Doing More with Less: Accurate and Scalable Ligand Free Energy Calculations by Focusing on the Binding Site
David Alencar Araripe, Alejandro Díaz-Holguín, Antti Poso, Gerard J. P. van Westen, Johan Åqvist, Hugo Gutiérrez-de-Terán, Willem Jespers

TL;DR
QligFEP is a new tool that makes drug binding predictions faster and cheaper by focusing simulations on the binding site.
Contribution
QligFEP v2.1.0 introduces a scalable, accurate, and computationally efficient workflow for ligand free energy calculations.
Findings
QligFEP achieves comparable accuracy to commercial tools while using fewer computational resources.
Each perturbation leg simulates around 6250 atoms and completes in under 2 hours on standard clusters.
The method is cost-effective, with simulations costing less than $1 on AWS spot instances.
Abstract
Predicting how chemical modifications affect drug binding is central to rational drug design. Free energy perturbation (FEP) calculations provide accurate estimates of these binding affinity changes, but existing methods often require substantial computational resources and expert knowledge. Here, we present QligFEP v2.1.0, a flexible open-source workflow based on a graphical and command-line interface for calculating relative binding free energies using spherical boundary conditions, which dramatically reduces simulation system size by confining simulations to a focused region around the binding site. QligFEP features a configurable restraint algorithm that automatically handles diverse chemical transformations, streamlined setup procedures, and enhanced analysis tools. We validated the method using industry benchmarks comprising 16 protein targets and 639 ligand transformations.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1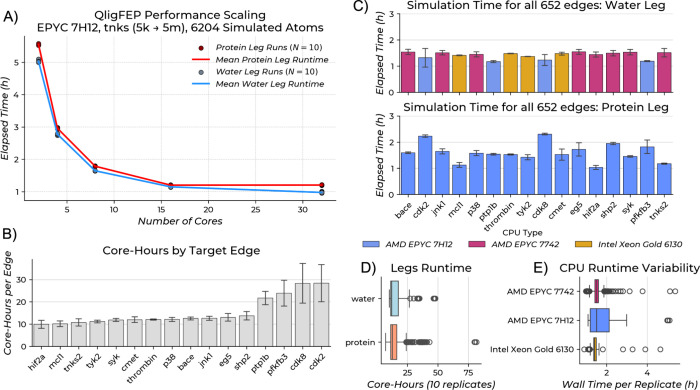 Figure 2
Figure 2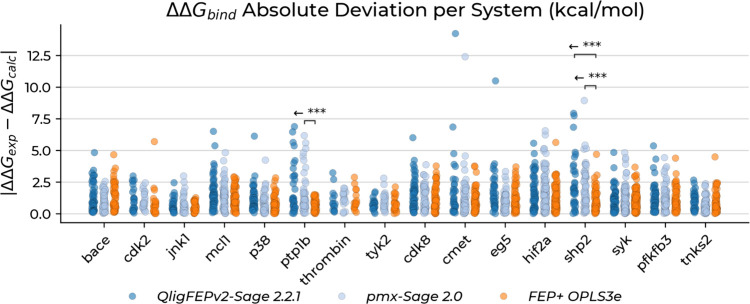 Figure 3
Figure 3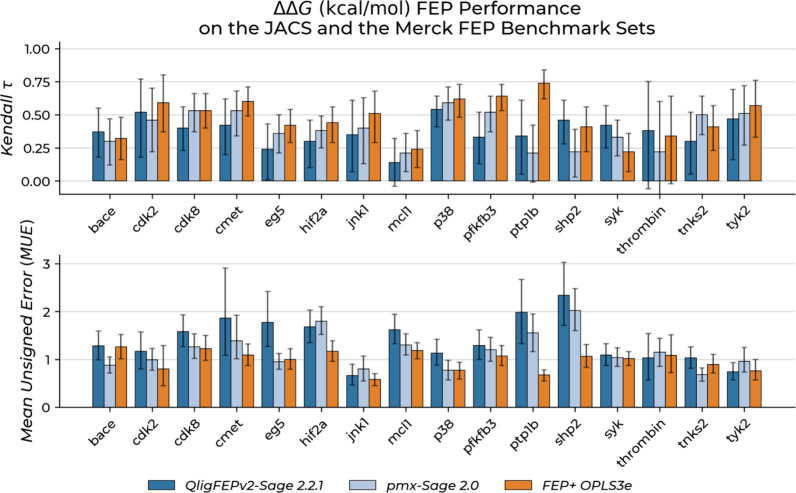 Figure 4
Figure 4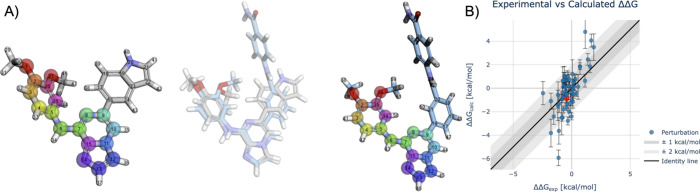 Figure 5
Figure 5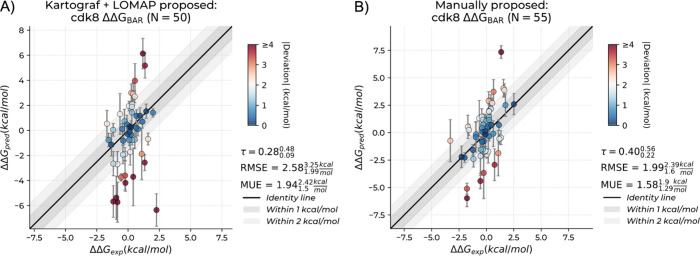 Figure 6
Figure 6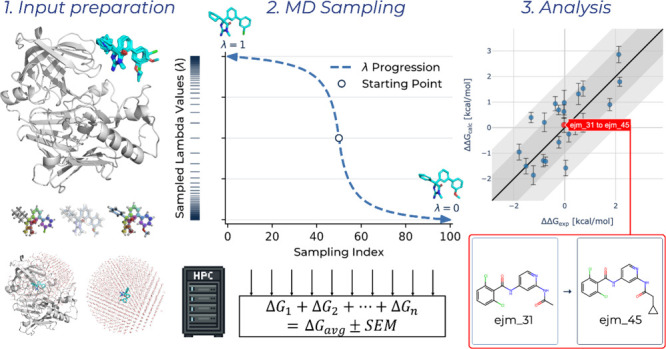 Figure 7
Figure 7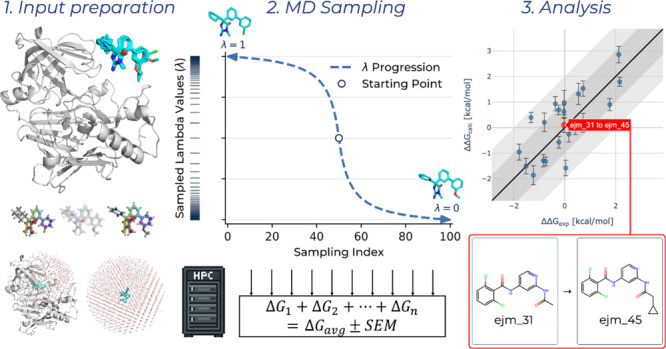 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComputational Drug Discovery Methods · Protein Structure and Dynamics · Protein Degradation and Inhibitors