memod-s: a standardised workflow to explore and analyse prokaryotic methylation patterns for Nanopore sequencing data
Alessia Marotta, Lapo Doni, Alessia Avesani, Iacopo Passeri, Camilla Fagorzi, Alessio Mengoni, Jaime Martinez-Urtaza, Frederico M Batista, Luigi Vezzulli, Emanuele Bosi

TL;DR
memod-s is a new workflow that simplifies analyzing bacterial DNA methylation patterns using Nanopore sequencing data.
Contribution
memod-s introduces a unified, user-friendly pipeline for prokaryotic methylation analysis with integrated visualization and statistics.
Findings
memod-s combines basecalling, assembly, and methylation analysis into one pipeline.
The workflow provides genome-wide methylation profiles with visualizations and statistics.
memod-s is open source and available for use in bacterial epigenomic studies.
Abstract
Understanding the bacterial epigenome is increasingly recognised as essential for uncovering key mechanisms of gene regulation, host-pathogen interactions, and adaptation to environmental changes. Third-generation sequencing technologies, such as Oxford Nanopore, now enable the direct detection of DNA modifications, making genome-wide epigenomic investigations both feasible and cost-effective. However, analysing Nanopore sequencing data remains computationally intensive and requires multiple steps, which can be complex to integrate. Currently, no existing workflow combines these steps in a single, easy-to-use pipeline. Additionally, many available tools lack automated genome-wide methylation profiling with integrated visualisations and statistics. Here, we present memod-s, a Snakemake-based workflow that integrates multiple state-of-the-art tools to address these challenges. memod-s is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
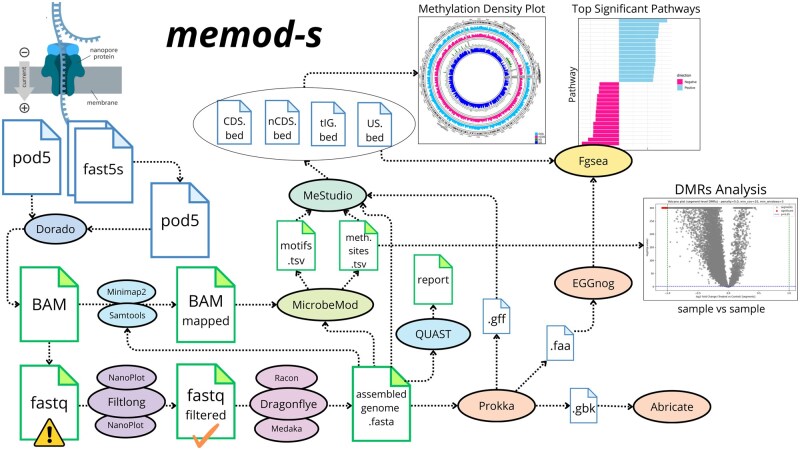 Figure 1
Figure 1| Feature |
| MehyloMiner | backmethy | modkit | Microbemod | mestudio |
|---|---|---|---|---|---|---|
|
|
| ✔ | ✖ | ✔ | ✔ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✔ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✔ | ✖ | ✖ | ◐ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✔ | ✖ | ✔ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✔ | ✖ |
|
| ✔ | ✖ | ✔ | ✖ | ✖ | ✔ |
|
| ✔ | ✖ | ✔ | ✖ | ✖ | ✔ |
|
| ✔ | ✖ | ✔ | ✖ | ✖ | ✔ |
|
| ✔ | ✖ | ✔ | ✖ | ✖ | ✔ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✔ | ✖ | ✖ | ✖ | ✔ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✔ | ✖ | ✖ | ✖ | ✖ | ✖ |
|
| ✖ | ✔ | ✖ | ✖ | ✖ | ✖ |
- —National Recovery and Resilience Plan (NRRP)
- —European Union—NextGenerationEU
- —Concession Decree
- —Italian Ministry of University and Research
- —Seedcorn funding from the Centre for Environment
- —Fisheries and Aquaculture Science
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Genomics and Phylogenetic Studies · Bacteriophages and microbial interactions
1 Introduction
Advances in sequencing technologies enabled the direct detection of DNA methylation and other base modifications by sequencing native DNA molecules. Third-generation platforms, such as single-molecule real-time sequencing by Pacific Biosciences (Eid et al. 2009) and Nanopore sequencing by Oxford Nanopore Technologies (Deamer et al. 2016), have overcome read-length limitations, achieving ultra-long reads and single-base resolution at a genome-wide scale. By sequencing native DNA, these technologies preserve the integrity of the molecule and retain epigenetic information, facilitating the comprehensive detection of base modifications across entire genomes. Although epigenomics is recognized as a key regulatory layer in cell biology, genome-wide studies of DNA methylation have started growing only recently, gaining significant traction in recent years. This trend is driven by increased sequencing accuracy, reduced costs, and growing interest in the biological roles of epigenetic modifications. As a result, the volume of available high-quality methylation data is expanding significantly, witnessing the start of what could be named as a golden age of epigenomics. This stands even more for bacteria, as the functional implications of epigenetic regulation in these organisms are still obscure, and their relatively simple genome structure, coupled with the feasibility of their maintenance, culturing and genetic manipulation, could provide new and important insights into the biological role of DNA modifications. It has been long recognized that DNA methylation in prokaryotes plays numerous crucial roles ranging from well-known restriction-modification systems defending against exogenous DNA to cell cycle regulation, DNA repair, phase variation, and gene expression regulation, including virulence and stress response (Seong et al. 2021). Despite these premises, research exploring this direction is still far from its full potential. Here, we present memod-s, a first step towards a comprehensive analysis of bacterial methylome that can be further improved to allow for the integration of gene expression data, i.e. transcriptomics and proteomics, to detail more precisely the functional consequences of DNA methylation, and their implications for various phenotypic traits of interest, enhancing our interpretation and understanding of the roles and implications of methylation in bacterial adaptation and evolution.
2 Motivation
Nanopore sequencing data analysis involves numerous steps and can be computationally intensive and time-consuming, involving several tools to produce interpretable results from reads. This complexity can be a barrier to entry for researchers interested in epigenomic studies. The primary goal of memod-s workflow is to streamline this process from squiggle Nanopore data and encompass all essential steps, including basecalling, genome assembly, quality check, quality filtering, annotation and methylation analysis, while minimising user efforts. The workflow not only generates comprehensive results but also provides genome-wide methylation profiles supported by graphical visualisations, all achievable with a single command-line execution. Additionally, the workflow is modular and highly customisable to meet specific user requirements. It leverages Conda environments to manage software dependencies, avoiding version conflicts and eliminating the need for manual installation. By significantly reducing the technical burden associated with Nanopore data analysis, memod-s makes epigenomic investigations more accessible and efficient for the broader scientific community.
3 Implementation
memod-s is a modular workflow implemented in Snakemake v8.27.1 (Mölder et al. 2021) which integrates several bioinformatic tools (Supplementary Table 1) and related features (Table 1). Users need to provide input data in FAST5, POD5 or BAM format, while memod-s automatically generates the sample table (.tsv format) and the configuration file (.yaml format) based on the parameters specified in the command line at runtime, allowing users to customise settings dynamically. The workflow is implemented in eight core steps: (i) Basecalling (ii) Filtering and quality check, (iii) Assembly, polishing and assembly evaluation, (iv) Annotation, (v) Methylation calling, (vi) Methylation analysis, (vii) Methylation density plots, (viii) Gene set enrichment analysis and (ix) Differentially methylated regions analysis (Fig. 1).
Schematic workflow diagram of memod-s.
Basecalling: The basecalling module converts raw Nanopore signal data into nucleotide sequences using the basecaller Dorado (https://github.com/nanoporetech/dorado.git) for Oxford Nanopore reads. Beyond traditional basecalling of A, T, C, and G nucleotides, Dorado also enables the detection of base modifications. Prior to basecalling, the workflow automatically handles the selection and retrieval of dataset-specific Dorado models. By default, memod-s relies on Dorado’s model auto-discovery mechanism, using the basecalling model recommended by the installed Dorado version. To ensure robustness and reproducibility, the workflow implements a controlled fallback strategy: if the auto-discovered model is unavailable or incompatible, users can customize these models via a command-line parameter when launching memod-s. The basecalling step is optional and can be skipped when pre-basecalled data are already available. In such cases, memod-s can start directly from externally generated BAM files. When basecalling is performed within memod-s, raw FAST5 files are converted to POD5 format if required, and an unmapped BAM file containing base modification information for each read is produced and used in subsequent methylation analyses. Filtering and quality check: To mitigate errors introduced during sequencing, the workflow includes a quality check step. In FASTQ files, each base is assigned a Phred quality score. The BAM file from basecalling is converted to FASTQ format, and NanoPlot (De Coster and Rademakers 2023) is employed to generate a visual and statistical report on read quality. Reads are then filtered using Filtlong (https://github.com/rrwick/Filtlong.git) with default parameters (as specified in the tool documentation), with the option for user-defined customization via command-line flags. A second NanoPlot report is generated to compare quality metrics before and after filtering. Assembly, polishing and evaluation: For the de novo genome assembly, memod-s allows users to select among multiple long-read assemblers, including Flye-dragonflye (https://github.com/rpetit3/dragonflye), Canu (https://github.com/marbl/canu) and Raven (https://github.com/lbcb-sci/raven). By default, assembly can be followed by multiple rounds of polishing with Racon (https://github.com/isovic/racon.git). Users can adjust polishing parameters via a command-line flag. The quality assessment of the genome assembly is performed using QUAST (Gurevich et al. 2013). Annotation: Genome annotation is performed with Prokka (Seemann 2014), while functional annotation of predicted coding sequences is achieved using EggNOG-mapper (Huerta-Cepas et al. 2019, Cantalapiedra et al. 2021) with DIAMOND (Buchfink et al. 2015). The amino acid sequences generated by Prokka are provided as input to eggNOG in FASTA format. As an optional step, genes related to virulence are mapped using the Virulence Factors Database (Chen et al. 2016) with Abricate (https://github.com/tseemann/abricate). Methylation calling: To map basecalled reads to the assembled genome, including methylation metadata, a bash-based script using Samtools (Danecek et al. 2021) and Minimap2 (Li 2018) is implemented. The script requires the BAM file from basecalling and the previously assembled genome in FASTA format. The assembled genome and its corresponding read mapping are then used as input for MicrobeMod to determine methylation status. The MicrobeMod “call_methylation” command generates several output files, including two main tab-separated tables: one detailing methylated sites and the other describing methylated motifs (Crits-Christoph et al. 2023). Additionally, a comprehensive table is generated listing all candidate genes involved in restriction-modification systems within the provided genome, ranked based on their operonic context, and including all subtype and homolog information. Methylation analysis: We developed a custom python parser to adapt Nanopore methylation data to MeStudio’s input format, originally designed for PacBio data; the previously identified nucleotide motifs are categorised based on the features extracted from the annotation file. Categories include: protein-coding genes on the sense strand (CDS), protein-coding genes on the antisense strand (nCDS), true intergenic regions located between annotated genes (tIG), and upstream regions relative to the reading frame of a gene on the sense strand (US) (Riccardi et al. 2022). Methylation density plots: For each identified motif, MeStudio generates tabular files in BED format with the methylation status for each category. These data are used to visualise methylation density distributions for each feature in the genome using the Circlize R package (Gu et al. 2014). Gene Set Enrichment Analysis: Gene Set Enrichment Analysis is performed using the fgsea R package (Korotkevich et al. 2021). The required pathway annotation was derived from EggNOG-mapper, generating a mapping of KEGG pathways to associated genes. The KEGG Orthology database (Kanehisa et al. 2016) is used to assign functional orthologs, facilitating pathway reconstruction and high-level functional interpretation. Significantly enriched pathways are identified based on the normalised enrichment score (NES) and adjusted P-value (padj). Differentially Methylated Regions Analysis: To identify differentially methylated regions in the genome, where methylation patterns vary between experimental conditions—such as stress responses to temperature shifts, antibiotic exposure or across different time points in a time-course experiment—we aggregated methylation counts into genomic windows. We then applied segmentation to detect consistent shifts between control and treated samples, followed by statistical testing of each segment to assess significance.
4 Case study
To better illustrate this software utility, we applied memod-s to a real case study, i.e. the analysis of Vibrio aestuarianus (Vaes) genome and its nucleotide modification patterns, representing, to the best of our knowledge, the first epigenomic investigation of a representative of the genus Vibrio. Vaes isolates were obtained from oysters in the Republic of Ireland between 2008 and 2015 during mortality events. Vaes is recognised as one of the most important pathogens in oyster infections (Doni et al. 2025), with adult mortality rates linked to this bacterium reaching up to 30% by the end of the farming cycle (Doni et al. 2023). For whole-genome sequencing, genomic DNA from Vaes was extracted using the QIAamp DNA Mini Kit. Library preparation was carried out with the Ligation Sequencing Kit (Q20+, SQK-Q20EA), and sequencing was performed with MinION Mk1C using the R10.4.1 flow cell (FLO-MIN114). The sequencing yielded a total of 266,643 reads (0.654 Gb) with an average length of 2,452.4 bp, representing a coverage of 151x of the genome of Vaes. The analyses were performed on a workstation equipped with: CPU Intel^®^ Core™ i9-13900 (13th Gen), 24 cores/32 threads; RAM: 128 GB; GPU: NVIDIA RTX A5000 (24 GB VRAM); Operating system: Ubuntu 24.04.1 LTS. The RTX A5000 GPU was used for accelerated basecalling (Dorado), while all downstream steps of the memod-s workflow were executed on CPU.
memod-s completed the analysis in 757 minutes, including the basecalling step. Annotation of putative virulence factors with Abricate identified 8 genes associated with flagella and 2 with chemotaxis. Methylation analysis step identified a total of two methylated motifs, YGATCR and CGSCG, with one exhibiting 6 mA and the other 5mC modification. Methylation density plots revealed distinct methylation patterns for each category within each motif (Fig. S1). For example, upstream regions displayed a higher density of methylation compared to tIG regions, indicating a potential role of methylation in promoter regions. To test the differential methylation analysis, a simulated treated sample was generated by introducing and altering specific methylation sites. A volcano plot was generated with the x-axis representing the methylation difference between treated and control samples (log_2_FC) and the y-axis representing statistical significance (-log_10_(FDR)), allowing visualization of the number of regions with substantial methylation changes, the distribution of hyper- and hypo-methylation, and the segments exhibiting the greatest statistical robustness (Fig. S2). The detailed results are reported in the Supplementary Material.
Supplementary Material
vbag072_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Buchfink B , Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12:59–60.25402007 10.1038/nmeth.3176 · doi ↗ · pubmed ↗
- 2Cantalapiedra CP , Hernández-Plaza A, Letunic I et al egg NOG-Mapper v 2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 2021;38:5825–9.34597405 10.1093/molbev/msab 293PMC 8662613 · doi ↗ · pubmed ↗
- 3Chen L , Zheng D, Liu B et al VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res 2016;44:D 694–7.26578559 10.1093/nar/gkv 1239 PMC 4702877 · doi ↗ · pubmed ↗
- 4Crits-Christoph A , Kang SC, Lee HH et al 2023. Microbe Mod: a computational toolkit for identifying prokaryotic methylation and restriction-modification with nanopore sequencing. bio Rxiv, 10.1101/2023.11.13.566931, 14 November, preprint: not peer reviewed. · doi ↗
- 5Danecek P , Bonfield JK, Liddle J et al Twelve years of SA Mtools and BC Ftools. Giga Science 2021;10:giab 008. 10.1093/gigascience/giab 008. · doi ↗
- 6Deamer D , Akeson M, Branton D. Three decades of nanopore sequencing. Nat Biotechnol 2016;34:518–24.27153285 10.1038/nbt.3423 PMC 6733523 · doi ↗ · pubmed ↗
- 7De Coster W , Rademakers R. Nano Pack 2: population-scale evaluation of long-read sequencing data. Bioinformatics 2023;39:btad 311. 10.1093/bioinformatics/btad 311. · doi ↗
- 8Doni L , Taviani E, Bosi E et al Milestones in vibrio science and their contributions to microbiology and global health. Ann Glob Health 2025;91:23. 10.5334/aogh.471140385502 PMC 12082447 · doi ↗ · pubmed ↗