Viral Infections and Immunology Research
Methicillin-resistant Staphylococcus aureus (MRSA) remains a leading cause of healthcare-associated infections worldwide. Although bloodstream infections decreased by approximately 13% between 2005 and 2012, progress has since plateaued [1]. Mortality remains high despite optimal therapy, making MRSA the primary driver of antimicrobial resistance-associated deaths globally [2].
Rapid PCR-based assays such as Xpert MRSA/SA Blood Culture (Cepheid) and Blood Culture Identification 2 (BCID2, BioFire) facilitate early MRSA detection and timely initiation of therapy that improve patient outcomes [3]. These assays detect mecA and species-specific markers (eg, spa), but rely on conserved sequences within SCCmec (Xpert) or the SCCmec–orfX junction (BCID2) to link mecA to S. aureus [4]. Sequence variation at this junction can disrupt primer binding, causing false negative assay results and impaired clinical decision-making [4, 5]. Knowing the prevalence and molecular basis of such MRSA variants is therefore critical to evaluating test performance.
Clustered regularly interspaced short palindromic repeats (CRISPRs) and associated Cas proteins mediate phage defense [6]. In MRSA, CRISPR-Cas systems occur within the SCCmec element and are likely acquired via horizontal gene transfer from coagulase-negative staphylococci (CoNS), given their high prevalence in CoNS (approximately 35%), but rarity in S. aureus (approximately 2%) [7]. Their impact on molecular MRSA detection is unknown.
We report a case of MRSA bacteremia in which resistance was undetected by rapid testing due to a CRISPR-Cas–associated SCCmec variant. Subsequent biobank screening revealed additional variants, including strains circulating in hospitals. Although currently infrequent, these variants compromise diagnostic accuracy, thus underscoring the need for genomic surveillance that shifts detection from reactive investigation of diagnostic failures to prospective monitoring of emerging diagnostic-escape variants as S. aureus evolves.
METHODS
Bacterial Isolates
The S. aureus isolate from the case patient was collected at NYU Langone Health's (NYULH) Long Island campus. The institutional biobank includes 9440 genome-sequenced isolates—5997 methicillin-susceptible S. aureus (MSSA) and 3443 MRSA—from 6670 patients over 2 years (March 2022−March 2024). Biobank isolates originated from NYULH Brooklyn (n = 2773) and Manhattan (n = 6667); Long Island isolates are not included. MRSA isolates were obtained from clinical and routine admission screening cultures, and MSSA were from blood and screening cultures only; all were handled identically before sequencing. MRSA was identified using Vitek 2 (BioMérieux, France) for clinical cultures and CHROMagar MRSA II (BD Diagnostics, NJ) for screening cultures. The study was approved by the NYULH Institutional Review Board (s24-01872).
Genomic Analysis
Genomic DNA was extracted using MagMax DNA Multisample 2.0 (Thermo Fisher Scientific, Waltham, MA). All strains except 139_092 were sequenced and assembled using Illumina [8] (coverage 120–520x; N50 = 46–847 kbp). A subset of strains (Figure 1) underwent additional Nanopore sequencing (76–104x; Plasmidsaurus, Eugene, OR); assemblies were polished with Illumina reads using pypolca v0.3.1 (“–careful” mode; https://github.com/gbouras13/pypolca), yielding hybrid assemblies. Strain 139_092 underwent both Nanopore and Illumina sequencing and assembly by Plasmidsaurus. All hybrid assemblies yielded closed chromosomes. Details of genome annotation, typing, phylogenetic analysis, and transmission events are provided in Supplementary Methods.
Structural comparison of SCCmec-associated III-A CRISPR-Cas elements in S. aureus. A, Comparison of six representative CRISPR-Cas elements from mecA-positive strains (90_005, 2_067, 6_063, 139_092 [index case], 22_044, 29_028), and 3 related clones (58_054, 43_003, 86_019). Subclones and related clones were selected based on SCCmec type, clonal complex, CRISPR-Cas subtype, and associated phage defense gene profiles (DefenseGroups A–F; Supplementary Table 1 and Supplementary Methods). Elements are ordered by overall similarity; gray shading highlights conserved regions. Annotations: SCCmec components (orange); CRISPR-Cas genes (blue); mecA (red); and SCCmec–orfX junction (red bars). Genomic regions spanning orfX to the CRISPR-Cas locus were defined by positional features, compared pairwise (blastn, default settings), and visualized with genoPlotR v0.8.11 (bit score <10 excluded). Junction sites and duplications were annotated via tblastn (query: GenBank ADC79452.1; PGAP CDS as subject; ≥95% query coverage) and blastn of the final 15 nt of orfX queried against the assembly (≥80% identity, 100% query coverage, word size 5, e value ≤100). Strains predicted in silico to evade detection [9] by SCCmec–orfX primer sets [10] are color coded (green, detectable; scarlet, undetectable; see also Supplementary Table 1). These predictions matched empirical SCCmec/junction detection by commercial Xpert and BCID2 assays, with one exception: the element from strain 2_067 were detected by both assays despite being undetectable by research primers, highlighting variability in primer performance. B, Comparison of mecA-negative strains 94_051 and 44_058 with their closest mecA-positive match from panel (A) (29_028). Labels, color schemes, and annotation conventions as in panel (A). Presence of pls and cstB/R, along with phylogenetic context (Figure 2), suggests mecA loss from an ancestral MRSA background.
Rapid Diagnostic Assays
Detectability of MRSA isolates carrying CRISPR-Cas SCCmec elements used BCID2 (bioMérieux, France) and Xpert (Cepheid, Sunnyvale, CA) assays. Tests were performed blinded according to manufacturers’ protocols, using a 0.5 McFarland suspension of S. aureus colonies in Vitek sterile saline (bioMérieux, France), diluted 10^4^-fold before inoculation.
RESULTS
Case
An 88-year-old man developed nosocomial bacteremia at NYULH-Long Island. Blood cultures grew gram-positive cocci in clusters, and the BCID2 assay reported S. aureus “Detected” but “Not Detected” for the “mecA/C and MREJ (mec right extremity junction)” target used to assess the presence of the SCCmec–orfX junction. Based on this result, empiric vancomycin was de-escalated to oxacillin for approximately 48 hours. When culture and susceptibility testing later confirmed MRSA, therapy was switched to daptomycin plus ceftaroline for 6 weeks for treatment of complicated bacteremia, leading to clinical resolution.
Genomic Analysis
This case prompted investigation into the probability of false-negative BCID2 results at our hospitals, beginning with genome sequencing of the index isolate. The strain (139_092; Supplementary Table 1), a clonal complex 5 (CC5) hospital-associated MRSA, harbored mecA but lacked typical SCCmec type II or IV cassettes associated with CC5 [11]. Instead, it contained a variant SCCmec element encoding a Class 1 Type IIIA CRISPR-Cas system (Figure 1A).
Methicillin-resistant coagulase-negative staphylococci (MR-CoNS) frequently carry SCCmec, serving as both a source and a reservoir for SCCmec in S. aureus [7]. To avoid false positive results when MR-CoNS and MSSA coexist in a sample, some rapid MRSA assays, such as BCID2, do not exclusively target mecA. Instead, they also detect the SCCmec-orfX integration site to specifically link mecA to S. aureus. Sequence analysis revealed that the CRISPR-Cas–associated SCCmec element contained alterations at the orfX-SCCmec junction that likely prevent detection by standard assay primers (Figure 1A).
Prevalence of CRISPR-Cas SCCmec Elements
Finding a CRISPR-Cas–associated SCCmec variant as the likely cause of diagnostic failure in our index case prompted an investigation into whether similar undetectable elements are circulating more broadly. To assess their prevalence—and their potential impact on the performance of rapid diagnostic assays—we screened our biobank of >9000 genome sequences of S. aureus isolates from NYULH's Manhattan and Brooklyn campuses for CRISPR-Cas systems. We identified 5 MSSA and 59 MRSA isolates from 5 and 40 patients, respectively, carrying CRISPR-Cas systems. Among 41 unique MRSA strains (one per patient, except for one patient from whom two distinct variants were recovered), we identified 39 SCCmec-associated CRISPR-Cas Class 1, Type IIIA systems and one Class 2 system—the latter, to our knowledge, the first reported in S. aureus (Supplementary Table 1). Six variant subclones (DefenseGroups A–F; Supplementary Table 1, Methods, and Figure 1A) were defined based on unique combinations of Clonal Complex, SCCmec type, CRISPR-Cas subtype, and DefenseCombinationID—the last reflecting the arrangement of associated antiphage defense genes. Among the five MSSA strains, at least three (94_051, 44_058, and 36_018) retained presumptive residual SCCmec genes (eg, cstB/R; Figure 1B). Together with sequence homology and phylogenetic context (Figure 2), these features suggest mecA loss. Although SCCmec excision has been described as a mechanism for mecA loss in MRSA, its occurrence in CRISPR-Cas–associated SCCmec elements is novel and raises the possibility that these elements destabilize mecA, a hypothesis warranting further study given the mutagenic potential of type III-A CRISPR-Cas systems in staphylococci [12].
Maximum likelihood phylogeny of isolates carrying SCCmec-associated III-A CRISPR-Cas elements. The phylogeny includes one representative isolate per patient (n = 46), except for one patient from whom two distinct variants were recovered (43_003 and 44_029; see Supplementary Table 1). Sequence type (ST) and clonal complex (CC) were assigned by multilocus sequence typing; CCs are indicated when the ST is the recognized founder of that clonal complex (eg, CC5 for ST5). Colored blocks denote CC, putative transmission events (<20 SNPs with epidemiological link), predicted detectability by Xpert and BCID2, mecA status, SCCmec type, and CRISPR-Cas Class (light blue, Class 2; dark blue, Class 3). Scale bars indicate substitutions per site. Core genome size: 2 345 248 bp; variable sites 65 220 bp.
CRISPR-Cas systems were concentrated primarily in CC5 (14 strains and 13 patients) and CC45 (26 strains and 26 patients) (Figure 2 and Supplementary Table 1). Each clonal complex was associated largely with a specific phage defense gene profile. This pattern, supported by phylogenetic analysis (Figure 2), suggests multiple independent acquisitions followed by vertical inheritance with limited horizontal transfer.
Impact on Diagnostic Assay Performance
When we tested representative isolates of the six mecA-positive CRISPR-Cas subclones (DefenseGroups A–F; Supplementary Table 1 and Supplementary Methods), we found that diagnostic escape was restricted to CC5, a dominant hospital-associated lineage [11]: the two CC5 subclones had SCCmec or junction sites undetectable by Xpert and BCID2 assays, respectively. Based on replicate results with related clones (Supplementary Table 1 and Supplementary Methods), we estimate that 11 of the 40 SCCmec/junctions in mecA-positive isolates would fail detection by at least one platform, while 31 would be identified. As with our case patient, test failures were associated with polymorphisms in SCCmec or the junction region that likely disrupt assay recognition by preventing primer binding (Figure 1A).
Notably, the Xpert assay yielded results of “MRSA Positive/SA Positive” even without detection of SCCmec, indicating that its rules-based algorithm conditions were met for mecA and spa cycle threshold (Ct) values (Supplementary Table 1). As a result, the Xpert system may be more robust than BCID2 for detecting certain CRISPR-Cas–associated SCCmec MRSA variants. However, the apparent advantage of the Xpert system may reflect our controlled conditions and bacterial inoculum; head-to-head evaluation in clinical blood cultures and real-world settings is needed to determine whether this performance difference holds in practice.
Healthcare Transmission and Geographic Distribution
CRISPR-Cas SCCmec–containing S. aureus strains were identified predominantly in patients residing in Brooklyn (29 out of 45), including most CC45 clones (22 out of 26) Supplementary Table 1). Many affected patients had a history of prior hospitalization and/or healthcare exposure (29 of 45), particularly in long-term care facilities, where 20 strains were isolated from residents. Clustering by healthcare exposure and geography, coupled with restricted clonality, suggests that CRISPR-Cas SCCmec variants are circulating in healthcare settings among highly susceptible hosts.
To assess transmission, we applied spatial-temporal overlap criteria [13], identifying a genomically and epidemiologically linked cluster of 12 patients (direct transmissions, Supplementary Table 1). Eleven resided in Brooklyn; nine transmissions occurred at NYULH Brooklyn and three at NYULH Manhattan, consistent with intranetwork spread. All cases involved CC45, and all patients lived in long-term care facilities or the same senior apartment complex. Using relaxed criteria—allowing acquisition after the index case had left the ward [13] (Indirect transmissions, Supplementary Table 1)—we identified additional CC45 events and a CC5 cluster involving nondetectable strains. Recovery of near-identical isolates from linked hosts supports nosocomial spread, including secondary transmission.
Screening Public Genomes for CRISPR-associated SCCmec Variants
Screening 70597 mecA-positive MRSA assemblies from GenBank identified cas systems in 1.4% (n = 1000; nearly all Class 1, subtype IIIA [n = 978]), a percentage similar to our biobank. Forty-seven assemblies met our stringent mecA-associated CRISPR-Cas colocalization criterion (Supplementary Methods), representing a conservative lower bound of high-confidence analogs for our NYULH variants (Supplementary Table 2). Additional examples are likely masked by fragmented assemblies. These isolates span diverse sequence types, including CC5 and CC45, and geographic regions, suggesting independent acquisitions. In silico primer analysis predicted that 24 (51%) would evade detection. Thus, SCCmec-linked CRISPR systems capable of diagnostic escape are widely distributed.
DISCUSSION
Our findings indicate that evolution of MRSA can yield variants that evade widely used rapid molecular diagnostics. We identified CRISPR-Cas–associated SCCmec elements, including the first Class 2 CRISPR-Cas system reported in S. aureus, that disrupt a key target of current assays rendering certain affected strains undetectable. This mechanism of diagnostic escape is distinct from novel mec homologs and small-scale SCCmec/orfX junction polymorphisms. Phylogenetic analysis suggests multiple independent emergences, now concentrated in CC45 and CC5, with evidence of ongoing circulation in healthcare settings. That diagnostic escape is concentrated in CC5—the dominant hospital-associated lineage [11 ]—is particularly concerning, since it suggests that the most clinically relevant clone may become the hardest to detect. Continued spread, potentially accelerated by selective pressure from surveillance and control measures [14], for example targeted screening and decolonization that treats only detectable strains, could compromise the reliability of rapid MRSA diagnostics guiding early therapy.
These results highlight a broader challenge: the continual threat that pathogen evolution places on diagnostic accuracy. The clinical consequences of escape variants depend on local prevalence and the extent to which rapid test results influence antibiotic selection. While missed detection of MRSA colonization may hinder de-escalation and infection control, a false-negative in bloodstream infection risks delaying definitive effective therapy. Local assessment of escape variant frequency is therefore essential, both to refine current detection methods and to inform the design of alternative testing platforms. In settings where such variants are circulating, clinicians should interpret negative rapid MRSA results in invasive infections with caution.
Early recognition of such variants depends on the vigilance of clinical laboratories and front-line physicians, who are often the first to encounter diagnostic anomalies. However, colonizing and most nonbloodstream MRSA isolates are not routinely subjected to molecular testing due to cost and logistical complexity. Consequently, variants can circulate undetected. Outside our index case, only 4 of 40 patients harboring potentially undetectable MRSA variants had isolates evaluated by PCR-based blood culture systems—one by BCID2, which did not report resistance markers, and three via Xpert, which did. These findings illustrate how easily such variants can evade routine surveillance and spread silently within healthcare settings.
In summary, our results underscore the need for genomic surveillance [15] and in silico primer screening [14] to detect diagnostic-escape variants before they become widespread. Ongoing monitoring for new variants and collaboration with diagnostic manufacturers to update molecular targets are essential to preserve early MRSA detection.
Supplementary Material
jiaf575_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kourtis AP, Hatfield K, Baggs J, et al Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections— - United States. Morb Mortal Wkly Rep 2019; 68:214–9.10.15585/mmwr.mm 6809 e 1PMC 642196730845118 · doi ↗ · pubmed ↗
- 2Antimicrobial Resistance C . Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022; 399:629–55.35065702 10.1016/S 0140-6736(21)02724-0PMC 8841637 · doi ↗ · pubmed ↗
- 3Bauer KA, West JE, Balada-Llasat JM, Pancholi P, Stevenson KB, Goff DA. An antimicrobial stewardship program's impact with rapid polymerase chain reaction methicillin-resistant Staphylococcus aureus/S. aureus blood culture test in patients with S. aureus bacteremia. Clin Infect Dis 2010; 51:1074–80.20879856 10.1086/656623 · doi ↗ · pubmed ↗
- 4Garcia-Alvarez L, Holden MT, Lindsay H, et al Meticillin-resistant Staphylococcus aureus with a novel mec A homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect Dis 2011; 11:595–603.21641281 10.1016/S 1473-3099(11)70126-8PMC 3829197 · doi ↗ · pubmed ↗
- 5Shore AC, Deasy EC, Slickers P, et al Detection of staphylococcal cassette chromosome mec type XI carrying highly divergent mec A, mec I, mec R 1, bla Z, and ccr genes in human clinical isolates of clonal complex 130 methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 2011; 55:3765–73.21636525 10.1128/AAC.00187-11PMC 3147645 · doi ↗ · pubmed ↗
- 6Makarova KS, Wolf YI, Alkhnbashi OS, et al An updated evolutionary classification of CRISPR-cas systems. Nat Rev Microbiol 2015; 13:722–36.26411297 10.1038/nrmicro 3569 PMC 5426118 · doi ↗ · pubmed ↗
- 7Hossain M, Aslan B, Hatoum-Aslan A. Tandem mobilization of anti-phage defenses alongside SC Cmec elements in staphylococci. Nature Commun 2024; 15:8820.39394251 10.1038/s 41467-024-53146-z PMC 11470126 · doi ↗ · pubmed ↗
- 8St John A, Perault AI, Giacometti SI, et al Capsular polysaccharide is essential for the virulence of the antimicrobial-resistant pathogen Enterobacter hormaechei. m Bio 2023; 14:e 0259022.36779722 10.1128/mbio.02590-22PMC 10127600 · doi ↗ · pubmed ↗