Genomic characterisation and antimicrobial resistance of Klebsiella species isolated from small stock and environmental sources in South Africa
Tshepang Motlhaping, Deidré Van Wyk, Oriel Thekisoe, Henriette Van Heerden, Kgaugelo E. Lekota, Tsepo Ramatla

TL;DR
This study identifies antibiotic-resistant Klebsiella species in South African rural environments, highlighting their public health risks and the need for better surveillance.
Contribution
The study provides genomic evidence that rural agricultural settings are reservoirs of multidrug-resistant Klebsiella species.
Findings
Klebsiella isolates from sheep and water sources showed resistance genes like fosA, oqxAB, and blaLEN24/16.
K. pneumoniae strains contained plasmid AB595 with tetracycline, sulphonamide, and aminoglycoside resistance genes.
Virulence genes for adhesion, siderophores, and motility were identified, emphasizing their role in antibiotic resistance.
Abstract
Klebsiella species pose a significant public health concern due to their association with various infections and the rising levels of antibiotic resistance. This study examined the antibiotic resistance profiles of nine Klebsiella species obtained from the culture collection at North-West University’s Potchefstroom campus in South Africa using whole genome sequencing. The average nucleotide identity (ANI), plasmid types, and the identification of antibiotic resistance and virulence genes were performed on Klebsiella species. The nine sequenced Klebsiella spp. isolates were identified as K. pneumoniae (n = 2) originated from sheep faeces, K. variicola (n = 2) and K. michiganensis (n = 5) isolated from water (river stream). The genomes of K. variicola and K. michiganensis contained antibiotic resistance genes for fosfomycin (fosA), nalidixic acid (oqxAB), and β-lactamase (blaLEN24/16 or…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
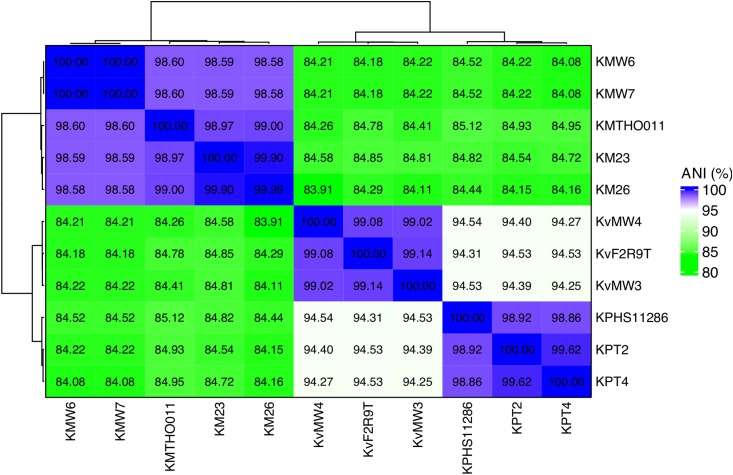 Figure 1
Figure 1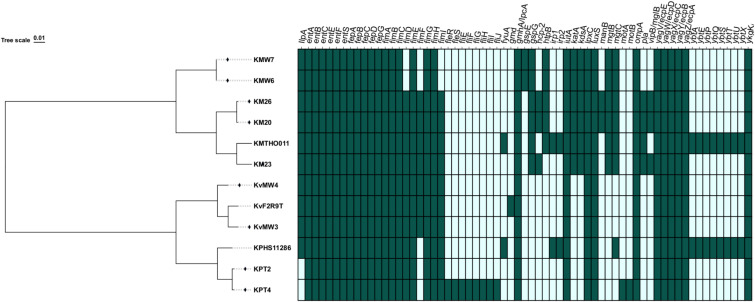 Figure 2
Figure 2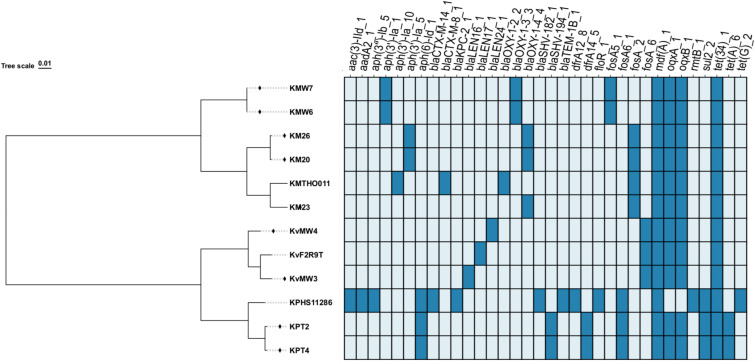 Figure 3
Figure 3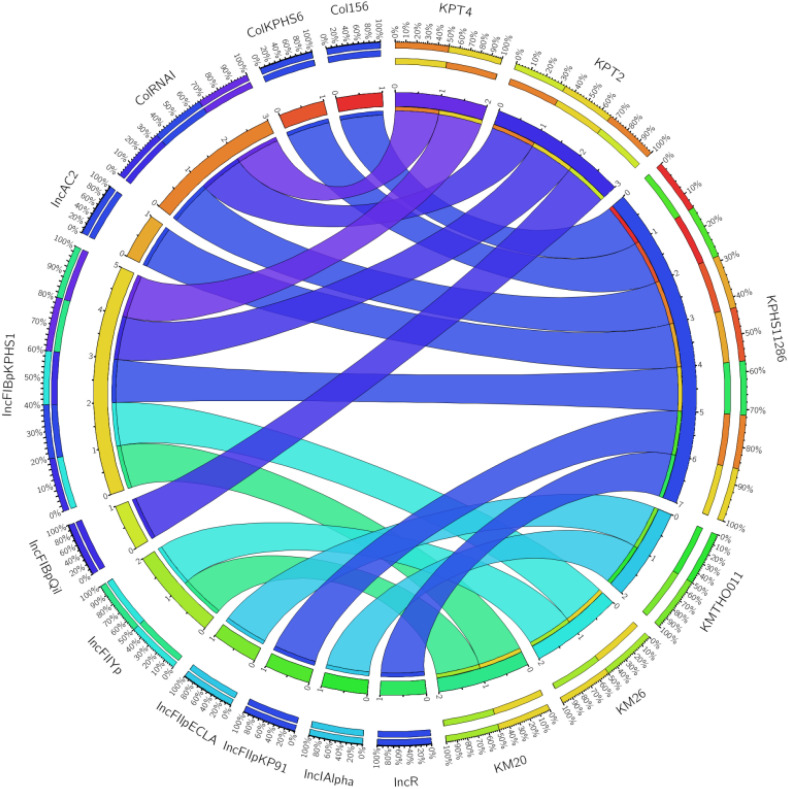 Figure 4
Figure 4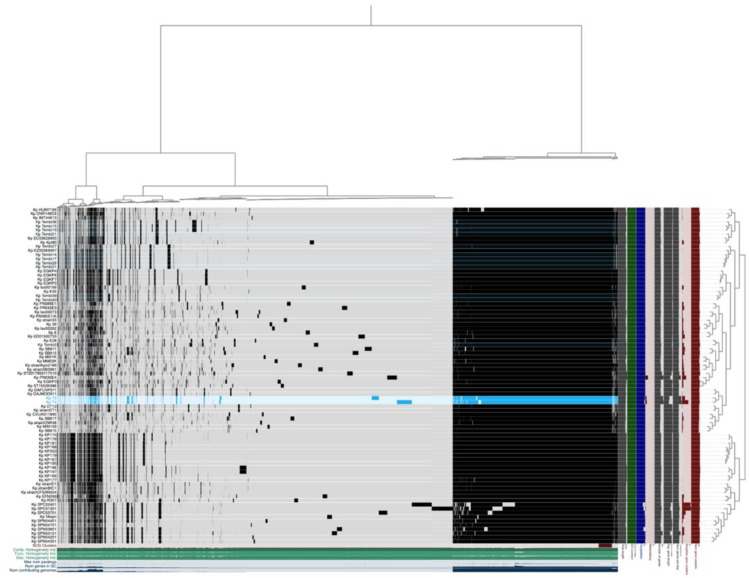 Figure 5
Figure 5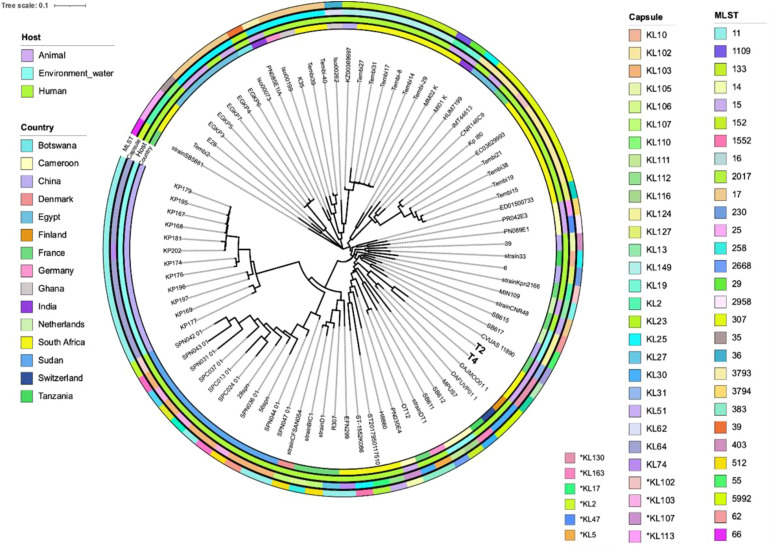 Figure 6
Figure 6| Aminoglycoside | Quinolone | Cephalosporin | Carbapenem | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Isolates | AMP | K | NA | KZ | ETP | MEM | IPM | DOR | |
|
| KMW3 |
|
|
|
|
|
|
|
|
| KMW4 |
|
|
|
|
|
|
|
| |
|
| KMW6 |
|
|
|
|
|
|
|
|
| KMW7 | + | + | + | – | – | – | – | – | |
| KM20 | + | + | + | + | – | – | – | – | |
| KM26 | + | + | + | + | – | – | – | – | |
| KM23 | + | – | + | + | – | – | – | – | |
|
| KPT2 |
|
|
|
|
|
|
|
|
| KPT4 |
|
|
|
|
|
|
|
| |
| Contigs stats | KM20 | KM23 | KM26 | KMTHO011* | KMW6 | KMW7 | KPHS11286* | KPT2 | KPT4 | KvF2R9T* | KvMW3 | KvMW4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total length | 6,447,561 | 5,217,832 | 6,550,179 | 6,041,841 | 5,335,245 | 5,335,245 | 5,682,322 | 5,427,021 | 5,432,140 | 5,521,194 | 5,288,060 | 4,908,316 |
| Num contigs | 162 | 344 | 264 | 2 | 35 | 35 | 7 | 4 | 7 | 1 | 72 | 31 |
| GC% | 555 | 55.5 | 56.5 | 55.1 | 56.5 | 56.5 | 57.2 | 57.5 | 57.5 | 57.6 | 58 | 57.5 |
| Num contigs > 100 kb | 20 | 0 | 23 | 2 | 20 | 20 | 4 | 2 | 2 | 1 | 19 | 16 |
| Num contigs > 50 kb | 41 | 9 | 44 | 2 | 27 | 27 | 4 | 3 | 2 | 1 | 32 | 21 |
| Num contigs > 20 kb | 58 | 93 | 56 | 2 | 31 | 31 | 4 | 3 | 4 | 1 | 44 | 28 |
| Num contigs > 10 kb | 65 | 168 | 67 | 2 | 33 | 33 | 4 | 3 | 5 | 1 | 48 | 30 |
| Num contigs > 5 kb | 70 | 261 | 72 | 2 | 34 | 34 | 4 | 4 | 7 | 1 | 51 | 31 |
| Num contigs > 2.5 kb | 75 | 315 | 82 | 2 | 35 | 35 | 6 | 4 | 7 | 1 | 53 | 31 |
| Longest contig | 471.72 | 77.505 | 411.148 | 5,935,402 | 503.373 | 503.373 | 5,333,942 | 5,241,790 | 5,249,345 | 5,521,194 | 429.209 | 930.887 |
| Shortest contig | 207 | 1.5 | 200 | 106.439 | 4.773 | 4.773 | 1.308 | 8.92 | 6.515 | 5,521,194 | 1.501 | 5.812 |
| Num genes (prodigal) | 6.135 | 5.1 | 6.281 | 5.557 | 4.921 | 4.921 | 5.528 | 7.359 | 7.851 | 5.156 | 4.961 | 4.597 |
| L50 | 12 | 68 | 14 | 1 | 8 | 8 | 1 | 1 | 1 | 1 | 11 | 5 |
| L75 | 27 | 137 | 30 | 1 | 15 | 15 | 1 | 1 | 1 | 1 | 21 | 12 |
| L90 | 43 | 217 | 45 | 1 | 23 | 23 | 1 | 1 | 1 | 1 | 33 | 18 |
| N50 | 155,285 | 25,84 | 137,614 | 5,935,402 | 236,863 | 236,863 | 5,333,942 | 5,241,790 | 5,249,345 | 5,521,194 | 176,312 | 318,064 |
| N75 | 81,609 | 12,841 | 79,853 | 5,935,402 | 124,429 | 124,429 | 5,333,942 | 5,241,790 | 5,249,345 | 5,521,194 | 94,536 | 160,257 |
| N90 | 41,102 | 7,329 | 41,679 | 5,935,402 | 84,444 | 84,444 | 5,333,942 | 5,241,790 | 5,249,345 | 5,521,194 | 44,415 | 86,723 |
| Transfer RNAs | 84 | 45 | 83 | 83 | 64 | 64 | 88 | 86 | 87 | 85 | 73 | 70 |
| Sequence Type | ST547 | ST547# | ST547 | ST817 | ST860# | ST860# | ST11 | ST5992# | ST5992# | ST2263 | – | *- |
| Capsule Type | KL116 | KL186 | KL116 | KL152 | KL41 | KL41 | KL103 | KL30 | KL30 | KL16 | KL166 | KL35 |
| Plasmids | AC125 | AB595 | AA023 | AC125 | AB595 |
| Size | 110000 bp | 8000 bp | 65000 bp | 110000 bp | 8000 bp |
| GC content | 48.97% | 54.98% | 52.53% | 48.97% | 54.97% |
| Replicon types |
|
|
|
|
|
|
| |||||
| Coding sequences (CDS) | 152 | 12 | 86 | 160 | 12 |
| tRNA | 2(75 bp) | 0 | 0 | 2 (75 bp) | 0 |
| Antibiotic resistance genes (ARGs) present | No ARGs |
| No ARGs | No ARGs |
|
|
|
| ||||
|
|
| ||||
|
|
| ||||
|
| |||||
| Total mobile genetic elements (MGEs) | 42 | 3 | 31 | 53 | 2 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Antimicrobial agents and applications · Salmonella and Campylobacter epidemiology
Introduction
1
The Klebsiella genus belongs to the Enterobacteriaceae family, comprising non-motile, Gram-negative, rod-shaped bacteria capable of thriving in aerobic and anaerobic environments (Bengoechea and Sa Pessoa, 2019). They are opportunistic pathogens with a global distribution (Alcántar-Curiel et al., 2018) and are commonly found in the microflora of animals and humans, playing a significant role in intestinal health (Holt et al., 2015). Currently, the Klebsiella genus includes many species, such as K. pneumoniae, K. oxytoca, K. ozaenae, K. rhinoscleromatis, K. terrigena, K. planticola, K. michiganensis, K. variicola, and K. ornithinolytica (Gómez et al., 2021; Palusiak et al., 2025).
Members of this genus typically produce two types of cell surface antigens: lipopolysaccharide (LPS) and capsular polysaccharide (CPS) (Choi et al., 2020). These antigens are utilised for serotyping: the CPS structure determines K-typing, while the O-specific polysaccharide (O-SP) structure of LPS determines O-typing (Podschun and Ullmann, 1998). Antibodies targeting the capsule protect against encapsulated strains (Salazar et al., 2023). Although 12 O-antigen serotypes were initially described, subsequent detailed chemical analyses revealed that four of these shared identical structures. Consequently, only eight distinct O-antigen serotypes are currently recognized (Slater et al., 2024).
Klebsiella spp. are known to cause various infections, including nosocomial bronchopneumonia, septicaemia, and urinary tract infections, many of which are resistant to multiple antibiotics (Idomir and Nicoara, 2023; Le et al., 2023). This poses a serious healthcare issue, as Klebsiella spp. can evade the immune system and present significant challenges in medical treatment due to rising antibiotic resistance and highly virulent strains (Ballén et al., 2021). They are becoming increasingly resistant to many antibiotics, most recently carbapenems (Mancuso et al., 2023). Several studies indicate that Klebsiella strains often show resistance to cephalosporins, levofloxacin, lincomycin, sulfamethoxazole, and ampicillin (Idomir and Nicoara, 2023; Tran et al., 2023). Importantly, Klebsiella isolates demonstrate susceptibility to colistin, carbapenems, gentamicin, and ceftiofur (Sivaramakrishnan et al., 2022). The World Health Organization (WHO) has classified Klebsiella pneumoniae as a critical pathogen owing to its high resistance to multiple drugs and the urgent need for new treatment options (WHO, 2022). Among Klebsiella isolates, resistance due to extended-spectrum β-lactamases (ESBL) is most prevalent, followed by ampicillin class C (ampC) and carbapenemase production (Garcia-Fierro et al., 2022). In addition, virulence factors such as fimbrial adhesin H (fimH), mannose-resistant Klebsiella-like adhesin D (mrkD), and enterobactin synthase component B (entB) are found in K. pneumoniae isolates, contributing to their pathogenicity and increasing the risk of mortality in infected individuals (Mirzaie and Ranjbar, 2021).
Identifying antibiotic resistance and virulence genes in Klebsiella spp. is crucial for understanding their pathogenicity and guiding therapeutic approaches. Studies have demonstrated varying antibiotic resistance patterns in K. pneumoniae strains, as well as the recurrent presence of carbapenem resistance genes such as β-Lactamase K. pneumoniae carbapenemase-2 (blaKPC-2) (Liu et al., 2023; Bonda et al., 2023; Li et al., 2023). The genetic diversity and evolutionary relationships of Klebsiella species isolated from animals and water sources can be elucidated through comparative genomics. Whole genome analysis of Klebsiella strains from these distinct environments can reveal genetic variants that highlight differences in antibiotic resistance, pathogenic potential, and adaptive processes. This method elucidates potential transmission pathways between aquatic habitats and animals, providing insight into the risks of zoonotic diseases and environmental contamination. Understanding these genetic variations aids in evaluating public health implications and developing targeted strategies for surveillance and managing Klebsiella infections in diverse contexts.
Klebsiella species are increasingly implicated in both hospital- and community-acquired infections, and their growing resistance to multiple antibiotics, especially in low-resource and under-surveilled environments, poses a critical threat to global health. Despite growing concerns about antimicrobial resistance (AMR), genomic surveillance of Klebsiella species in animals and the environment remains critically underexplored, especially in rural South African communities like Matlwang village, Potchefstroom, North West Province. This knowledge gap hampers our ability to trace the genetic drivers of resistance and virulence, particularly in areas where humans, livestock, and water sources interact closely. Without comprehensive whole-genome data, emerging resistant strains remain undetected, posing a silent threat to animals and public health. To address this, our study sought to uncover the genomic profiles of Klebsiella spp. circulating in these environments by characterizing their antibiotic resistance genes, virulence factors, and genetic diversity through whole genome sequencing. By filling these knowledge gaps, the study aims to inform targeted interventions, highlight the role of rural environments in AMR dissemination, and contribute to more inclusive and geographically representative AMR databases and strategies.
Materials and methods
2
Bacterial isolates and genomic DNA extraction of the Klebsiella spp.
2.1
The 9 Klebsiella spp. isolates, comprising 5 from K. michiganensis, 2 from K. pneumoniae, and 2 from K. variicola, were selected based on the available strains from the Microbiology Biobank at North-West University’s Potchefstroom Campus in South Africa. The original faecal samples were collected directly from the rectum of sheep. In contrast, water samples were collected from river streams using sterile bottles as part of the AMR surveillance research collected from a rural communal farm in Matlwang, Potchefstroom. The original specimens, comprising fresh faecal material from sheep and water samples, were aseptically obtained between February 2022 and April 2022 and transported to the laboratory under cold-chain conditions. Previously stored at -80°C in nutrient broth with 15% sterile glycerol, the isolates were revived by subculturing on Eosin Methylene Blue (EMB) agar and incubating at 37°C for 24 hours.
Identification of the isolates using the SensiTitre system
2.2
The SensiTitre system was used in this study to identify Gram-negative bacterial isolates. Bacterial colonies were first cultured on nutrient agar and incubated overnight at 37°C. Isolated colonies were then emulsified in sterile saline (0.85% NaCl) to achieve a turbidity equivalent to a 0.5 McFarland standard, which was confirmed using a densitometer. The prepared bacterial suspension was dispensed into the wells of a Gram-negative SensiTitre identification panel, containing specific biochemical substrates, following the manufacturer’s instructions (Thermo Fisher Scientific Inc., Waltham, MA, USA). The plates were sealed and incubated at 37°C for 18–24 hours under aerobic conditions. After incubation, the results were either visually read or interpreted using an automated reader. Biochemical reactions, such as colour changes and gas production, were recorded and compared against the SensiTitre database using the system’s software or manual reference tables for species identification.
Genomic extraction of the isolates
2.3
The genomic DNA was extracted from overnight culture broths of Klebsiella spp. isolates using a ZymoBIOMICS DNA extraction kit following the manufacturer’s instructions (Zymo Research Corp., Irvine, California, USA). The Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, California, USA) was used for genomic DNA quantification. Finally, the genomic DNA was stored at −20°C until further analysis.
Molecular identification of Klebsiella using the rpoB gene
2.4
This study subjected the isolates to PCR with primer sets specific to Klebsiella targeting the rpoB gene. The PCR assay was conducted with the forward primer 5’-CAA CGG TGT GGT TAC TGA CG-3’ and the reverse primer 5’-TCT ACG AAG TGG CCG TTT TC-3’ (Bivand et al., 2024), yielding a 108 bp amplicon band size. The amplification reaction was carried out in a total volume of 25 μL that consisted of 10 µM of both forward and reverse primers, 12.5 μL of a 2X DreamTaq Green PCR Master Mix [4 mM MgCl_2_, loading buffer and 0.4 mM each of dATP, dCTP, mM dGTP, mMdTTP] (Thermo Fisher Scientific Inc., Waltham, MA, USA) and 5 ng of template DNA and nuclease-free water. The PCR conditions consisted of initial denaturation at 95°C for 5 minutes and then 35 cycles of denaturation at 94°C for 30 seconds, annealing at 55°C for 30 seconds, extension at 72°C for 30 seconds and a final extension at 72°C for 7 minutes. PCR products were identified by electrophoresis on 1% agarose gel stained with ethidium bromide and visualised under ultraviolet on a ChemiDoc imaging system (Bio-Rad ChemiDocTM MP Imaging System, Hertfordshire, United Kingdom). K. pneumoniae ATCC Type strain was used as a positive control, while RNase nuclease-free PCR water was used as a negative control.
Antimicrobial susceptibility testing for Klebsiella isolates
2.5
Antibiotic testing was carried out using the Kirby-Bauer disc diffusion method on Müller-Hinton (MH) agar plates. Pure cultures of isolates from faecal samples from sheep and water from the river stream were utilised to assess antibiotic resistance patterns. Nine different antibiotics from five distinct classes, including ampicillin (AMP; 10 μg), kanamycin (K; 30 μg), nalidixic acid (NA; 30 μg), cephazolin (KZ; 30 μg), ertapenem (ETP; 10 μg), meropenem (MEM; 10 μg), doripenem (DOR; 10 μg), and imipenem (IPM; 10 μg), were tested for antibiotic resistance on Klebsiella isolates following the Clinical and Laboratory Standards Institute (CLSI) antimicrobial susceptibility testing standards (CLSI, 2023). The MH plates were incubated at 37°C for 24 hours. After incubation, the isolates were classified as Resistant (R), Intermediate (I), or Susceptible (S) based on the size of the zone of bacterial growth inhibition as stipulated by the CLSI guidelines. Multidrug resistance (MDR) was defined as resistance to three or more classes of antibiotics tested (Magiorakos et al., 2012).
Library preparation and sequencing
2.6
Nine Klebsiella species isolates from sheep faeces and river stream were whole genome sequenced for downstream genomic analysis. Genomic DNA from K. pneumoniae isolates (n = 2) underwent library preparation with the ligation sequencing gDNA-Native barcoding kit (SQK-NBD114.96) from Oxford Nanopore Technology (ONT). Initially, the DNA was repaired using the FFPE DNA repair mix, followed by end preparation with the NEBNext Ultra II End Repair/dA-tailing Module from New England Biolabs (Teffera and Biolabs, 2019). The end-prepped DNA was then used for native barcode ligation employing the NEB Blunt/TA Ligase master mix and the NEBNext Quick Ligation Module in accordance with the manufacturer’s instructions. Subsequently, the barcoded DNA libraries were purified using Ampure XP beads, and native sequencing adapters were ligated. The libraries were further purified and quantified using the Qubit high-sensitivity kit from Thermo Fisher Scientific before being loaded for sequencing on the MinION 9.4.1 flow cell using the MinION 1KB platform from Oxford Nanopore. Library preparation for K. pneumoniae (n = 2), K. variicola (n = 2), and K. michiganensis (n = 5) was performed using the MGIEasy FS DNA Prep Kit (BGI, China) according to the manufacturer’s protocol. The prepared libraries were sequenced on the BGI MGISEQ-2000 platform (BGI Shenzhen, China) using a paired-end 150 nt strategy.
QC and bacterial genome assembly
2.7
Guppy base caller version 5.0.17 was employed to convert raw data in fast5 format into base-called data in fastq format. All reads with a quality score (Q) below 7.5 were excluded from subsequent data analysis. The quality of the trimmed data was evaluated using NanoPlot version 1.18.1 (De Coster et al., 2018). Quality filtering was performed using FiltLong version 0.2.0 (https://github.com/rrwick/Filtlong). The filtered Oxford Nanopore Technology (ONT) reads of Klebsiella strains were assembled de novo with Flye version 2.3.3 (Kolmogorov et al., 2019) and subsequently polished using the Medaka (https://github.com/nanoporetech/medaka) consensus pipeline. The quality of the sequenced reads generated by the MGI platform was assessed using FastQC software v0.10.1 (Andrews et al., 2010). Trimmomatic version 0.39 (Bolger et al., 2014) was used to trim ambiguous reads and remove sequenced adapters. The paired-end trimmed reads were assembled de novo using the MEGAHIT version 1.2.9 pipeline (Li et al., 2015). CheckM version 1.2.4 (Parks et al., 2015) was utilised to identify potential contaminants within assembled genomes. QUAST version 2.3 (Gurevich et al., 2013) was used to evaluate the draft genome assemblies of the strain. Subsequently, the assembled contigs were annotated employing the NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) (Tatusova et al., 2016). Average Nucleotide Identity of the Klebsiella species was computed using skani v0.3.1 (Shaw and Yu, 2023) and visualized on R-studio v 4.5.1 using heatmap package. The taxonomic classification of bacterial strains was performed in silico using multilocus sequence typing (MLST) (Carattoli et al., 2014) and pathogenWatch (https://pathogen.watch/). Moreover, MLST from the Institut Pasteur (https://bigsdb.pasteur.fr/cgi-bin/bigsdb/bigsdb.pl?db=pubmlst_klebsiella_isolates&page=plugin&name=GenomeComparator) was used to type the sequenced genomes using the Klebsiella oxycota and K. pneumoniae complex databases.
Antibiotic resistance, virulence gene detection, and plasmid replicon determination
2.8
The ABRicate pipeline, together with AMRFinderplus v 4.0.23 (Feldgarden et al., 2021), was utilized to detect antibiotic resistance and virulent genes in the genomes of the nine Klebsiella spp. strains. Antibiotic resistance determinants were identified in the assembled genome using the ResFinder database, with minimum identity and coverage thresholds set at 70% each, respectively. The comprehensive antibiotic resistance database (CARD) was also consulted to identify antibiotic resistance genes. ABRicate v 1.0.0 was further utilised to identify efflux pump-coding genes and virulence factors in the sequenced genome, using the Virulence Factor Database (VFDB) with minimum identity and coverage thresholds of 70% each, respectively. Plasmid replicons were identified in the sequenced genomes using the plasmid finder database (Carattoli and Hasman, 2020) through ABRicate v.1.0.0. MobileOG-db v.2.0 (Brown et al., 2022) was also utilised to identify mobile genetic elements. The capsule types were identified using Kaptive (https://kaptive-web.erc.monash.edu/jobs), and sequence types were detected using Pathogenwatch (Pathogenwatch A Global Platform for Genomic Surveillance). Circos circular v0.63–10 data visualization software (Krzywinski et al., 2009) and chiplot (Ramírez et al., 2014) were used to visualise the profiles of antibiotic resistance genes (ARGS), virulence genes, and plasmid replicon types. Moreover, complete plasmids were generated using the Proksee server (https://proksee.ca). FastANI version 1.33 (Hernández-Salmerón and Moreno-Hagelsieb, 2022). and Clincker version 0.0.31 (Gilchrist and Chooi, 2021) were used to compare the plasmids of closely related replicons.
Pangenomics analysis
2.9
Pangenome analysis was conducted on the K. pneumoniae genomes determined using Roary v3.6.8 (Page et al., 2015) and Anvio-7.1 (Eren et al., 2015). Pangenome analysis was restricted to K. pneumoniae because it is the clinically dominant and most epidemiologically relevant species within the K. pneumoniae species complex (KpSC), particularly in relation to antibiotic resistance. A total of 84 downloaded K. pneumoniae genomes from 15 countries were retrieved and compared with the two sequenced K. pneumoniae strains T2 and T4 isolated from sheep faeces in South Africa (Supplementary Table 1). The selection was based on global genomes related to sequence types in this study as well asl including some of the South African genomes. The genomes were annotated using Prokka version 1.14 (Seemann, 2014) and Prodigal (Hyatt et al., 2010). For similarity searches between the coding domain sequences (CDSs) of assembled genomes Pairwise BLASTp and the Markov Cluster Algorithm (MCL) were utilized as described previously by Magome et al. (2024). The phylogenetic tree of the genomes was visualized using ITOL (Letunic and Bork, 2021) and FigTree v1.4.4 (Rambaut, 2014).
Whole genome data availability
2.10
The genome sequences of K. pneumoniae, K. variicola and K. michiganensis isolates used in this study have been deposited in the National Center for Biotechnology Information (NCBI) GenBank under accession numbers (JBIPSE000000000, JBIPSD000000000, JBBMKX000000000, JBBMLA000000000, JBBMKZ000000000, JBBMKY000000000, JBPPKQ000000000, JBPPKR000000000 and JBPPKS000000000).
Results
3
Identification and antibiotic resistance profiles of Klebsiella species
3.1
The SensiTitre system and PCR amplification of the rpoB gene identified these nine isolates as Klebsiella species. A total of two isolates from sheep faeces were confirmed as K. pneumoniae, two from the river stream as K. variicola and five from the river stream as K. michiganensis. These identifications were confirmed using PCR assays targeting the rpoB housekeeping gene, and whole genome sequencing.
Klebsiella isolates exhibiting resistance to three or more antibiotic classes were classified as multidrug resistant (MDR) (Table 1). All Klebsiella strains showed resistance to ampicillin, and high resistance rates were also observed to nalidixic acid (88.8%), kanamycin, and cephazolin (55.5%). Notably, no carbapenem resistance was detected in this study. All isolates exhibited MDR, harbouring resistance to three or more antibiotic classes (Motlhaping et al., 2025).
Genomic features and assembly metrics of Klebsiella species
3.2
A total of 12 Klebsiella isolates were sequenced and assembled, comprising Klebsiella michiganensis (n = 5), Klebsiella pneumoniae (n = 3), and Klebsiella variicola (n = 4) (Table 2). Three genomes, KMTHO011 (K. michiganensis), KPHS11286 (K. pneumoniae), and KvF2R9T (K. variicola), served as references based on their completeness and availability in RefSeq. The total genome sizes ranged from 4.91 Mb to 6.55 Mb. Among K. michiganensis strains, KM26 (6.55 Mb) and KM20 (6.45 Mb) exhibited the largest genome sizes, while KM6, KM7, and KM23 had smaller genomes (5.22-5.33 Mb) but were the most fragmented, with 344 contigs and an N50 of 25.8 kb. In contrast, the reference strain KMTHO011 was highly contiguous, consisting of only two contigs with an N50 of 5.93 Mb and L50 of 1, consistent with high assembly quality. The K. pneumoniae strains, particularly KPHS11286, KPT2, and KPT4, showed high assembly continuity, with ≤7 contigs and N50 values exceeding 5.2 Mb. KPT4 and KPT2 had the highest predicted gene counts (7,851 and 7,359, respectively), in line with their relatively large genome sizes (~5.43 Mb). K. variicola isolates displayed moderate to high assembly variability. The reference genome KvF2R9T was assembled into a single contig, with an N50 of 5.52 Mb and L50 of 1, indicating a complete genome. In contrast, KvMW3 and KvMW4 exhibited draft assemblies (72 and 31 contigs, respectively) and smaller genome sizes (5.29 and 4.91 Mb). GC content across all isolates ranged from 55.1% to 58.0%, with species-specific trends observed. K. michiganensis strains exhibited lower GC content (55.1-56.5%), while K. variicola strains had the highest GC values (up to 58.0%). The number of protein-coding genes predicted by Prodigal ranged from 4,597 (KvMW4) to 7,851 (KPT4). Transfer RNA (tRNA) counts varied from 45 (KM23) to 88 (KPHS11286), with reference genomes generally showing higher tRNA content, reflecting their improved assembly completeness.
MLST and capsule type of Klebsiella species
3.3
The K. michiganensis strains (KMW6 and KMW7) were identified as having the closest sequence type 860 (ST860), which best matches capsule KL41. K. michiganensis strain KM20 and KM26 are assigned sequence types that best match sequence type 547 (ST547), which best matches KL116, while strain KM23 has a closest ST of 547, with an unknown capsule type that best matches KL186. In addition, the K. michiganensis strain KMTHO011 used as a reference has an ST of 817 and which best matches KL152. Moreover, the K. variicola strain KvMW3 best matches capsule KL166, while strain KvMW4 best matches KL41. The reference K. variicola strain KvF2R9T was assigned sequence type 2263 (ST2263) and capsule type KL16. The two sequenced K. pneumoniae strains, KPT2 and KPT4, present with STs, that are closely related to ST5992, and both have the capsule type KL30. Their closest STs are K. pneumoniae strains DAFUVP011 and DAJMOO011, both classified as ST5992 and isolated from humans in Switzerland and Finland, respectively. The reference strain KPHS11286 was assigned sequence type 11 (ST11), which best matches capsule KL103 (Table 2).
Average nucleotide identity of Klebsiella species
3.4
The average nucleotide identity (ANI) analysis in 12 Klebsiella genomes revealed clear genomic distinctions among the Klebsiella isolates (Figure 1). The K. michiganensis strains KM20 and KM26 showed extremely high ANI values (> 99.8%), indicating they are nearly identical and likely belong to the same strain or a closely related lineage. The K. michiganensis strain KM23 also clustered closely with strains KM20 and KM26, sharing over 99% ANI, forming a tight group that likely represents a clonal lineage. In contrast, K. michiganensis strain KMTHO011, KMW6, and KMW7 formed a separate cluster, showing ANI values ranging between 86-87% compared to the K. michiganensis group. Another distinct group consisted of K. pneumoniae strains KPHS11286, KPT2, and KPT4, which exhibited high within-group ANI values of 85%-100% but much lower similarity to the K. michiganensis groups, confirming their genomic distinctness. Similarly, K. variicola strain KvF2R9T grouped with strains KvMW3 and KvMW4, sharing moderate to high ANI values (80-100%) within the cluster, yet remaining distinct from the clinical isolates. These findings, visually supported by the heatmap in Figure 1, demonstrate at least four major genomic groups among the Klebsiella isolates, highlighting both species-level diversity and potential clonal relationships within Klebsiella populations.
Average nucleotide identity (ANI) of the sequenced Klebsiella species strains. Strains KMTHO011, KvF2R9T and KPHS11286 were used as references based on the completeness of the sequenced genomes available on RefSeq. KP represents K. pneumoniae, while Kv as K. variicola, and Km as K. michiganensis.
Identification of virulence genes in the Klebsiella species
3.5
The nine Klebsiella spp. strains were analysed for virulence genes (Figure 2). The K. pneumoniae strains KPT2 and KPT4 from sheep contained 30 virulence genes linked to adhesin and invasion (ompA), as well as enterobactin for iron absorption (entA, entB, entC, entD, entE, entF, entS, fepA, fepB, fepC and fepG), fimbrial adhesin (fimA, fimB, fimC, fimD, fimE, fimF, fimG, fimH, fimI, yagV/ecpE, yagW/ecpD, yagX/ecpC, yagY/ecpB, yagZ/ecpA and ykgK/ecpR), LPS core biosynthesis (gmhA/lpcA), Iron uptake (iutA), Lipid A synthesis (lpxC) and Quorum sensing via AI-2 (luxS). Strain KPT4 exhibited 10 additional flagellar virulence genes: fleS, fleR, fliE, fliF, fliG, fliH, fliI, fliJ, motA, and motB. The shared virulence genes between strains KPT2 and KPT4 encoded proteins for adhesin, invasion, enterobactin, iron absorption, and adhesion.
Virulence genes identified among Klebsiella species: Dark green colour= presence of the genes; Light green colour=absence of the gene.
Each of the K. variicola strains, KvMW3 and KvMW4, isolated from a river stream, harboured 32 virulence-associated genes. These included genes involved in iron acquisition (e.g., entA, entB, entC, entE, entF, entS, fepA, fepB, fepC, fepD, fepG), fimbrial assembly (fimA–fimI), outer membrane integrity (ompA), quorum sensing (luxS), and other functions such as lipopolysaccharide biosynthesis (gmhA/lpcA, lpxC) and siderophore uptake (iutA). Additional genes associated with the ecp operon (yagV/ecpE to ykgK/ecpR) were also consistently present.
In contrast, K. michiganensis strains KMW6 and KMW7 carried 36 virulence genes, encompassing all those found in KMW3 and KMW4, with the addition of genes such as gspE, gspG, htpB, katA, kdsA, mgtB, and mgtC, suggesting enhanced stress response and secretion capabilities. Strains KM20 and KM26 of K. michiganensis exhibited the most extensive virulence gene repertoire, with 42 genes, including those found in KMW6 and KMW7, as well as hcp-2, manB, pla, and vipB/mglB, indicating potential for increased pathogenicity and environmental adaptability. KM23 harboured 40 virulence genes, lacking only htpB and manB, compared to KM20 and KM26. Across all genomes analysed, 29 virulence genes were universally present (100%), including entA–entF, fepA–fepG, fimA–fimH, gmhA/lpcA, iutA, lpxC, luxS, ompA, and the ecp cluster (yagV to ykgK). These genes can be grouped into functional clusters such as iron acquisition, fimbrial assembly, membrane integrity, and secretion systems. Conversely, the least prevalent genes, fleR, fleS, fliE, fliF, fliG, fliH, fliI, fliJ, gmd, motA, and motB were detected only in strain K. pneumoniae KPT2, suggesting limited distribution or strain-specific roles.
Identification of antibiotic resistance genes in the Klebsiella species
3.6
Analysis of antibiotic resistance genes (ARGs) in the nine Klebsiella genomes (Figure 3) reveals that K. pneumoniae strains KPT2 and KPT4 harbour 10 ARGs. The core ARGs identified in both strains include tet(A)_6 and tet(34)_1, which encodes tetracycline resistance; sul2 (sulphonamide resistance); dfrA14_5 (trimethoprim resistance); aph(6)-id (aminoglycosides resistance); blaSHV-194 (β-lactamase production); fosA6 (fosfomycin resistance); oqxB and oqxA (quinolones resistance); and mdf(A)_1 (multidrug efflux pump).
Antibiotic resistance genes identified among Klebsiella species: Dark blue colour= presence of the genes; Light blue colour=absence of the gene; ARGs=Antibiotic resistance genes.
In contrast, the K. variicola strains from the river stream contained 6 ARGs, including oqxB, oqxA, fosA, mdf(A), tet34 and either blaLEN24 or blaLEN16. In addition, the K. michiganensis strains KM20 and KM26 exhibited 7 ARGs, which included aph(3’)-Ia_5, blaOXY-1-4_4, fosA, mdf(A)_1, oqxB, oqxA and tet(34). The K. michiganensis strain KM23 exhibited all genes present in strain KM20 and KM26 except for the aph(3’)-Ia gene. Furthermore, the K. michiganensis strains KMW6 and KMW7 harboured 7 ARGs. The core ARGs identified in both K. michiganensis strain KMW6 and KMW7 include aph(3’)-Ia_1, blaOXY-1-3, fosA5, mdf(A), oqxA, oqxB and tet(34). Among the examined sequenced genomes, the most prevalent ARGs identified were mdf(A)_1 and tet(34)_1, present in 100% (n = 12) of the strains, including the reference strains used in this study. The second most common ARGs were detected in 92% (n = 11) of strains, including oqxA and oqxB. The least ARGs identified included aac(3)-IId_1, aadA2_1, aph(3’’)-Ib_5, aph(3’)-Ia_10, blaCTX-M-14_1, blaCTX-M-8_1, blaKPC-2_1, blaLEN16_1, blaLEN17_1, blaLEN24_1, blaOXY-1-2_2, blaSHV-182_1, blaTEM-1B_1, dfrA12_8, floR_1, rmtB_1 and tet(G)_2, which were present in 8% (n = 1) of the genomes examined.
Plasmid replicon types across 12 Klebsiella species strains
3.7
The plasmid replicon types were analysed across 12 genomes of Klebsiella species, including the nine sequenced strains compared with the reference strains used for ANI (Figure 1). A total of 11 plasmid replicon types were identified across all the Klebsiella spp. genomes (Figure 4). In both K. pneumoniae strains KPT2 and KPT4, two plasmid types were detected: ColRNAI_1 and IncFIB(pKPHS1_1_pKPHS1), with strain KPT2 also harbouring the additional replicon type IncFIB(pQil_1_pQil). In K. michiganensis strain KM20 and KM26, two plasmid replicon types were detected, which included IncFIB(pKPHS1_1_pKPHS1 and IncFII(Yp)_1. No replicon types were detected in K. variicola strains KvMW3 and MW4, and in K. michiganensis strains KMW6, KMW7, and KM23. Among the genomes examined, the most prevalent plasmid type identified was IncFIB (pKPHS1_1_pKPHS1), present in 41% (n = 5) of the strains, including K. michiganensis strains KM20 and KM26, as well as K. pneumoniae strains KPT2, KPT4 and KPHS11286. The second most common plasmid type ColRNAI_1 was detected in 25% (n = 3) strains, exclusive to K. pneumonaie strains KPT2, KPT4 and KPHS11286.
Circos plots of the plasmid replicon types in Klebsiella species genomes. Kp, K. pneumoniae and Km, K. michiganensis.
Plasmid genetic features of K. pneumoniae strains KPT2 and KPT4
3.8
Three plasmids (AC125, AA023, and AB595) were identified in K. pneumoniae strain KPT2, and two plasmids (AC125 and AB595) in K. pneumoniae strain KPT4 (Table 3). No plasmids were detected in other Klebsiella species. The 110 kbp plasmid AC125 found in both strains comprises ColRNAI and IncFIB(pQil) replicon types, with a GC content of 48.97%. This plasmid contains 152 coding sequences (CDS) and two transfer RNAs (tRNA 75 bp) along with a significant number of mobile genetic elements (MGEs). The annotated MGEs include 42 genes, which consist of (n = 1) integration/excision, (n = 10) replicon/recombination/repair, (n = 22) phages, (n = 7) stability/transfer/defence, and (n = 2) transfer in strain KPT2 (Supplementary Figure 1A). The plasmid lacks antibiotic resistance genes. Similarly, many MGEs were identified in plasmid AC125 of K. pneumoniae strain KPT4, which also lacks ARGs (Supplementary Figure 1B). The plasmid exhibits 98.3% identity with K. pneumoniae strain CRKP-35’s unnamed plasmid (accession CP107355.1), according to ANI. The carbapenem resistance gene (blaKPC-2) is present in strain CRkp-35’s unnamed plasmid, whereas this gene is absent from the plasmids of strains KPT2 and KPT4 (Supplementary Figure 1C).
The plasmid AB595, identified in these two strains, is 98% similar and consists of the replicon-type IncFIB with a GC content of 54.98% (Table 3). This plasmid contains 12 coding sequences (CDS). The plasmid AB595 in strain KPT2 has three mobile genetic elements annotated as integration/excision genes (tnpA) and exc1 transfer genes (Supplementary Figure 2A). It carries aminoglycoside aph(6)-Id, trimethoprim dfrA14, tetracycline tetR and tetA, and sulphonamide sul2 resistance genes (Supplementary Figure 2A). Plasmid AB595 in K. pneumoniae strain KPT4 contains 12 coding sequences (CDS), with two mobile genetic elements identified (Supplementary Figure 2B). This plasmid harbours ARGs, including aph(6)-Id, sul2, and three copies of tetA. The gene orientation in this plasmid places dfrA14 and aph(6)-Id in close proximity. Based on ANI, the closest related plasmid AB595 is Shigella sonnei strain 202108535-plasmid_p202108535-7 (accession OP038299:1-8375), which consists of ARGs such as sul2, dfrA14, and aph(6)-Id. However, the dfrA14 gene’s percentage identity and positioning differ within the plasmid AB595 cluster of the two sequenced genomes in this study (Supplementary Figure 2C). Other hypothetical protein genes are present before the aph(6)-Id gene in strain KPT2. The aph(6)-Id is more conserved and identical (100%) among these compared plasmids.
A 65-kbp plasmid AA023 was identified exclusively in the K. pneumoniae strain KPT2, which boasts a GC content of 52.53%. This plasmid carries an IncFIB replicon with no ARGs detected. It comprises 86 coding sequences (CDS) and 31 MGEs, including (n = 11) for integration/excision, (n = 5) for replication/recombination/repair, (n = 14) for stability/transfer/defence, and (n = 1) for transfer (Supplementary Figure 3). This plasmid shows an ANI greater than 95.5%, identical to plasmid AA022 (CP133155.1) from K. variicola strain T2. Plasmid AA022 was isolated from wastewater treatment plant B in the North West Province of South Africa.
Pangenomes and average nucleotides identity of K. pneumoniae genomes
3.9
The pangenome of the 86 K*. pneumoniae* genomes, including the two sequenced strains, (highlighted in blue in Figure 5), consist of 34–305 total gene clusters. The analysis of dispensable genomes was conducted using the Roary and Anvi’o tools. This collection includes genomes from 23 South African strains, 22 from other African countries, and the rest from non-African countries (Supplementary Table 1). Roary identified pangenome clusters shared across all strains, classifying 229 gene clusters as core genes present in all strains. About 2553 gene clusters were categorized as soft-core genes, found in 95-99% of strains, while 3819 clusters were classified as shell genes, which are present in some strains. The majority of the gene clusters, totalling 27,704, were identified as cloud genes, which are found in only a few strains. Across the 86 genomes, 34305 genes were observed using the Anvi’o tool. Several accessory genes were identified in the sequenced T4 strain, including fliE, fliF, fliG, fliH, fliI, fliJ, fleS and fleR, along urease alpha subunit and including hypothetical proteins. Average nucleotide identity (ANI) analysis showed that the South African strains T2 and T4 had 98% genetic similarity, as they were isolated from two different sheep from the same farm. These strains clustered with K. pneumoniae strains DAFUVP011 and DAJMOO011, isolated from humans in Switzerland and Finland, respectively, which also share an ANI of 98%.
Global pangenome of the K. pneumoniae strains (n = 86), including the two sequenced strains highlighted in blue in this study visualized using ANVI’O. Based on core genes, the layers depict individual genomes arranged according to their phylogenomic relationships. The pangenome was computed using 34305 gene clusters found across the genomes. Dendrogram clustering of samples is ordered by gene cluster presence/absence. Items order: presence absence (D, Euclidean; L, Ward).
Phylogenetic analysis based on pangenomics analysis revealed that the South African strains (Tembi40 and Tembi39) shared the KL25 capsule type, while strains from Egypt (EGKP5, EGKP6, EGKP7, and EGKP4), Ghana (Iso00199) and China (K35), grouped together, designated as ST-17 with KL25 capsule type (Figure 6). Multilocus sequence typing (MLST) analysis of the 86 K*. pneumoniae* genomes revealed 45 distinct sequence types (STs). The most prevalent sequence type was ST11 (n = 16), commonly found in Sudan, France, China and Ghana. This was followed by ST307 (n = 8) from South Africa, France and Germany, and ST152 (n = 7) from South Africa and China. A total of 35 capsule types were identified, while 10 capsule types were classified as unknown (Figure 6). The most common capsule type, KL64 (n = 11), was predominantly found in water-derived isolates from China and was associated with ST11. Other prevalent capsule types included KL25 (n = 9), followed by KL102 (n = 8), KL149 (n = 7), and KL103 (n = 7), observed in countries such as Germany, France, Botswana, Sudan, South Africa, Egypt, Ghana, and China. Several South African genomes, isolated from human and animal strains (Tembi15, Tembi38, Tembi19 and Tembi21), were assigned the KL102 capsule type, which is associated with ST307. Other South African strains (Tembi27, Tembi31, Tembi7, Tembi8, Tembi14, and Tembi29) exhibited the KL149 capsule type, belonging to ST152. ST307 was also found in other countries, such as Germany (strain IMT44613) and France (strain CNR146C9). The two sequenced South African strains, T2 and T4, presented capsule type that best matched KL130 (Figure 6).
Classification of MLST sequence and capsule types identified in the 86 compared K. pneumoniae strains including the two sequenced strains in this study. The phylogenetic tree is based on maximum likelihood computed from pangenome analysis. The inner circular ring represents the country, followed by host, capsule types and sequence types. * Indicated closest capsule types.
Discussion
4
This study identified and characterized nine Klebsiella isolates recovered from sheep faeces and river stream samples in a communal village in the North West Province of South Africa. Through integrated genomic and phenotypic analyses, the study highlights the role of rural agricultural environments as reservoirs and potential amplifiers of antimicrobial resistance (AMR) and virulence determinants. The isolates were taxonomically classified as Klebsiella pneumoniae, K. variicola, and K. michiganensis, reflecting both species-level diversity and evidence of clonal expansion within the local ecosystem. The detection of K. pneumoniae in sheep faeces and K. variicola and K. michiganensis in adjacent river streams underscores the close interface between livestock and environmental compartments in rural settings. Such environments, where untreated water is commonly used for livestock, human consumption, and irrigation, facilitate microbial exchange and may promote the dissemination of pathogenic and resistant strains. Notably, the identification of clonally related K. michiganensis strains (KM20, KM23, and KM26), sharing >99% average nucleotide identity (ANI), across distinct water samples suggests environmental persistence and potential for long-term survival and transmission. These findings emphasize Klebsiella species’ ecological adaptability and zoonotic potential in rural agricultural landscapes. This study significantly contributes to understanding Klebsiella species in small ruminant-associated environments in South Africa by applying whole-genome sequencing (WGS). The study offers a comprehensive genomic and phenotypic characterization encompassing the resistome, virulome, plasmid content, and phylogenetic relationships by examining isolates from sheep faeces and adjacent river streams.
The use average nucleotide identity (ANI) analysis of Klebsiella genomes revealed clear genomic groupings reflecting species-level differences and possible clonal relationships. K. michiganensis strains KM20 and KM26 were nearly identical, sharing over 99.8% ANI, strongly suggesting they are part of the same clonal lineage. The K. michiganensis strain KM23 also grouped closely with these two strains, showing more than 99% similarity, which points to a shared evolutionary background (Hernández-Salmerón and Moreno-Hagelsieb, 2022). On the other hand, K. michiganensis strains KMTHO011, KMW6, and KMW7 showed much lower similarity (86-87% ANI) when compared to strain KM20 and KM26, indicating notable genomic divergence. This is consistent with earlier studies reporting high genomic plasticity and the presence of multiple K. michiganensis lineages in both environmental and clinical contexts (Simoni et al., 2023; Yamada et al., 2024). The K. pneumoniae strains KPHS11286, KPT2, and KPT4 formed a distinct genomic cluster, sharing high similarity (85-100%ANI), yet clearly separating from the K. michiganensis group, confirming species-level differentiation (Liu et al., 2012; Lekota et al., 2024). They highlight the value of ANI-based genomic surveillance for distinguishing between Klebsiella species and tracking clonal lineages, particularly in complex environments where different strains may circulate silently.
Strains T2 and T4 were identified as non-hypervirulent K. pneumoniae strains, though they carried virulence genes related to invasion, enterobactin, capsule formation, iron-absorption and adhesive, which are key factors for pathogenesis in “One Health” settings (Monteiro et al., 2024). The two strains, T2 and T4, had >98% ANI, indicating they were isolated from two different sheep on the same farm. Based on phylogenomic placement, they group closely with isolates DAFUVP011 and DAJMOO011, isolated from humans in Switzerland and Finland, respectively. All four strains mentioned above were defined with ST5992. This study reports the first sequence type identified in livestock, and it has not been reported in South Africa in other hosts. It is therefore evident that this sequence can also be found in humans, as demonstrated by comparative genomics in this study.
Pangenomic analysis of K. pneumoniae strain KPT4 revealed the presence of unique accessory genes, including hypothetical proteins and the urease alpha subunit, which are essential for survival in diverse environments (Duran Ramirez et al., 2022). Additionally, genes associated with flagellar motor assembly (fliE, fliF, fliG, fliH, fliI, and fliJ) and motility (motA and motB) were identified, demonstrating their roles in biofilm formation and survival under antibiotic stress (Sharma et al., 2019). Such environmental fitness genes improve the survival and persistence of these bacteria in shared water sources and animal habitats, heightening the potential for cross-species transmission and chronic contamination in “One Health” contexts. This study showed that blaSHV-194, which confers β-lactamase resistance, was present in the two K. pneumoniae genomes studied and is commonly associated with ampicillin resistance in K. pneumoniae. This gene is underreported among South African K. pneumoniae strains, particularly among ESBL-producing strains. However, the gene was reported in 91.4% of K. pneumoniae strains from several hospitals in northern Iran (Farhadi et al., 2021).
Whole-genome sequencing of K. pneumoniae isolates revealed the presence of multiple antibiotic resistance genes (ARGs) located on plasmids, notably plasmid AB595. The co-localization of these genes on plasmid AB595 suggests its potential role as a multidrug resistance vector, particularly if it possesses conjugative capabilities (Khedkar et al., 2022). Mobile genetic elements (MGEs), often adjacent to ARGs, facilitate horizontal gene transfer and stable maintenance within bacterial populations (San Millan, 2018; Stephens et al., 2020). In this study, plasmid AB595 harboured such MGEs, supporting its role in resistance dissemination. Notably, aph(6)-Id and sul2 were found within the same plasmid or operon, indicating a genetic linkage that may enhance co-transfer and expression (Venkatesan et al., 2023). Comparative analysis revealed that AB595 shares structural similarity with plasmid p202108535–7 from Shigella 202108535 (Lefèvre et al., 2023) and plasmid pABC-3 (Miranda et al., 2016), suggesting interspecies plasmid exchange. The presence of such multidrug-resistant plasmids in K. pneumoniae from livestock and environmental water sources poses a significant public health risk, particularly in rural settings where direct contact, contaminated water, or food may facilitate transmission, an issue of concern within the “One Health” framework. The tet(A) gene, carried by AB595, encodes an efflux pump that actively expels tetracycline from bacterial cells, reducing intracellular antibiotic concentrations and promoting survival (Xu et al., 2021; Wang et al., 2024). This mechanism is especially concerning in rural areas where tetracyclines are widely used in human and veterinary medicine, increasing the likelihood of resistance selection. Furthermore, the dfrA14 gene, conferring resistance to trimethoprim, was identified in strains KPT2 and KPT4. Although detected in only 17% of Klebsiella strains in this study, dfrA14 is more prevalent in clinical isolates, often co-occurring with sul2 (Xu et al., 2021). Its global emergence following the introduction of trimethoprim into bacterial populations underscores the need for continued surveillance and molecular characterization of resistance determinants in environmental and animal reservoirs.
The primary plasmid replicon types belong to the diverse incompatibility (Inc) family, primarily affecting Enterobacteriales (Chen et al., 2024). Plasmid AC125 showed 98.3% identity to an unnamed plasmid in K. pneumoniae strain CRkP-35 (Hu et al., 2023), suggesting that AC125 may be a prophage plasmid rich in MGEs, particularly in prophage regions. Similar plasmids lacking ARGs have been reported, such as the P3 prophage plasmid found in K. pneumoniae strains from patients diagnosed with relapsed acute myeloid leukaemia (Yoshino et al., 2021). A relatively exclusive plasmid AA023 was identified in strain KPT2, exhibiting 95.55% identity to plasmid AA022 (CP133155.1) found in K. variicola strain KPT2, isolated from the North-West Province, South Africa. Both AA022 and AA023 appear to be related phage-like plasmids with minimal ARGs and encode many uncharacterised proteins. Both plasmids contain over 90% hypothetical proteins with unknown functions.
The antimicrobial resistance (AMR) gene profiles identified in Klebsiella species from water sources associated with small livestock farming highlight the environmental persistence and potential spread of multidrug-resistant Klebsiella strains. The presence of oqxAB efflux pumps is particularly concerning, as these genes are associated with reduced susceptibility to last-resort antibiotics such as tigecycline and fluoroquinolones, and have been frequently detected in environmental reservoirs, including swine manure, hospital wastewater, and aquatic systems (Wyres and Holt, 2018; Wang et al., 2019; Yang et al., 2024). In contrast, while blaLEN variants naturally occur in K. pneumoniae, their association with resistance to last-resort antibiotics is limited, and evidence of their prevalence in environmental reservoirs remains minimal (Dingiswayo et al., 2024). The K. michiganensis strains displayed an even broader resistance profile, with up to seven AMR genes. The commonly detected genes included aph(3’)-Ia, blaOXY-1, fosA, mdf(A), oqxAB, and tet(34), suggesting a conserved resistome across Klebsiella strains (Kang et al., 2020; Zhang et al., 2022). The K. michiganensis strain KM23 lacked only the aph(3’)-Ia gene. These findings align with earlier studies that identified K. michiganensis as an emerging multidrug-resistant opportunistic pathogen in clinical and environmental settings (Simoni et al., 2023; Long et al., 2024). The consistent detection of fosA, oqxAB, and mdf(A) genes in Klebsiella strains from livestock-associated water environments suggests persistent selective pressure likely driven by antibiotic runoff and manure contamination (Kang et al., 2020). These findings reinforce the role of such environments as reservoirs of transferable AMR genes and underscore the importance of genomic surveillance within a “One Health” framework (Zalewska et al., 2021). The virulence gene profiles of K. variicola and K. michiganensis strains isolated from a river near livestock farming areas reveal their strong environmental adaptability and potential to cause disease. K. variicola strains KvMW3 and KvMW4 carried 32 virulence genes, including iron-acquisition systems like ent and fep genes, fimbrial adhesion genes (fimA-I), and key outer membrane components such as ompA and lpxC. These features are essential for helping bacteria secure nutrients, adhere to surfaces, and evade the host immune response (Paczosa and Mecsas, 2016; Holt et al., 2015).
The K. michiganensis strains KMW6 and KMW7 had more diverse virulence profiles, with 36 genes identified. These included stress response genes (katA and htpB), magnesium transporters (mgtB and mgtC), and components of the type II secretion system (gspE and gspG), suggesting that these strains are metabolically flexible and capable of surviving harsh environmental conditions (Martin and Bachman, 2018). The K. michiganensis strains KM20 and KM26 were even more enriched, carrying 42 virulence genes, including hcp-2, pla, and vipB/mglB, which are linked to bacterial competition and increased toxicity (Yamada et al., 2024). The K. michiganensis strain KM23 had a similar profile but lacked htpB and manB, suggesting some differences in stress tolerance and capsule production. Across all Klebsiella strains, 29 virulence genes were consistently present. Antibiotic resistance genes like ent, fep, fim, gmhA/lpcA, iutA, luxS, and ompA were dominant, supporting bacterial colonization, biofilm development, and survival in low-nutrient environments (Follador et al., 2016). On the other hand, motility-related genes (fle, fli motA and motB) were rarely detected, which aligns with the generally non-motile nature of Klebsiella species observed in both clinical and environmental isolates (Wyres et al., 2020; Podschun and Ullmann, 1998; Holt et al., 2015).
Combining these virulence traits with antimicrobial resistance genes reinforces the potential of these bacteria to act as zoonotic threats within livestock-associated water systems (Zalewska et al., 2021). In these sequenced strains, virulence gene profiles revealed notable similarities between K. variicola and K. michiganensis. Several adhesive genes (yagV/ecpE, yagW/ecpD, yagX/ecpC, yagY/ecpB, and yagK/ecpR) and the iron-absorption gene (fepC) were present in both K. variicola and K. michiganensis. These adhesive genes are critical for colonization and infection, as they assist bacteria in adhering to host tissues and surfaces (Martin and Bachman, 2018). The iron-absorption genes, essential for infection in iron-limited environments, were absent in K. michiganensis strains (Chen et al., 2020; Kumar et al., 2024). The mgtB gene, which encodes a magnesium transporter and supports bacterial survival in low-magnesium environments and stress responses, was found only in K. michiganensis (Liu et al., 2024).
Comparative genomics demonstrate that the blaLEN gene is prevalent in 75.3% of K. variicola strains (Lekota et al., 2024; Yang et al., 2024). In contrast, the blaOX-1-3 β-lactamase gene was discovered in K. michiganensis strains, marking the first report of blaOX-1–3 in K. michiganensis from a water stream in South Africa. However, the blaOX-181 carbapenemase-producing K. michiganensis isolate from hospital effluent in South Africa has been previously reported (King et al., 2021). The aph(3’)-Ia_1 gene that confers resistance to streptomycin, which is one of the aminoglycoside resistances, was found exclusively in the K. michiganensis strain in this study and had been previously identified in South African hospital effluent (King et al., 2021). Both K. pneumoniae, K. variicola and K. michiganensis sequenced genomes contained the nalidixic acid resistance gene oqxAB, which encodes a multidrug efflux pump. This gene is found in the clinical groups of Enterobacteriaceae and plays a vital role in the resistance of bacteria to antibiotics and antimicrobial agents, including detergents and disinfectants (Li et al., 2019). The presence of these genes highlights the role of environmental water sources as reservoirs of antibiotic-resistant bacteria (Gomi and Adachi, 2025). These findings raise public health concerns and underscore the importance of genetic antibiotic testing, particularly for K. michiganensis and K. variicola, which are underreported and understudied from a “One Health” perspective in South Africa (Simoni et al., 2023).
Plasmid replicon analysis of Klebsiella genomes showed a diverse distribution of plasmid types, highlighting their critical role in spreading antimicrobial resistance. These IncF-replicon types are well recognized for carrying key resistance genes, such as blaKPC and blaNDM, and for promoting horizontal gene transfer in clinical and environmental settings (Carattoli, 2009; Bi et al., 2018). In K. michiganensis strains KM20 and KM26, the presence of IncFIB(pKPHS1_1_pKPHS1) and IncFII(Yp)_1 replicons suggest they share plasmid structures commonly associated with multidrug-resistant Klebsiella strains (Zhang et al., 2023). Interestingly, no identifiable replicon types were detected in K. variicola and K. michiganensis strains KMW6, KMW7, and KM23. A key limitation of this study is that several genomes particularly K. variicola isolates were untypeable using standard multilocus sequence typing (MLST) schemes due to missing loci in their assemblies. This incomplete representation of allele profiles may have reduced the accuracy of sequence-type assignment and potentially limited comparative genomic analyses.
Limitations
4.1
A key limitation of this study is that sequence types could not be assigned to some genomes due to missing MLST alleles, particularly among K. variicola strains. This incompleteness may reflect limitations inherent to short-read sequencing and draft genome assemblies, where fragmented contigs can lead to missing allele recovery. Alternatively, it is also possible that certain Klebsiella genomes genuinely lack these alleles due to genomic variation within the species. Consequently, this may have affected the resolution of population structure and comparative analysis. Future studies may incorporate long-read sequencing technologies to generate more complete assemblies, which could help clarify whether the missing alleles are due to technical constraints or represent true biological absence in these strains.
Conclusion
5
This study offers critical insights into the genetic and antimicrobial resistance profiles of Klebsiella species isolated from animal and environmental sources within a communal farming setting in the North West Province, South Africa. Detecting multidrug-resistant K. pneumoniae, K. variicola, and K. michiganensis strains, some of which carry antimicrobial resistance genes, underscores the complex and dynamic nature of bacterial populations at the human-animal-environment interface. High-risk clones such as mobile genetic elements like the AB595 plasmid in faecal isolates suggest potential zoonotic and environmental transmission pathways in shared-resource communities. These findings underscore the urgent need for integrated “One Health” approaches in rural South African communities to monitor, prevent, and mitigate the emergence and spread of antibiotic-resistant and potentially pathogenic Klebsiella species. Public health interventions must include regular environmental and veterinary microbiological assessments, improved sanitation infrastructure, and stewardship programmes o target antimicrobial use in livestock and protect both animal and human health in communal agricultural ecosystems.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alcántar-Curiel M. D. Ledezma-Escalante C. A. Jarillo-Quijada M. D. Gayosso-Vázquez C. Morfín-Otero R. Rodríguez-Noriega E. . (2018). Association of antibiotic resistance, cell adherence, and biofilm production with the endemicity of nosocomial Klebsiella pneumoniae. Bio Med. Res. Int. 2018, 7012958. doi: 10.1155/2018/7012958, PMID: 30345305 PMC 6174813 · doi ↗ · pubmed ↗
- 2Andrews S. Krueger F. Segonds-Pichon A. Biggins L. Krueger C. Wingett S. (2010). Fast QC: A quality control tool for high throughput sequence data [Software]. (Cambridge, UK: Babraham Bioinformatics, Babraham Institute. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 3Ballén V. Gabasa Y. Ratia C. Ortega R. Tejero M. Soto S. (2021). Antibiotic resistance and virulence profiles of Klebsiella pneumoniae strains isolated from different clinical sources. Front. Cell. Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.738223, PMID: 34540722 PMC 8440954 · doi ↗ · pubmed ↗
- 4Bengoechea J. A. Sa Pessoa J. (2019). Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol. Rev. 43, 123–144. doi: 10.1093/femsre/fuy 043, PMID: 30452654 PMC 6435446 · doi ↗ · pubmed ↗
- 5Bi D. Zheng J. Li J. J. Sheng Z. K. Zhu X. Ou H. Y. . (2018). In silico typing and comparative genomic analysis of Inc FIIK plasmids and insights into the evolution of replicons, plasmid backbones, and resistance determinant profiles. Antimicrob. Agents Chemother. 62, 10–128. doi: 10.1128/AAC.00764-18, PMID: 30012771 PMC 6153814 · doi ↗ · pubmed ↗
- 6Bivand J. M. Dyrhovden R. Sivertsen A. Tellevik M. G. Patel R. KommedalØ. (2024). Broad-range amplification and sequencing of the rpo B gene: a novel assay for bacterial identification in clinical microbiology. J. Clin. Microbiol. 62, e 00266–e 00224. doi: 10.1128/jcm.00266-24, PMID: 38884485 PMC 11324016 · doi ↗ · pubmed ↗
- 7Bolger A. M. Lohse M. Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, pp.2114–2120. doi: 10.1093/bioinformatics/btu 170, PMID: 24695404 PMC 4103590 · doi ↗ · pubmed ↗
- 8Bonda N. A. Stoma I.О. Osipkina O. V. Ziatskov A. A. Shaforost A. S. Karpova E. V. . (2023). Molecular genetic markers of resistance and virulence of invasive Klebsiella pneumoniae strains according to whole genome sequencing data. Health Ecol. Issues. 20, 7–15. doi: 10.51523/2708-6011.2023-20-1-01 · doi ↗