Calcium-Sensing Receptor Regulation of Gastrointestinal Hormone Secretion
Javad Anjom-Shoae, Simon Veedfald, Arthur D Conigrave, Michael Horowitz, Christine Feinle-Bisset

TL;DR
The calcium-sensing receptor (CaSR) in the gut helps regulate hormone release in response to nutrients like amino acids and calcium, which could lead to new treatments for obesity and diabetes.
Contribution
This review highlights how CaSR activators, including calcium and aromatic amino acids, stimulate gut hormones and reduce energy intake and blood sugar.
Findings
CaSR in enteroendocrine cells responds to L-Trp and L-Phe to stimulate gut hormone secretion.
Extracellular calcium enhances the hormone-releasing effects of L-Trp in humans.
Activating CaSR may offer new strategies for managing obesity and type 2 diabetes.
Abstract
The interaction of dietary nutrients with chemoreceptors in the gastrointestinal tract after a meal stimulates the secretion of gut hormones, which trigger the key processes of digestion and absorption, and also regulate energy intake and postprandial glycemia. One of these receptors, first recognized for its capacity to gauge extracellular calcium (Ca2+), is the calcium-sensing receptor (CaSR). Subsequent to its cloning, the CaSR was found to sense not only Ca2+, but also L-amino acids (AAs) and, based on solved protein structures, distinct binding sites have been reported for Ca2+ ions and the aromatic AA, L-tryptophan (L-Trp). In the stomach and small intestine, the CaSR is expressed in enteroendocrine cells, and a substantial body of preclinical work has demonstrated that it mediates gut hormone secretion in response to L-Trp and another aromatic AA, L-phenylalanine (L-Phe), and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
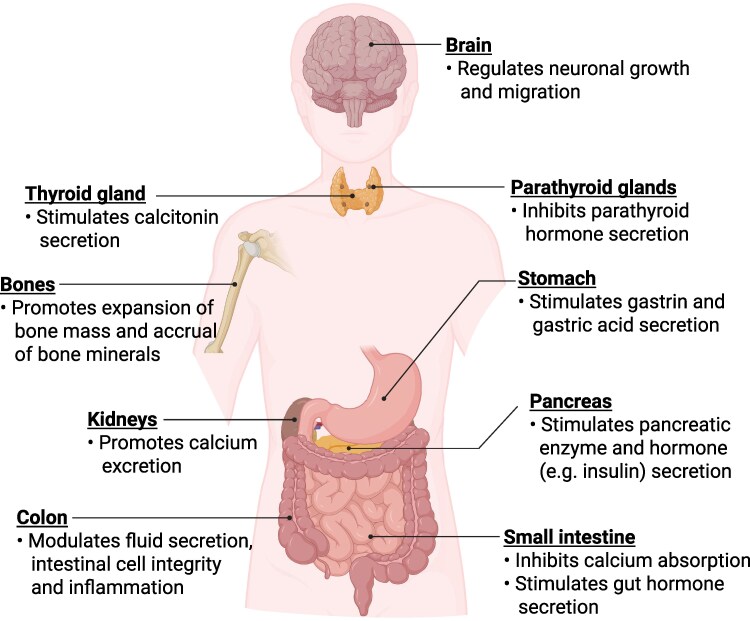 Figure 1
Figure 1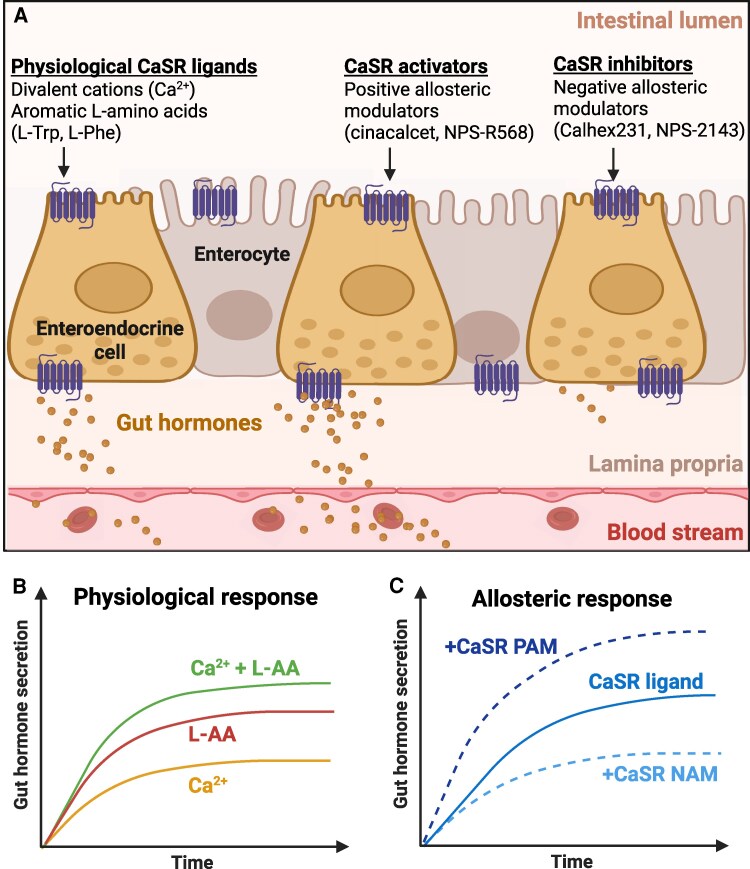 Figure 2
Figure 2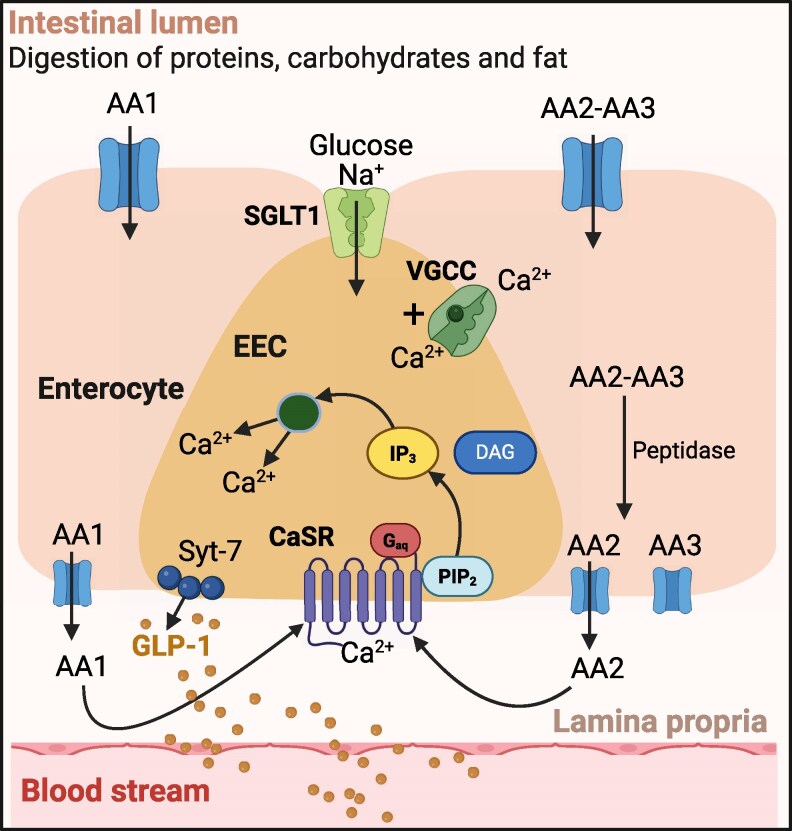 Figure 3
Figure 3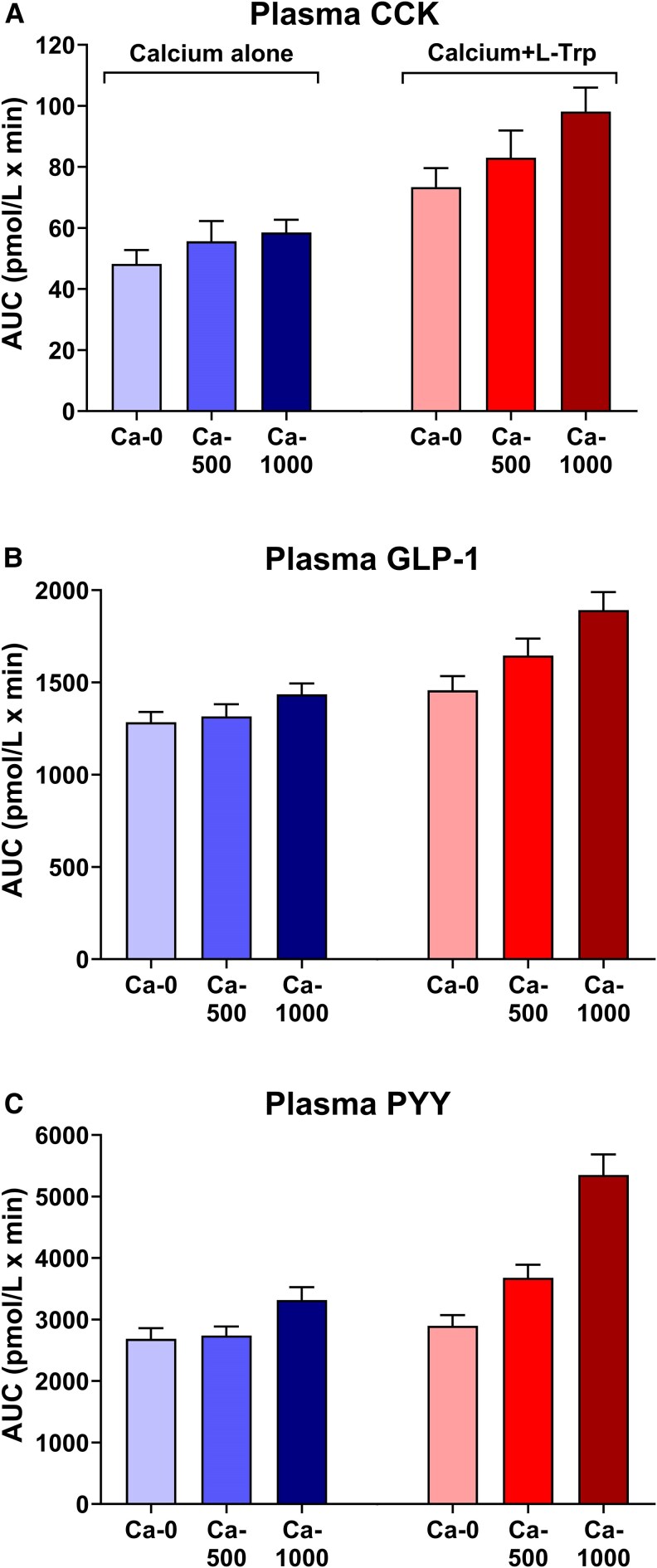 Figure 4
Figure 4| Experimental model | AA type | Gut hormones | Treatment | Ref | |||
|---|---|---|---|---|---|---|---|
| AA alone | AA + CaSR activator | AA + CaSR inhibitor | AA + extracellular Ca2+ | ||||
| STC-1 cells | L-Phe | CCK | ↑ | NR | ↓ | ↑↑ | ( |
| CaSR-null mice (in vivo) | L-Phe | Gastrin | ↑ | ↑↑ | ↓ | ↑↑ | ( |
| Duodenal tissue (mice) | L-Trp | CCK | ↑ | NR | ↓ | NR | ( |
| L-Phe | CCK | ↑ | ↓ | ||||
| Duodenal tissue (mice) | L-Phe | CCK | ↑ | NR | NR | ↑↑ | ( |
| Intestinal tissue (rats) | L-Trp | GIP | ↑ | ↑↑ | ↓ | ↑↑ | ( |
| GLP-1 | ↑ | ↑↑ | ↓ | ↑↑ | |||
| PYY | ↑ | ↑↑ | ↓ | ↑↑ | |||
| L-Phe | GIP | ↑ | ↑↑ | ↓ | ↑↑ | ||
| GLP-1 | ↑ | ↑↑ | ↓ | ↑↑ | |||
| PYY | ↑ | ↑↑ | ↓ | ↑↑ | |||
| Oral administration (rats, mice) (in vivo) | L-Phe | GLP-1 | ↑ | NR | ↓ | NR | ( |
| PYY | ↑ | ↓ | |||||
| Intestinal tissue (pig) | L-Trp | Gastrin | ↑ | ↑↑ | ↓ | ↑↑ | ( |
| L-Phe | Gastrin | ↑ | ↑↑ | ↓ | ↑↑ | ||
| Intestinal tissue (pig) | L-Trp | CCK | ↑ | NR | ↓ | ↑↑ | ( |
| GIP | ↑ | ↓ | ↑↑ | ||||
| STC-1 cells | L-Phe | GLP-1 | ↑ | NR | ↓ | NR | ( |
| Intestinal tissue (pig) | L-Phe | CCK | ↑ | NR | ↓ | ↑↑ | ( |
| GIP | ↑ | ↓ | ↑↑ | ||||
| Intraduodenal infusion (rats) (in vivo) | L-Trp | GLP-1 | ↑ | NR | ↓ | NR | ( |
| PYY | ↔ | ↔ | |||||
| Study population |
| Age (y) | BMI (kg/m2) | Dose of calcium (mg) | Type of intervention | Duration | Outcomes | Ref | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Gut hormones | Energy intake (kcal) | Plasma glucose (mmol/L) | ||||||||
| Healthy overweight | 18 (M) | 25 ± 1 | 27 ± 1 | 850 | Oral mixed-nutrient meal | 7 h | ↔ CCK | ↔ | ↔ | ( |
| ↑GLP-1 | ||||||||||
| ↔ PYY | ||||||||||
| Metabolic syndrome | 38 (M/F) | 52 ± 2 | 32 ± 1 | 1400/day | Oral mixed-nutrient meal | 12 w | ↔ GIP | ↔ | ↔ | ( |
| ↑GLP-1 | ||||||||||
| ↑PYY | ||||||||||
| Healthy lean | 10 (M) | 25 ± 3 | 26 ± 2 | 1200 | Oral mixed-nutrient meal | 4 h | ↑GIP | NR | ↔ | ( |
| ↑GLP-1 | ||||||||||
| Healthy lean | 13 (M) | 26 ± 4 | 24 ± 1 | 1000/day | Oral supplement | 2 w | ↔GIP | NR | NR | ( |
| ↔GLP-1 | ||||||||||
| Healthy lean | 9 (M) | 27 ± 4 | 23 ± 2 | 1100/day | Oral mixed-nutrient meal | 3 w | ↔GIP | NR | ↔ | ( |
| ↑GLP-1 | ||||||||||
| Healthy lean | 20 (M/F) | 22 ± 1 | 22 ± 1 | 1170 | Oral mixed-nutrient meal | 1 h | ↔GIP | ↔ | NR | ( |
| ↔GLP-1 | ||||||||||
| Healthy lean | 20 (M/F) | 25 ± 4 | 23 ± 3 | 1000 | Oral mixed-nutrient meal | 2 h | ↑GIP | NR | ↔ | ( |
| ↑GLP-1 | ||||||||||
| ↑PYY | ||||||||||
| Study population | N | Age (y) | BMI (kg/m2) | Dose (g) | Type of intervention | Duration | Outcomes | Ref | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Gut hormones | Energy intake (kcal) | Plasma glucose (mmol/L) | ||||||||
|
| ||||||||||
| Healthy lean | 24 (M/F) | 21 | 24 ± 2 | 1.4 | Oral | 180 | ↑ GLP-1 | +11 | −0.3 | ( |
| Healthy lean | 10 (M) | 26 ± 8 | 22 ± 2 | 3.3 | ID infusion | 90 | ↑ CCK | −219 | NR | ( |
| ↑ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
| Healthy lean Obesity | 10 (M/F) | 24 ± 1 | 21 ± 1 | 1.5 | IG bolus | 120 | ↑ CCK | NR | <−0.5 | ( |
| ↑ GLP-1 | ||||||||||
| 10 (M/F) | 27 ± 8 | 40 ± 4 | 1.5 | IG bolus | ↑ CCK | |||||
| ↑ GLP-1 | ||||||||||
| Healthy lean Obesity | 16 (M) | 31 ± 3 | 22 ± 1 | 3 | IG bolus | 90 | ↔ CCK | 98 | −0.4 | ( |
| 16 (M) | 32 ± 3 | 33 ± 1 | 3 | ↔ CCK | −48 | ∼−1.0 | ||||
| Healthy lean | 16 (M) | 24 ± 1 | 22.9 ± 2 | 2.2 | ID infusion | 90 | ↑ CCK | +37 | NR | ( |
| ↔ GLP-1 | ||||||||||
| Healthy lean Obesity | 12 (M) | 30 ± 3 | 23 ± 1 | 3 | IG bolus | 150 | ↑ CCK | −217 | NR | ( |
| 13(M) | 31 ± 3 | 33 ± 1 | 3 | IG bolus | ↑ CCK | −237 | ||||
| Healthy lean | 12 (M) | 28 ± 7 | 23.8 ± 2 | 1.1 | ID infusion | 45 | ↔ CCK | NR | <−0.5 | ( |
| ↔ GLP-1 | ||||||||||
| Healthy lean | 15 (M) | 26 ± 7 | 22 ± 2 | 1.8 L-Trp + 500 mg Ca2+ | ID infusion | 150 | ↔ gastrin | −147 | NR | ( |
| ↑ CCK | ||||||||||
| ↔ GIP | ||||||||||
| ↑ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
| 1.8 g L-Trp + 1000 mg Ca2+ | ↔ gastrin | −186 | ||||||||
| ↑ CCK | ||||||||||
| ↓GIP | ||||||||||
| ↑ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
| Obesity | 15 (M) | 27 ± 8 | 30 ± 2 | 1.8 L-Trp + 500 mg Ca2+ | ID infusion | 150 | ↔ gastrin | −70 | NR | ( |
| ↑ CCK | ||||||||||
| ↔ GIP | ||||||||||
| ↑ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
| 1.8 g L-Trp + 1000 mg Ca2+ | ↔ gastrin | −172 | ||||||||
| ↑ CCK | ||||||||||
| ↔ GIP | ||||||||||
| ↑ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
|
| ||||||||||
| Healthy lean | 12 (M) | 19-34 | <25 | 5 | ID infusion | 90 | ↑ CCK | NR | NR | ( |
| Healthy lean | 5 (M) | 25 ± 1 | 21 ± 2 | 20 | ID infusion | 40 | ↑ CCK | NR | ↔ | ( |
| Healthy lean | 6 (M/F) | 30 ± 1 | 20-24 | 10 | Oral | 40 | ↑ CCK | −498 | NR | ( |
| Healthy lean | 16 (M) | 23 ± 2 | 25 ± 1 | 10 | ID infusion | 60 | ↑ CCK | NR | NR | ( |
| Healthy lean | 10 (M) | 23 ± 1 | 22 ± 1 | 10 | IG bolus | 90 | ↑ CCK | −184 | <−1.0 | ( |
| ↔ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
| Healthy lean | 11 (M/F) | 30 ± 1 | 22 ± 2 | 10 | Oral | 150 | ↑ GIP | −89 | ∼−1.0 | ( |
| ↔ GLP-1 | ||||||||||
| ↑ PYY | ||||||||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsParathyroid Disorders and Treatments · Regulation of Appetite and Obesity · Biochemical Analysis and Sensing Techniques
Essential Points
The calcium-sensing receptor (CaSR), located on enteroendocrine cells, has the capacity to sense, and bind, both extracellular calcium (Ca^2+^) and aromatic L-amino acids (particularly L-tryptophan (L-Trp) and L-phenylalanine (L-Phe)), owing to its complex dimeric structurePreclinical studies have established that CaSR activation mediates the effects of Ca^2+^, L-Trp, and L-Phe on the secretion of gastrointestinal hormones, including gastrin, cholecystokinin (CCK), glucose-dependent insulinotropic polypeptide (GIP), glucagon-like peptide-1 (GLP-1), and peptide YY (PYY)L-Trp- or L-Phe-induced secretion of these hormones is augmented by increases in extracellular Ca^2+^ and is substantially reduced in the absence of extracellular Ca^2+^CaSR-mediated stimulation of GI hormones by L-Trp and L-Phe, or direct activation of the CaSR with CaSR agonists, has been shown to reduce food intake and lower postprandial plasma glucose in animalsRecent clinical studies have demonstrated that Ca^2+^ also enhances the effects of L-Trp to stimulate CCK, GLP-1, and PYY secretion and suppress energy intake in humansFurther research is needed to establish whether the CaSR mediates L-Trp-induced gut hormone release in humans, using CaSR antagonists, and to characterize the effects of selective agonists to evaluate their therapeutic potential
Dietary nutrients (eg, monosaccharides, L-amino acids (AAs), and fatty acids) activate specialized chemoreceptors that are expressed on gut hormone-producing (enteroendocrine) cells throughout the gastrointestinal (GI) mucosa, to stimulate the release of more than 20 hormones (1-3). While the precise sites of GI hormone release remain uncertain, specific regions of the GI tract are recognized as primary sites of their secretion. Accordingly, (i) gastrin is released primarily from the stomach, (ii) cholecystokinin (CCK) and glucose-dependent insulinotropic polypeptide (GIP) from the proximal small intestine, and (iii) glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) from the distal small intestine (4), although there is now evidence for co-localization of multiple hormones within the same enteroendocrine cell type (5). The release of these hormones is central to the regulation of key gut functions, including gastric acid secretion, gastric emptying, intestinal digestion, and absorption of nutrients, as well as important systemic effects, including regulation of appetite, energy intake, and postprandial plasma glucose (6-8).
One key chemoreceptor that is expressed in the gut is the calcium-sensing receptor (CaSR). The CaSR was first cloned from a bovine parathyroid mRNA library in the early 1990s based on its capacity to sense extracellular divalent and trivalent cations (9), and it is now understood to be critical to Ca^2+^ homeostasis (Fig. 1) via (i) inhibitory control over parathyroid hormone (PTH) secretion from parathyroid chief cells and renal Ca^2+^ reabsorption; (ii) stimulatory control of calcitonin secretion from parafollicular C-cells of the thyroid glands; as well as (iii) a range of effects on bone cells (10). In the GI tract, the CaSR also exerts local inhibitory control on intestinal Ca^2+^ absorption (11). These discoveries led to the development of the currently used CaSR positive allosteric modulators, now widely used in the treatment of several hypercalcemic disorders, including various forms of primary and secondary hyperparathyroidism, typically caused by inactivating mutations and downregulation of the CaSR, respectively (12, 13). In addition, the therapeutic potential of CaSR negative allosteric modulators is being investigated for hypocalcemic disorders, such as autosomal dominant hypocalcemia (14, 15).
Physiological roles of the calcium-sensing receptor. Figure created with BioRender.com.
Shortly after the development of CaSR-expressing human embryonic kidney (HEK-293) cells as a convenient cell expression model for studies of the wild-type and mutant CaSRs (16), and in the presence of suprathreshold extracellular Ca^2+^ concentrations, the CaSR was unexpectedly found to act as a nutrient sensor for both aromatic and aliphatic AAs, of which L-tryptophan (L-Trp) and L-phenylalanine (L-Phe) were the most potent activators (17, 18). This work stimulated several predictions regarding the role of the CaSR as a multimodal sensor in the GI tract (19) and raised questions regarding the molecular basis by which 2 distinct classes of nutrients interact to promote receptor activation.
Subsequent preclinical studies, using CaSR inhibitors or genetically modified animal models (eg, CaSR knockout mice), provided direct evidence that L-Trp and L-Phe engage the CaSR to stimulate the secretion of several GI hormones, including gastrin (20, 21), CCK (22-26), GIP (25-27), GLP-1 (27-30), and PYY (27, 28, 30). Moreover, L-Trp- or L-Phe-induced secretion of these hormones was shown to be augmented by increases in extracellular Ca^2+^ (20-22, 24-27); in contrast, their stimulatory effects, particularly on CCK, GIP, and GLP-1, were compromised in vitro upon withdrawal of extracellular Ca^2+^ (22-24). CaSR-mediated stimulation of GI hormone release by these AAs, or in response to other activators, reduces food intake as well as the postprandial glucose response in several animal models (28, 30-33).
In humans, while the evidence for CaSR-mediated stimulation of Ca^2+^- or AA-induced GI hormone release is incomplete (although reported in human organoids (34, 35)), in part due to a paucity of studies using CaSR inhibitors, both Ca^2+^ and AAs have the capacity to stimulate the secretion of gastrin, GIP, GLP-1, and PYY, associated with reductions in energy intake and/or postprandial plasma glucose. For example, in 2 recent studies, intraluminal administration of Ca^2+^, in the form of a CaCl_2_ solution, increased plasma GLP-1 and PYY levels in both healthy individuals (36), and those with obesity (37). In some other studies, which have reported effects of Ca^2+^-containing meals on gut hormone secretion, however, the role of Ca^2+^ has been less clear, because it was primarily administered as a component of a test drink/meal (eg, milk or yogurt), in which other macronutrients were present (38-44), rather than as a defined supplement. There is also evidence that Ca^2+^ enhances the effect of aromatic AAs to increase plasma levels of gut hormones and to suppress energy intake. For example, in both healthy individuals and those with obesity, L-Trp-stimulated increases in CCK, GLP-1, and PYY secretion and reductions in energy intake were enhanced by the addition of Ca^2+^ (36, 37). Thus, in human studies, Ca^2+^ and AAs act together to stimulate GI hormone release, consistent with the 2-site engagement of the CaSR described in preclinical studies. They also negatively modulate energy intake and lower postprandial plasma glucose.
This review provides an overview of the outcomes of preclinical and clinical studies regarding the role of the CaSR in mediating the release of gut hormones in response to Ca^2+^ in the absence, or presence, of aromatic AAs. Although other AAs (eg, L-tyrosine, L-histidine, L-alanine, L-threonine or analogues of L-arginine (17, 45, 46)) also engage the CaSR, we focus herein on the effects of 2 aromatic AAs, L-Trp and L-Phe, which are the most potent AA activators in HEK-293 cells (17) and human parathyroid cells (47). Some studies were designed to test whether the CaSR is the molecular target of Ca^2+^ and AAs, either alone or in combination, with respect to GI hormone release (eg, with the use of selective CaSR inhibitors). Other studies have solely focused on whether Ca^2+^ or AAs, or a combination of Ca^2+^ and AAs, were effective at elevating plasma GI hormone levels, as would be predicted for cell types that express the CaSR protein. This review considers both types of studies and also the impacts of the CaSR activators, L-Trp and L-Phe, in the absence and presence of Ca^2+^, on GI hormone levels in humans (48-63), and whether the 2 distinct nutrient classes of CaSR ligands interact positively to reduce energy intake and lower postprandial plasma glucose profiles.
In considering the effects of aromatic AAs, it is important to note that a distinct class A G protein–coupled receptor, known as GPR142, has been de-orphanized and found to be expressed in some, but not all, enteroendocrine cells (34). While this receptor responds to L-Trp, L-Phe, and L-tyrosine, it does not appear to respond more broadly to AA mixtures that arise from protein digestion (46, 64), or in a Ca^2+^-dependent manner, as observed for the CaSR. While this review focuses on the CaSR for the reasons stated above, we also draw attention to settings in which GPR142 may contribute to GI hormone release, particularly in the case of GIP.
The Calcium-Sensing Receptor
The CaSR was first cloned from a bovine parathyroid mRNA library (9) and, subsequently, from rat kidney (65) and human adenomatous parathyroid (66) mRNA in the early-mid 1990s. The mRNA encodes a class C G protein–coupled receptor (GPRC2A), which forms homodimers when expressed in cell membranes (67-69). In humans, each subunit is composed of 1078 AA residues, taking the form of an N-terminal extracellular region (formed from a venus fly trap [VFT] domain, a cysteine-rich domain [CRD], and a C-terminal linker), a heptahelical bundle (ie, a 7-transmembrane domain unit), and an intracellular C-terminal domain (67-69). The intracellular loops (iCLs), particularly iCL2 and iCL3, in collaboration with the intracellular C-terminus, couple intracellularly to several G proteins, including G_q/11_, G_i_, G_0_, and, in some cases, Gs, and thereby mediate cellular signal transduction pathways (67-69). This structure provides the CaSR with diverse binding sites to respond to a surprisingly extensive list of activators which, in addition to its principal agonist, Ca^2+^, include (i) divalent cations, such as Mg^2+^, and trivalent cations, such as Gd^3+^ (70); (ii) natural positive modulators that bind in the VFT domain (eg, AAs (17, 71), γ-glutamyl peptides (72, 73), and spermine (74)); and (iii) high-potency synthetic positive allosteric modulators (eg, cinacalcet and NPS-R568) and negative allosteric modulators (eg, Calhex231 and NPS-2143) (75). In addition to these pharmacological activators and inhibitors, receptor activity is also promoted by low ionic strength and high pH (76, 77). It is now known that the receptor undergoes repetitive transitions between 2 main states, an inactive and an active state (67-69). These 2 states impact the dimer interface at the level of the VFT domains, proximal CRDs, and heptahelical bundle and act to either facilitate or obstruct the creation of a single G protein binding site per dimer formed by the intracellular loops, transmembrane domain-3, and proximal C-terminus. Agonists and positive modulators promote the active state, while antagonists and negative modulators promote the inactive state.
Structural Basis of Ligand Binding and Activation of the CaSR
Recent studies have provided critical insights into the nature of the CaSR's distinct binding sites for Ca^2+^ and AA, and the mechanism(s) by which they interact. Thus, several solved (ie, high-resolution) structures of CaSR homodimers have been reported in both inactive and active states, in the presence of low and high Ca^2+^, and in the absence and presence of L-Trp (67-69). These studies have led to the identification of a single AA binding site per subunit of the CaSR, which is located in the bilobed cleft of the receptor's N-terminal VFT domain. This corresponds to the canonical class C agonist binding site for the AA, glutamate, in metabotropic glutamate receptors and the AA analogue, γ-aminobutyrate, in GABA_B_ receptors (78). The structure studies have also led to the identification of 4 Ca^2+^ binding sites per subunit. Of these sites, “Site-4,” which is located at the extreme C-terminus of the VFT and at the N-terminus of the CRD, which follows it in tandem, stabilizes the active form of the receptor by electrostatic interactions across the dimer interface. As the receptor's dimer interface extends from VFT Lobe-1 to include VFT Lobe-2 upon activation, the extreme C-termini of the dimeric CRDs are drawn toward each other. Because the CRDs are rigid, caliper-like structures, this has the effect within the membrane of apposing the pair of 7-transmembrane domain units and inducing mutual rotations to reposition TM helices 6 and 7 at the dimer interface, leading to the formation of a single G protein binding site per receptor dimer. Thus, L-Trp, and presumably other AA molecules, promote closure of the bilobed cleft of the receptor's VFT and induce extended apposition of the dimeric subunits involving VFT Lobe-2 and the CRD. In addition, Ca^2+^ interacts positively with the AA-bound (VFT closed) form of the receptor by stabilizing the extended dimer interface.
Physiological Roles of the CaSR
The CaSR plays a role in Ca^2+^ homeostasis, in which it acts to lower the circulating Ca^2+^ concentration (10), as well as in energy metabolism, where it promotes digestion and absorption of nutrients, and, via downstream effects, modulates plasma nutrient concentrations and food intake (17-19). With respect to its critical role in Ca^2+^ homeostasis, the CaSR is highly expressed in chief cells of the parathyroid glands, where it mediates extracellular Ca^2+^-induced feedback inhibition of PTH secretion (79), and in cortical thick ascending limb cells of the renal tubules, where it mediates extracellular Ca^2+^-dependent inhibition of Ca^2+^ reabsorption, to promote urinary Ca^2+^ excretion (80). These 2 key effects to lower circulating Ca^2+^ are supported by the expression of the CaSR in thyroid parafollicular C-cells, which mediates extracellular Ca^2+^-stimulated calcitonin release to inhibit bone resorption and promote bone formation (81). In addition, the CaSR is expressed in cells that regulate skeletal turnover, including bone-resorbing osteoclasts and bone-forming osteoblasts, and thereby favors an expansion of bone mass and accrual of bone mineral (82).
Although expressed at low levels in intestinal epithelial cells, when compared with its levels in the parathyroid, the renal thick ascending limb, and indeed enteroendocrine cells, the CaSR mediates local feedback control of Ca^2+^ absorption from luminal contents, whereby elevations in circulating Ca^2+^ on the contraluminal (ie, blood-facing) surface of intestinal epithelial cells inhibit intestinal Ca^2+^ absorption (11).
The CaSR is also expressed in organs and tissues that are not associated with Ca^2+^ metabolism, including the brain, liver, pancreas, and enteroendocrine cells, indicative of potential roles in other physiological processes (83). Beyond its physiological role, CaSR expression can also be altered in various benign and malignant tumors, including parathyroid, breast, prostate, and colon cancers, where it may contribute to tumorigenesis (84). However, discussion of CaSR overexpression, and/or its oncogenic or tumor-suppressive roles, is beyond the scope of the present review. In the GI tract, as we will discuss in detail, the CaSR acts as a nutrient sensor, particularly for AAs and Ca^2+^ (85). Its expression in enteroendocrine cells (86) enables its involvement in the release of gut hormones that impact energy metabolism in several phases. In the initial phase of the gut hormone response, the release of digestive enzymes is stimulated, leading to the breakdown of macronutrients and, subsequently, the absorption of their building blocks, ie, AAs, monosaccharides, and fatty acids. In the subsequent phases of the response, impacts are exerted on plasma nutrient profiles (eg, postprandial glucose levels) and regulation of appetite. Thus, due to its expression across gastric parietal cells (20, 87), pancreatic acinar (88), beta (33) and alpha (89) cells, and hepatocytes (90), the CaSR directly couples nutrient signals to gastric acid secretion, pancreatic enzyme, hormone (eg, insulin and glucagon), and bile secretion. In the case of pancreatic islet beta and alpha cells (89, 91), the CaSR operates alongside the second aromatic AA sensing receptor, GPR142, which is expressed at high levels in these cell types (92). The CaSR has also been reported to modulate the integrity of the intestinal epithelium (93), fluid secretion (94), and local inflammatory responses (95).
Role of the CaSR in Mediating Nutrient-Dependent GI Hormone Secretion
Preclinical findings, based on cell culture and animal models, have established a role for the CaSR in the nutrient-dependent stimulation of GI hormone release (20-30) (Table 1). Initial studies demonstrated that the CaSR mediates extracellular Ca^2+^-stimulated gastrin secretion (87), and there was also evidence for CaSR protein expression in acid-secreting parietal cells and pepsinogen-secreting mucous cells of gastric glands (96). Following the recognition that the CaSR also senses AAs, particularly of the aromatic subgroup (17, 18), roles for the CaSR in mediating AA-induced secretion of other GI hormones, including CCK, GIP, GLP-1, and PYY, have been reported (17-27). The finding that the CaSR is expressed on both the apical and basolateral membranes of enteroendocrine cells suggests that it responds to changes in the composition of both the luminal and contraluminal fluids to stimulate GI hormone release (19). It was also demonstrated that the addition of extracellular Ca^2+^ enhanced the stimulation of GI hormones by aromatic AAs (20-22, 24-27) (Fig. 2). While the role of the CaSR in mediating Ca^2+^- or AA-induced stimulation of GI hormones has not been investigated in humans, several studies have reported that administration of CaSR agonists (97, 98), oral Ca^2+^ supplements (99), or intravenous (IV) infusions of Ca^2+^, sufficient to raise plasma total Ca^2+^ concentration from approximately 2.2-2.4 mmol/L to 2.8-3.0 mmol/L (100), acutely increase plasma gastrin levels. In healthy individuals, in the case of IV infusion of Ca^2+^, plasma gastrin levels rose by 20% and gastric acid secretion by 40% (100). However, these effects in healthy individuals were not observed in patients with primary hyperparathyroidism, who characteristically have elevated plasma levels of gastrin as well as Ca^2+^ (99, 101). Interestingly, the CaSR positive modulator cinacalcet was reported to have no effect on plasma gastrin or CCK levels in patients with hyperparathyroidism (102), possibly because (i) the gastrin and CCK levels were already raised in response to the high prevailing plasma Ca^2+^ concentration; (ii) desensitization of the CaSR in enteroendocrine cells upon chronic exposure to high Ca^2+^; or (iii) the 2-hourly blood sampling protocol used in the study was most likely insufficient to detect short-term changes in plasma CCK, which usually occur during the first 60 minutes.
(A) In the gastrointestinal (GI) tract, the calcium-sensing receptor (CaSR) is expressed on both the apical and basolateral membranes of enteroendocrine cells to mediate the effects of physiological CaSR ligands, including extracellular Ca2+ and the aromatic L-amino acids (AAs), L-tryptophan (L-Trp), and L-phenylalanine (L-Phe), on the stimulation of GI hormones. The CaSR also responds to synthetic positive allosteric modulators (PAMs; eg, cinacalcet and NPS-R568) and negative allosteric modulators (NAMs; eg, Calhex231 and NPS-2143), which stimulate and inhibit GI hormone secretion, respectively. (B) The physiological response of the CaSR to either Ca2+ or AAs to stimulate GI hormones has been shown to be enhanced when the 2 ligands are administered in combination. (C) In addition, the stimulation of GI hormones by physiological CaSR ligands is allosterically augmented by CaSR PAMs and diminished by CaSR NAMs. Figure created with BioRender.com.
Cellular Mechanisms Linking CaSR Activation by AAs to Secretion of Hormones From Enteroendocrine Cells
In the proximal small intestine, luminal glucose triggers an early phase of GLP-1 and GIP release, and in turn, insulin secretion, dependent upon luminal membrane expression of sodium-glucose cotransporter 1 (SGLT1) in enteroendocrine L and K cells, respectively. The key events appear to be (i) glucose-dependent Na^+^ influx; (ii) depolarization and initiation of action potentials; and (iii) Ca^2+^ influx via voltage-gated Ca^2+^ channels (103). Exocytosis of hormone-containing large, dense-core, secretory vesicles follows via SNARE-mediated fusion with the plasma membrane (3). The identities of the Ca^2+^-binding proteins that promote fusion and exocytosis are not yet clear, but may include synaptotagmin-7, which is (i) expressed in enteroendocrine cells; (ii) regulated via high-affinity Ca^2+^-binding sites, like other canonical synaptotagmins; and (iii) required for glucose-dependent GLP-1 secretion (104).
The CaSR, on the other hand, is expressed primarily on the contraluminal (blood-facing) membranes of enteroendocrine cells, including gastrin-secreting G cells (87) and CCK-secreting I cells (23). Thus, the CaSR, like other nutrient-sensing receptors, is positioned to respond to protein-derived oligopeptides and AAs following intestinal absorption. The mechanisms by which dietary protein induces the secretion of GLP-1 and GIP are different to those described above for carbohydrates. In particular, the most powerful mechanisms for protein-induced GLP-1 and GIP secretion depend on the uptake of oligopeptides and free AAs across the luminal membranes of intestinal epithelial cells, followed by intracellular catabolism of oligopeptides to free AAs and the release of AAs across the basolateral membranes onto the interstitial surfaces of enteroendocrine cells. Thus, recent analyses using a vascularly-perfused rat proximal intestine model concluded that secretion of GLP-1 in response to either ingested peptone (a meat-derived extract rich in oligopeptides), or to mixtures of AAs, is dependent primarily upon absorption by intestinal epithelial cells, followed by CaSR-mediated detection on the blood-facing membranes of enteroendocrine cells (46, 64).
After delivery of AAs from the GI lumen to the intestinal interstitium, a subset of available AAs bind to the CaSR on the basolateral membranes of enteroendocrine cells (Fig. 3). As described above, aromatic AAs, including L-Trp and L-Phe, stabilize the active form of the CaSR's dimeric VFT, provided the extracellular Ca^2+^ concentration is sufficient (≥0.75 mM) to stabilize the dimer interface at the N-terminus of the receptor's Cys-rich domains. At higher interstitial Ca^2+^ concentrations (eg, from 1.2-1.5 mM), additional AAs are recruited to stabilize the active forms of more receptor molecules. After activation by AAs in the presence of extracellular Ca^2+^, the CaSR recruits diverse G proteins, including G_q_ and G_i_. With respect to the secretion of gut hormones, the key step appears to be G_αq_-dependent activation of PI-PLC with attendant generation of inositol 1,4,5-trisphosphate (IP_3_) and, in turn, Ca^2+^ release from ER membrane-bound intracellular stores to raise the intracellular Ca^2+^ concentration. Consistent with this, AAs induced elevations in intracellular Ca^2+^ (23, 35) and AA-dependent activation of GLP-1 release from the perfused rat intestine was markedly suppressed not only by vascular delivery of the CaSR inhibitor NPS-2143, but also by the PI-PLC inhibitor, U73122 (64). Whether the CaSR might also stimulate the secretion of gut hormones by raising the cytoplasmic cAMP concentration (103) either by coupling to G_s_ (105) or via G_βγ_-dependent activation of the adenylyl cyclase isoform, ADCY2, as described for the free FA receptor FFAR1 (106), is unknown.
Intracellular signaling cascade that evokes GLP-1 secretion in response to L-amino acid activation of the CaSR. Ingested protein is digested in the small intestinal lumen. Resulting free amino acids (AA1) and oligopeptides (eg, the dipeptide AA2-AA3) are taken up by specific transporters on the apical (luminal) membrane of intestinal epithelial cells. In the case of oligopeptides, cytoplasmic peptidases release free AAs, which are delivered into the interstitial space. There, AAs bind and activate CaSRs located on the basolateral membrane of enteroendocrine cells (EECs). The AA-bound CaSR elevates cytoplasmic free Ca2+ concentration via release from intracellular stores. The intracellular Ca2+ signal triggers exocytosis of GLP-1 (as well as other gut hormones)-containing secretory vesicles. Also shown is glucose-dependent Na+ uptake across the luminal membrane of EECs, which induces depolarization, the transmission of action potentials and Ca2+ influx via voltage-gated Ca2+ channels (VGCCs). Free fatty acids are also transported across intestinal epithelial cells to act on basolateral FFA receptors, eg, FFAR1 (not shown). Figure created with BioRender.com.
Effects of Ca2+ on GI Hormone Secretion, Energy Intake, and Blood Glucose
Ca^2+^-dependent stimulation of gastrin and gastric acid secretion has been recognized since the first half of the twentieth century (107). In contrast, the ability of Ca^2+^ to stimulate the secretion of other GI hormones, particularly those involved in the regulation of energy intake and blood glucose, including CCK, GIP, GLP-1, and PYY, has only received attention more recently. The following sections review the current understanding of the effects of Ca^2+^ on GI hormone secretion, energy intake, and blood glucose, from both preclinical and clinical studies.
Preclinical Outcomes
The recognition that intracellular Ca^2+^ serves as an essential messenger to mediate secretion of GI hormones has led to a particular interest in understanding the role of extracellular Ca^2+^ in this process (108-110). It is now well established, in both in vitro and in vivo studies, that extracellular Ca^2+^ plays a key role in the stimulation of gastrin and gastric acid secretion (111-118). For example, the removal of extracellular Ca^2+^ by different Ca^2+^-chelating agents (eg, EDTA or EGTA) diminished the stimulation of gastrin and gastric acid secretion (111, 114), while IV infusion of Ca^2+^ increased serum gastrin levels and gastric acid secretion (115). Subsequent studies provided evidence that extracellular Ca^2+^ might be critical in the secretion of other GI hormones (119-127). For example, stimulation of CCK secretion by different stimuli, including bombesin, an agent that increases intracellular Ca^2+^ levels (119, 122), monitor peptide (121), peptones (124), fatty acids (123, 127), aldehydes (125) or endogenous CCK-releasing factor (126), was reduced, if not totally inhibited, in the absence of extracellular Ca^2+^, achieved by using either EDTA or EGTA and/or voltage-gated Ca^2+^ channel blockers. In addition, both basal and/or glucose-induced secretion of GLP-1 from cell lines (128, 129) or a perfused rat intestine model (130) was inhibited by Ca^2+^ channel blockers. While these findings indicate the importance of extracellular Ca^2+^ in the stimulation of GI hormones, the precise underlying cellular mechanisms, particularly whether this effect involves the CaSR, or is achieved through other pathways, such as voltage-gated Ca^2+^ channels, which are highly expressed on enteroendocrine cells and underlie Ca^2+^ influx into the cells, remain to be fully understood (103).
A role for the CaSR in the stimulation of GI hormone secretion was first demonstrated in the late 1990s in in vitro studies, in which an elevation of the extracellular Ca^2+^ concentration or exposure to the CaSR activator spermine stimulated gastrin secretion from human antral G cells (87, 131, 132). In a subsequent in vivo study in transgenic mice, oral administration of Ca^2+^ or the CaSR agonist cinacalcet stimulated gastrin and gastric acid secretion in wild-type, but not CaSR-null, mice (20). In rats, oral Ca^2+^-induced stimulation of PYY secretion was reduced by pretreatment with the CaSR inhibitor, NPS-2143, by about 44% (31).
There is also preclinical evidence indicating that activation of the CaSR reduces food intake and/or blood glucose levels (31, 33). For example, in rats, oral administration of cinacalcet reduced food intake (31). Furthermore, IV administration of the CaSR agonist NPS-R568 increased plasma insulin and lowered postprandial blood glucose levels, whereas the CaSR inhibitor NPS-2143 blocked these effects (33). There have been no studies investigating the involvement of the CaSR in the effects of Ca^2+^ on food intake or blood glucose, but a number of animal studies indicate a link between dietary Ca^2+^ intake and energy metabolism, with high-Ca^2+^ diets potentially attenuating weight gain, at least in part, by simultaneously stimulating lipolysis and inhibiting lipogenesis during caloric restriction (133-135). Thus, further studies are required to clarify the role of the CaSR in this context.
Clinical Outcomes
As alluded to earlier, in humans, Ca^2+^ has long been known to increase plasma gastrin levels and gastric acid secretion (99, 101). However, its effect on other GI hormones is less clear. The majority of the available studies that investigated the acute or longer-term effects of Ca^2+^ have done so by incorporating Ca^2+^ in a meal or drink, that is, along with other nutrients, such as protein (Table 2) (38-44). For example, several studies used dairy products, which are also rich in whey and casein, known to stimulate GI hormone secretion (7). In contrast, few studies have evaluated the effects of Ca^2+^, in an isolated form, on gut hormone secretion (36, 37). Moreover, the duration of the Ca^2+^ intervention and the characteristics of the study population (eg, healthy lean, overweight, or with obesity), are also likely to be relevant. Outcomes of the different approaches are reviewed in the following sections.
Effects of acute (single-dose) dietary Ca2+ on GI hormones, energy intake, and blood glucose
Several studies have investigated the effects of dietary Ca^2+^ on the plasma levels of several GI hormones in healthy individuals. In one such study, consumption of a breakfast consisting of a porridge prepared with milk and supplemented with milk-extracted Ca^2+^ powder (Ca^2+^ dose: 1200 mg) resulted in higher postprandial levels of plasma GIP, GLP-1, and insulin, when compared with a porridge prepared with water (Ca^2+^ dose: 248 mg) (39). A second study compared the effects of a porridge prepared with water and supplemented with either 1170 mg milk-extracted Ca^2+^ or 25 g protein, or both, with the effects of a porridge that contained much lower amounts of Ca^2+^ (104 mg) and protein (4 g) (44). While the porridge, when supplemented with Ca^2+^ plus protein or with protein alone, increased plasma GLP-1 levels, the porridge that was enriched with Ca^2+^ alone had no effect (44), and none of the meals affected plasma GIP levels. A third study compared the effects of aqueous drinks, containing either 1000 mg of added Ca^2+^ in the form of calcium citrate or milk-extracted Ca^2+^ powder, or 50 g whey protein, or both Ca^2+^ and protein, on the stimulation of plasma GIP, GLP-1, and PYY (40). Interestingly, the drink that contained a combination of milk-extracted Ca^2+^ as well as protein resulted in significantly greater stimulations of GIP, GLP-1, and PYY, when compared with the effects of calcium citrate alone, or milk-extracted Ca^2+^ alone, protein alone, or water alone (40). When compared with water, ingestion of milk-extracted Ca^2+^ alone modestly increased plasma GLP-1 (40), while no effects were observed on plasma GIP or PYY levels.
A few studies have also investigated whether Ca^2+^-enriched foods acutely modulate energy intake (38, 44, 136, 137) and/or plasma glucose levels (38-40). For example, in one study, consumption of 600 mL skim milk, containing 1000 mg Ca^2+^, compared with 600 mL fruit juice, as part of a fixed-energy breakfast, induced greater postprandial fullness and was associated with a mean 8.5% reduction in energy intake at a subsequent ad-libitum lunch (136). In another study, in which healthy lean individuals consumed a porridge supplemented with either 1170 mg milk-extracted Ca^2+^ or 25 g protein, or both, only the porridge supplemented with both Ca^2+^ and protein significantly reduced ad-libitum energy intake by 169 kcal (17%), while smaller reductions in energy intake in response to Ca^2+^ alone or protein alone were not statistically significant (44).
The glucose-lowering effect of increased Ca^2+^ intake, if any, appears modest, and may be due to the effects of co-ingested nutrients. For example, in healthy individuals with overweight, isocaloric test meals, containing either 68, 350, or 793 mg of Ca^2+^ from dairy products, or 850 mg of Ca^2+^ in the form of a CaCO₃ supplement, had no effect on postprandial plasma glucose (38). Furthermore, no differences were evident in the plasma glucose response to a porridge containing either 1200 mg or 248 mg Ca^2+^ (39). In contrast, consumption of a drink, containing 1000 mg milk-extracted Ca^2+^ and 50 g whey protein, reduced postprandial plasma glucose (ie, primarily 60 minutes post-meal) more than drinks containing 1000 mg of milk-extracted Ca^2+^ or calcium citrate alone, although the absolute difference in plasma glucose was only 0.3 to 0.4 mmol/L (40).
Effects of longer-term Ca2+ supplementation on GI hormones, energy intake, and blood glucose
Several studies have investigated the longer-term effects of Ca^2+^ to evaluate whether the effects on GI hormones and/or energy intake, or plasma glucose, are sustained (41-43). For example, in healthy lean participants, consumption of bread enriched with 1100 mg Ca^2+^ daily for 3 weeks, as part of a controlled diet, significantly increased postprandial plasma levels of GLP-1, 30 minutes after the meal, more than non-enriched bread (42), although there were no significant effects on GIP, insulin, or postprandial glucose. Fasting GLP-1 concentrations were not affected by Ca^2+^ (42), suggesting that its primary effect is on meal-related GI hormone secretion. In another study in individuals with metabolic syndrome, supplementation of an energy-restricted diet with 1400 mg/day of Ca^2+^ from dairy products for 12 weeks had a greater stimulatory effect on postprandial plasma levels of GLP-1 and PYY, but not GIP, than 700 mg/day of Ca^2+^ (41), although no effects on energy intake or postprandial plasma glucose levels were evident. Consistent with these observations, the CaSR has been reported to be involved in the secretion of GLP-1 and PYY (28, 34), while secretion of GIP appears to involve GPR142 as well as the CaSR (35).
A few studies have reported the effect of Ca^2+^ supplementation on appetite perceptions, although not consistently and mostly under conditions of energy restriction, and findings have been inconclusive (137-140). For example, in a 6-month energy-restricted weight loss program in overweight women, Ca^2+^ supplementation in a dose of 1000 mg/d in the form of milk, reduced fasting levels of desire to eat and hunger, compared with a placebo supplement (140). A number of variables, including particularly the different nutritional composition of these foods, which may affect both the bioavailability and palatability of Ca^2+^ and, accordingly, its impact on GI hormone release, limit the ability to interpret the findings.
Effects of isolated Ca2+ on GI hormone secretion, energy intake, and blood glucose
To better characterize the potential effects of Ca^2+^ per se on GI hormone levels, 2 recent well-controlled studies in healthy lean males (36) and individuals with obesity (37), performed by our group, isolated Ca^2+^ from other nutrients by administering Ca^2+^, as a solution of CaCl_2_, directly into the duodenum. In both these studies, Ca^2+^, in a dose of 1000 mg, markedly increased plasma GLP-1 and PYY (Fig. 4), but not CCK or GIP, levels. As outlined above, whether these effects are mediated by the CaSR, and whether these effects of Ca^2+^ on GI hormone are associated with a reduction in energy intake and/or lowering of postprandial plasma glucose levels, now warrants investigation, for example, in the absence or presence of calcilytics.
Dose-related effects of intraduodenal administration of 0 mg (‘Ca-0’), 500 mg (“Ca-500”) or 1000 mg (“Ca-1000”) Ca2+, as a solution of CaCl2, alone (left columns in shades of blue) and in combination with L-tryptophan (L-Trp, right columns in shades of red) on areas under the curve (AUCs) of plasma concentrations of cholecystokinin (CCK) (A), glucagon-like peptide-1 (GLP-1) (B) and peptide YY (PYY) (C) in healthy lean males. Ca-1000 alone stimulated plasma GLP-1 and PYY, but not CCK. Both Ca-500 and Ca-1000 enhanced the effects of L-Trp, in a load of 0.1 kcal/min, to increase plasma CCK, GLP-1, and PYY. Data are expressed as means ± SEMs; n = 15. Figure is based on data from Anjom-Shoae et al (36).
Effects of L-Trp and L-Phe on GI Hormones, Energy Intake, and Blood Glucose
Various in vitro and in vivo studies have reported that the CaSR mediates L-Trp- or L-Phe-induced stimulation of GI hormone secretion (Table 1) (21-23, 26, 27). Consistent with these findings, human and mouse studies have demonstrated that L-Trp (55-61) and L-Phe (48-51) increase plasma concentrations of GI hormones, suppress energy intake, and reduce the rise in plasma glucose in response to a meal.
Preclinical Outcomes
As early as ∼1995, L-Phe-induced stimulation of CCK release from STC-1 cells was reported to be dependent on a Ca^2+^-dependent process (141). More recent in vitro studies have also demonstrated that the CaSR mediates L-Trp- or L-Phe-stimulated release of a number of GI hormones (21-23, 26, 27). For example, perfusion of pig gastric tissue with L-Trp or L-Phe increased expression of the CaSR (21), and the effects of L-Trp or L-Phe to stimulate gastrin secretion were enhanced by the CaSR activator cinacalcet and abolished by the CaSR inhibitor, NPS-2143 (21). The effects of L-Trp or L-Phe to stimulate CCK release from isolated mouse or pig small intestine were also inhibited in the presence of the CaSR inhibitors Calhex231 (by ∼55% for L-Trp and ∼25% for L-Phe) (23) or NPS-2143 (L-Trp- or L-Phe-induced peak plasma CCK was reduced from ∼0.20-0.25 ng/mL to ∼0.08-0.18 ng/mL) (25, 26). Furthermore, L-Phe-stimulated CCK secretion from the rat small intestine was completely blocked in the presence of NPS-2143 (22). Similar effects were also observed for other GI hormones. Thus, in isolated loops of rat intestine, the stimulatory effects of L-Trp or L-Phe on GIP, GLP-1, and PYY secretion were blocked by Calhex231 and augmented by the CaSR activator NPS-R568 (27). In porcine duodenal tissue, L-Phe also stimulated GIP release, and the effect was enhanced by cinacalcet, and attenuated by NPS-2143 (peak plasma GIP was reduced from ∼130 ng/mL to ∼70 ng/mL) (26).
There has been less attention to the potential role of the CaSR in mediating L-Trp- (30) or L-Phe-induced (20, 28) stimulation of GI hormone secretion in vivo, and whether these effects impact energy intake or blood glucose levels. In rats, for example, intraduodenal administration of L-Trp increased plasma GLP-1, but not PYY, levels (30), an effect associated with a modest reduction in the fasting blood glucose concentration (30). Both the elevation in plasma GLP-1 levels and reduction in fasting blood glucose were blocked by co-administration of the CaSR inhibitor NPS-2143 (30). Furthermore, in a study on transgenic mice, oral administration of L-Phe stimulated gastrin and gastric acid secretion in wild-type but not in CaSR-null mice (20), and the effect in wild-type mice was completely blocked by NPS-2143. In another study in rats and mice, administration of NPS-2143 blocked L-Phe-induced GLP-1 secretion and reversed L-Phe-induced reductions in food intake (28).
Clinical Outcomes
Findings in human studies are consistent with the preclinical data with respect to the effects of aromatic amino acids on GI hormones, but studies have not investigated how CaSR agonists/inhibitors modulate such effects; accordingly, a clear mechanistic role of the CaSR has not been established. In several studies, performed by us and other groups, L-Trp (55-62, 142) or L-Phe (48-53), administered mainly orally in capsules or directly into the small intestine or stomach (ie, via nasoduodenal or orogastric catheter), or IV, potently increased plasma GI hormone levels (Table 3). For example, IV infusion of L-Trp (in graded doses of 1.25-5 mmol/h) or L-Phe (in doses of 3.1-12.5 mmol/h), over 4 hours, increased plasma gastrin levels and also stimulated gastric acid secretion (143), and oral or rapid intragastric administration of L-Trp or L-Phe stimulated gastrin secretion in healthy individuals (144, 145). Moreover, oral consumption of 1.5 g L-Trp markedly elevated plasma CCK levels and modestly increased plasma GLP-1 in both healthy lean participants and individuals with obesity (59). In a further study, by our group, intraduodenal infusion of L-Trp for 90 minutes, in a caloric load of 0.15 kcal/min (providing 3.3 g over 90 minutes), but not 0.075 kcal/min (1.65 g over 90 minutes), increased plasma concentrations of CCK, GLP-1, and PYY (56). Similar effects on plasma CCK levels have been observed for L-Phe, when administered intraduodenally (in doses of 3.3 and 5 g) (48, 52), or intragastrically (53) or orally (49) (in a dose of 10 g). In contrast, L-Phe, administered alone either as an oral dose of 10 g (49), or as an intragastric dose of 5 or 10 g (53), did not significantly increase plasma GLP-1 levels.
L-Trp (56, 59, 61, 62, 142) or L-Phe (49, 50, 53, 54) have also been found to reduce energy intake. For example, we and others have reported that oral administration of L-Trp, in doses of 2 to 3 g, or intragastric administration in a dose of 3.3 g, reduced energy intake substantially in both healthy lean males (61, 146) and in individuals with obesity (61, 147), by up to 200 kcal. Similarly, oral administration of L-Phe (in a dose of 10 g) decreased energy intake in one study in healthy volunteers by 498 kcal (49), and in another study, intragastric or oral administration of L-Phe, in a dose of 10 g, reduced energy intake by 200 (53) or 89 (50) kcal, respectively.
In contrast, the glucose-lowering effects of L-Trp or L-Phe appear to be more modest (50, 53, 54, 62, 142). For example, intragastric administration of 3 g L-Trp as an aqueous solution, 30 minutes before a carbohydrate-containing drink, delayed the rise in plasma glucose in individuals with type 2 diabetes (142), and modestly attenuated the glycemic response in those with obesity by ∼1 mmol/L (62).
Interactions of Ca2+ With L-Trp and L-Phe on GI Hormone Secretion
A number of preclinical studies have investigated whether the stimulatory effects of L-Trp or L-Phe on GI hormone release are dependent on, or positively modulated by, extracellular Ca^2+^ concentration (22, 24-26). Two recent clinical studies have also provided the first evidence that Ca^2+^ enhances the stimulatory effects of L-Trp on GI hormone release in humans (36, 37). The following sections review evidence on the interaction between the effects of Ca^2+^ and the aromatic AAs, L-Trp, and L-Phe, on plasma levels of GI hormones, from both preclinical models and humans. It is important, however, to reiterate that such observations do not allow conclusions as to a direct role for the CaSR.
Preclinical Outcomes
The stimulatory effects of L-Trp or L-Phe on gastrin secretion from pig intestinal tissue were abolished in the absence of extracellular Ca^2+^ (21). On the other hand, L-Trp- and L-Phe-stimulated CCK secretion from isolated mouse or porcine small intestine was enhanced by increases in extracellular Ca^2+^ (22, 24-26), and co-perfusion of porcine duodenal tissue with Ca^2+^ enhanced the stimulatory effects of L-Trp or L-Phe on GIP secretion (25, 26). In isolated loops of rat intestine, luminal perfusion of AAs, including L-Phe or L-Trp, in the presence of a physiologically relevant Ca^2+^ concentration (1.25 mM), stimulated the secretion of GIP, GLP-1, and PYY, and these effects were blocked by the CaSR inhibitor Calhex231 (27). In addition, extracellular Ca^2+^ markedly enhanced the stimulatory effect of L-Phe in a concentration-dependent manner between 0.3 and 10 mmol/L (27), and the effect of L-Phe was abolished in the absence of extracellular Ca^2+^ (27). Taken together, these observations demonstrate that extracellular Ca^2+^ is required for, and positively modulates, the stimulatory effects of AAs, as previously reported for the CaSR (17, 19). It is, accordingly, possible that reports in which AAs have been observed to stimulate the secretion of some GI hormones, but not others, reflect differences in the Ca^2+^ concentration thresholds required for the effects. Consistent with this notion, in the perfused rat intestine model described above, the extracellular Ca^2+^ threshold concentrations for the stimulatory effect of L-Phe on different GI hormones were around 1.0mM for GIP, 0.3mM for GLP-1, and 0.1mM for PYY (27). Differences of this magnitude might reflect variations in the levels of CaSR expression and/or in the signaling pathways downstream of the CaSR in hormone-secreting cells. These observations may also explain why the administration of AAs has been less effective with respect to GIP than GLP-1 and PYY in some models, both preclinical and clinical, dependent on the prevailing intestinal luminal Ca^2+^ concentration.
Clinical Outcomes
In humans, the impact of Ca^2+^ to enhance the effect of L-Trp to stimulate GI hormone secretion and enhance the suppression of energy intake was investigated in 2 recent studies (36, 37). In these studies, the modest effects of intraduodenal infusion of L-Trp, in a load of 0.1 kcal/min, to increase plasma CCK, GLP-1, and PYY levels were shown to be enhanced by the addition of 500 or 1000 mg Ca^2+^ (as a solution of CaCl_2)_, in both healthy lean participants (36) and those with obesity (37) (Fig. 4). These effects were also associated with a substantial suppression of energy intake (36, 37), so that Ca^2+^ enhanced the suppressive effects of L-Trp on energy intake in a Ca^2+^ dose-related manner. In contrast to CCK, GLP-1, and PYY, the stimulation of GIP by L-Trp was reduced by Ca^2+^ (36). This apparent discrepancy is consistent with previous studies in humans, in which GIP secretion exhibited atypical responses to some nutrients, such as fructose (148, 149). Moreover, no effect on gastrin levels was observed, suggesting minimal, if any, interaction of Ca^2+^ or L-Trp with the G cells in the stomach in this study (36, 37). Moreover, as noted above with respect to the Ca^2+^ concentration thresholds for AA effects, it is possible that the Ca^2+^ concentration ranges over which positive interactions occur with AAs also differ between GI hormone-secreting cell types, and that the effects for GIP and gastrin were maximal in the presence of the baseline Ca^2+^ concentration.
Taken together, these findings indicate that Ca^2+^ has the potential to enhance the effect of L-Trp on the release of several GI hormones in humans, most notably CCK, GLP-1, and PYY, but not others (GIP and gastrin being notable exceptions), and that the outcomes are dependent on the Ca^2+^ dose. The findings support preclinical evidence that extracellular Ca^2+^ enhances the effects of L-Trp on these (22-30) and other GI hormones (20, 21). Clinical evidence for any positive effects of L-Phe, as well as the role of the CaSR in the above effects, is awaited with considerable interest.
Summary and Future Directions
Since it was first cloned, the CaSR has been the subject of intense research, in part, due to its multiple nutrient binding sites. The dimeric structure of the CaSR supports the binding of AAs as well as extracellular Ca^2+^, thereby displaying diverse physiological impacts. Preclinical studies have established that the CaSR mediates AA-induced secretion of several gut hormones, including gastrin, CCK, GLP-1, GIP, and PYY, with the most potent responses observed for aromatic AAs, including L-Trp and L-Phe. Moreover, these effects are dependent on extracellular Ca^2+^, with distinct threshold Ca^2+^ concentrations required for the effects of AAs on different hormones. Of these GI hormones, the secretion of GLP-1 and PYY are particularly sensitive to elevations in the Ca^2+^ and AA concentrations. Recent human data have also provided evidence that administration of Ca^2+^ dose-dependently enhances the effects of L-Trp to increase plasma levels of CCK, GLP-1, and PYY, associated with greater suppression of energy intake. These findings underscore the intriguing possibility that Ca^2+^ and L-Trp (along with other AAs) act synergistically in humans, as well as animal models, to achieve metabolic benefits through enhanced stimulation of GI hormone release.
While the advent and highly successful application of GLP-1 receptor agonists (150) and the dual GIP/GLP-1 receptor agonist, tirzepatide (151), attest to the therapeutic potential of targeting gut hormone pathways, current pharmacological treatments for the management of obesity and type 2 diabetes are not without limitations, including variable efficacy and high cost, and they are frequently associated with adverse effects, particularly GI symptoms (152), resulting in high discontinuation rates in clinical practice (153). Furthermore, cessation of successful obesity therapy is usually associated with rebound weight gain (154). Thus, targeted nutritional interventions designed to accentuate endogenous gut hormone release remain attractive and have several notable advantages. First, nutritional interventions are expected to concurrently stimulate the release of multiple hormones, including CCK, GLP-1, and PYY, thereby amplifying their collective impacts on appetite suppression and glycemic control (8), while minimizing nonphysiological adverse effects. Secondly, while GLP-1 receptor agonists act primarily via central mechanisms (155), nutrient-induced endogenous GLP-1 release is expected to engage beneficial local GI mechanisms, including locally via receptors on vagal afferent nerve endings, and indirectly through slowing of gastric emptying (156, 157). Finally, while nutrients evoke physiological patterns of hormone secretion and, in turn, receptor activation, sustained activation of GLP-1 receptors via drug agonists may promote receptor downregulation and thereby reduce efficacy in the longer term, so-called “tachyphylaxis.” Accordingly, physiologically informed, well-tolerated, and cost-effective nutritional strategies, either as standalone therapies or as adjuncts to pharmacological treatments, are required for the management of obesity and type 2 diabetes.
Future research should explore the dose-response relationship and longer-term effects of combined administration of Ca^2+^ with L-Trp, L-Phe, and other effective AAs, on energy intake, and postprandial glycemia, as well as body weight, in both healthy people and in metabolically impaired populations. Furthermore, more research is required in humans to directly investigate the role of the CaSR in mediating these effects. This includes evaluating whether nutrient-induced hormone release is CaSR-dependent in vivo, using CaSR antagonists in individuals expressing the wild-type CaSR and individuals with selected mutations of the AA binding site. Additionally, the development and testing of selective CaSR agonists (eg, recently described very high-potency calcimimetics (158)) may represent a novel therapeutic avenue for enhancing endogenous gut hormone secretion without systemic side effects.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol. 2016;78(1):277‐299.26442437 10.1146/annurev-physiol-021115-105439 · doi ↗ · pubmed ↗
- 2Steensels S, Depoortere I. Chemoreceptors in the gut. Annu Rev Physiol. 2018;80(1):117‐141.29029594 10.1146/annurev-physiol-021317-121332 · doi ↗ · pubmed ↗
- 3Davison A, Reimann F, Gribble FM. Molecular mechanisms of stimulus detection and secretion in enteroendocrine cells. Curr Opin Neurobiol. 2025;92:103045.40378579 10.1016/j.conb.2025.103045 · doi ↗ · pubmed ↗
- 4Gribble FM, Reimann F, Roberts GP. Gastrointestinal hormones. In: Said HM, ed. Physiology of the Gastrointestinal Tract. 6th ed. Academic Press; 2018:31‐70.
- 5Fothergill LJ, Furness JB. Diversity of enteroendocrine cells investigated at cellular and subcellular levels: the need for a new classification scheme. Histochem Cell Biol. 2018;150(6):693‐702.30357510 10.1007/s 00418-018-1746-x PMC 6447040 · doi ↗ · pubmed ↗
- 6Bany Bakar R, Reimann F, Gribble FM. The intestine as an endocrine organ and the role of gut hormones in metabolic regulation. Nat Rev Gastroenterol Hepatol. 2023;20(12):784‐796.37626258 10.1038/s 41575-023-00830-y · doi ↗ · pubmed ↗
- 7Anjom-Shoae J, Feinle-Bisset C, Horowitz M. Impacts of dietary animal and plant protein on weight and glycemic control in health, obesity and type 2 diabetes: friend or foe? Front Endocrinol (Lausanne). 2024;15:1412182.39145315 10.3389/fendo.2024.1412182 PMC 11321983 · doi ↗ · pubmed ↗
- 8Steinert RE, Feinle-Bisset C, Asarian L, Horowitz M, Beglinger C, Geary N. Ghrelin, CCK, GLP-1, and PYY (3–36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol Rev. 2017;97(1):411‐463.28003328 10.1152/physrev.00031.2014 PMC 6151490 · doi ↗ · pubmed ↗