Aging modulation of the immune system and immunotherapy efficacy in cancer
Yurong Wang, Mengjie Liang, Yichen Mao, Wei Zhu, Xiaofei Shen, Wenxian Guan

TL;DR
This review explores how aging affects the immune system and cancer immunotherapy outcomes in older adults.
Contribution
The paper systematically summarizes immunotherapy data in elderly patients and proposes sensitization strategies.
Findings
Immunosenescence contributes to an immunosuppressive tumor microenvironment.
Aging-related immune changes reduce immunotherapy efficacy in older adults.
Senescence-modulating drugs may improve immunotherapy response rates.
Abstract
Immunosenescence is characterized by immune decline and chronic inflammation. With advancing age, the incidence of tumors increases significantly. Understanding how immunosenescence influences the initiation and progression of tumors, as well as its implications for tumor immunotherapy, has become a matter of urgent importance. This review begins with an analysis of the phenotypic changes and underlying mechanisms associated with immune system and immune cell aging, and further explores the interplay between immunosenescence and tumorigenesis. Evidence indicates that cytokines, cell interactions and other mediators serve as critical links connecting aging and cancer, exerting complex anti-tumor and pro-tumor effects. However, in the context of immunosenescence, these factors collectively contribute to the formation of an immunosuppressive tumor microenvironment (TME) that facilitates…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
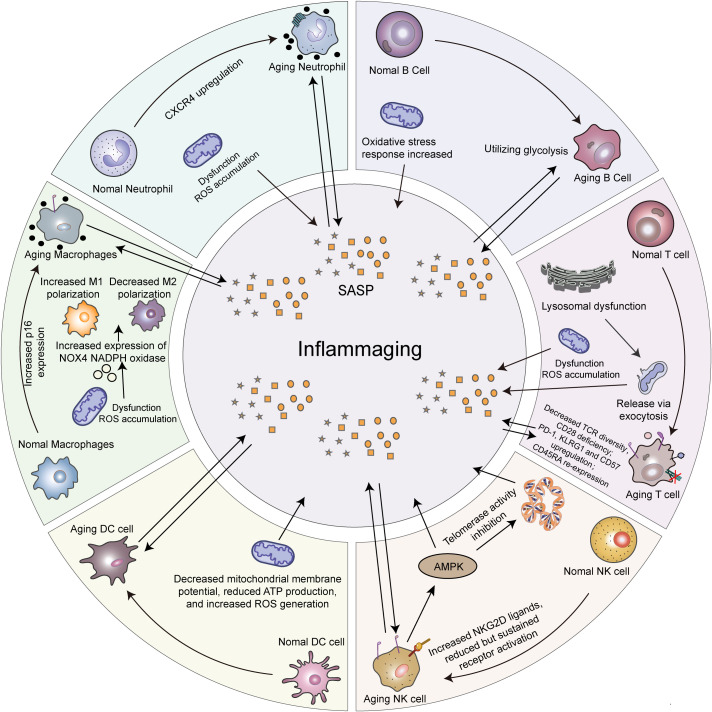 Figure 1
Figure 1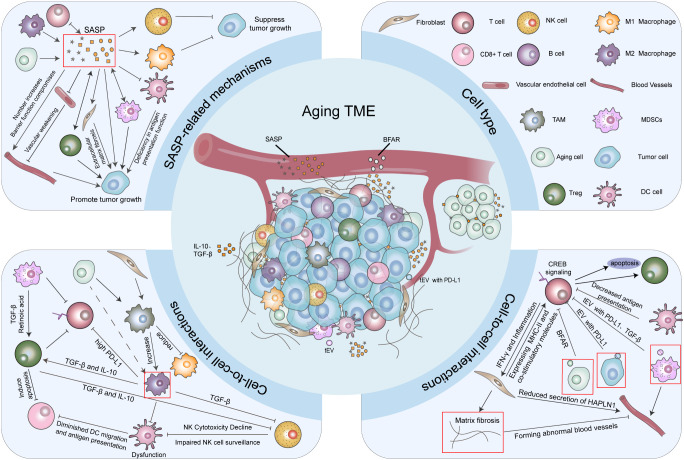 Figure 2
Figure 2| Tumor type | Year | Inclusion type | Population | Age | Medication | Conclusion | Ref |
|---|---|---|---|---|---|---|---|
| NSCLC* | 2022 | IIIB/C or IV | 1208 | <75 vs. ≥75 | ICI monotherapy; | ICI monotherapy improves survival, especially in younger patients. | ( |
| 2018 | / | 245 | <60 vs. 60–69 vs. | ICI monotherapy | similar OS and better PFS in age <80; shortest OS and PFS in age ≥80 | ( | |
| 2022 | IIIB/C or IV | 53719 | <55 | ICI monotherapy vs. ICI combined with chemotherapy | older patients have lower OS and 2-year survival rate in both group. | ( | |
| 2016 | PD-L1 positive (≥1%). | 1034 | Med*:63 | Pembrolizumab (10 or 2 mg/kg) vs. Docetaxel | Pembrolizumab provided superior OS regardless of age and better tolerance in elderly patients | ( | |
| 2022 | IIIB/C, IV or recurrence; PD-L1 TPS* ≥50%. | 26 | 75-90(Med:78) | Pembrolizumab 200mg q3w.* | certain tolerability. | ( | |
| 2019 | Locally advanced or metastatic;PD-L1 TPS* ≥1%. | 1274 | <65 vs.≥65 | Pembrolizumab 200mg q3w. vs. Platinum-based chemotherapy | Better survival and lower incidence of adverse events in pembrolizumab group, regardless of age | ( | |
| 2022 | Locally advanced or metastatic, III(unresectable)/IV NSCLC* | 466 | <65 vs. ≥65 | Cemiplimab or Placebo with Chemotherapy | Better OS and PFS of all patients; good efficacy and tolerance in elderly patients. | ( | |
| 2017 | Locally advanced or metastatic (stage IIIB or IV). | 1225(ITT:850) | <65 vs. ≥65 | Atezolizumab vs. Docetaxel | ICI has a better OS, especially in older patients (≥65) | ( | |
| 2021 | Chemotherapy-naive metastatic (stage IV) | 1202 | <65 vs 65-74vs. 75-84vs. ≥85 | ABCP vs. BCP and ACP vs. BCP | ABCP showed a better survival, particularly in patients under 65 | ( | |
| 2019 | / | 2612 | <75 vs. ≥75 | Pembrolizumab vs. chemotherapy | Pembrolizumab has a better OS and safety in in elderly patients(≥75). | ( | |
| SCLC | 2023 | stage III/IV, without radical radiotherapy or postoperative recurrence | 65 | <70 vs. ≥70 | Atezolizumab + carboplatin + etoposide | Excellent efficacy and acceptable toxicity in elderly patients | ( |
| Upper gastrointestinal tract cancer | 2022 | HER2(-), unresectable advanced or recurrent GC or gastric-esophageal junction cancer | 724 | / | Nivolumab or Placebo + chemotherapy | Certain therapeutic advantages in patients of different ages. | ( |
| 2021 | untreated, inoperable, HER(-) GC, gastroesophageal junction cancer or esophageal adenocarcinoma | 1581 | <65 vs. ≥65 | Nivolumab + Chemotherapy vs. Chemotherapy alone | Combination therapy had better survival and PFS, with acceptable safety, regardless of the age | ( | |
| 2017 | Advanced or recurrent gastric adenocarcinoma or adenocarcinoma of the gastroesophageal junction | 493 | <65 vs. ≥65 | Nivolumab vs Placebo | Nivolumab has shown survival benefits in patients of all age groups | ( | |
| Breast cancer | 2020 | untreated, non-metastatic TNBC | 1174 | <65 vs. ≥65 | Pembrolizumab or Placebo + chemotherapy | Better pCR*, more significant in elder group. | ( |
| Liver cancer | 2022 | Local advanced, metastatic or unresectable | 501 | <65 vs 65–74 vs ≥75 | Atezolizumab +Bevacizumab vs. Sorafenib | Combination therapy has better efficacy and acceptable safety, even in elderly patients. | ( |
| Kidney cancer | 2019 | Newly diagnosed or recurrent stage IV ccCRCC* | 861 | <65 vs. ≥65 | Pembrolizumab +Axitinib vs Sunitinib | Lower risk of death and better PFS in combination therapy, regardless of age. | ( |
| Melanoma | 2018 | advanced or metastatic | 99 | ≥75 | Pembrolizuma vs. Nivolumab vs. Ipilimumab | Efficacious and well-tolerated in elderly patients of ICIs, particularly PFS outcomes associated with pembrolizumab. | ( |
| 2018 | / | 538 | <62 vs. ≥62 | Pembrolizumab;Some patients have previously received treatment with MAPKi | Better response to PD-1 inhibitors in age ≥62 | ( | |
| 2016 | metastatic | 3 | ≥90 | Ipilimumab vs. Nivolumab vs. Pembrolizumab | Older patients can tolerate and benefit from ICIs. | ( | |
| CRC | 2022 | MSI-H or dMMR in mCRC | 307 | Med:63 | Pembrolizumab monotherapy vs. Chemotherapy | Better PFS, better objective response rate, and better safety than chemotherapy in patients with MSI-H/dMMR mCRC in all age. | ( |
| 2023 | dMMR- mCRC | 41 | <65 vs ≥65 | Pembrolizumab monotherapy | Good efficacy and safety in older patients. | ( | |
| Prostate cancer | 2025 | mCRPC* with disease progression after receiving second-generation ARPI treatment | 1030 | <65 vs ≥65 | Pembrolizumab +Docetaxel vs. Placebo+ Docetaxel | No significantly better efficacy than placebo group. No difference among different age groups. | ( |
| Drug/Agent | Tumor Type/Indication | Mechanism of Action & Target | Impact on Immune Cells | Impact on ICIs* & Clinical Stage | Ref |
|---|---|---|---|---|---|
| Regulators targeting the modulation of SASP | |||||
| Metformin | HNSCC* | Inhibits secretion of SASP factors (IL-6, IL-8, MCP-1, GRO) via the AMPK/mTOR pathway; delays tumor progression. | Enhances antitumor efficacy of CDK4/6 inhibitors; modulates the TME immune landscape. | Preclinical studies. | ( |
| Rapamycin | Wilms’ Tumor, Melanoma | Inhibits mTOR pathway, reducing IL-6, IL-8, TNF-α, and IL-1α secretion; autophagy activator; combined with TLR4/9 agonists. | Restores hematopoietic stem cell regenerative capacity; enhances influenza vaccine efficacy; reverses immune senescence; improves PD-1+ T cell function; activates autophagy to potentiate antitumor effects via immune modulation. | Enhances antitumor effects when combined with TLR4/9 agonists. Preclinical studies. | ( |
| Resveratrol | Colorectal Cancer, Lung Cancer | Inhibits MAPK pathway; reduces IL-6, IL-1β, and TNF-α secretion. | Inhibits SASP-associated protumorigenic effects in MRC5 fibroblasts. | Preclinical studies. | ( |
| Echinacea Extract | NSCLC* | Inhibits MAPK pathway; reduces IL-6, IL-8, and IL-1α secretion. | Inhibits formation of the senescence-associated secretory phenotype (SASP). | Preclinical studies. | ( |
| SB203580 | NSCLC* | Inhibits p38 MAPK; reduces secretion of IL-6, IL-8, IL-10, TGF-β, and TNF-α. | Inhibits SASP formation; improves the immune microenvironment. | Preclinical studies. | ( |
| BIRB796 | Glioblastoma | Inhibits p38 MAPK pathway; reduces IL-6 secretion. | Inhibits SASP formation. | Preclinical studies. | ( |
| SYQ-PA | Breast Cancer | Modulates NF-κB pathway via PPARγ inhibition; promotes macrophage polarization from M2 to M1 phenotype. | Enhances antitumor immunity; inhibits TNF-α secretion. | Preclinical studies. | ( |
| Biological agents targeting key inflammatory factors and receptors | |||||
| Siltuximab (CNTO328) | CRPC*, Multiple Myeloma, Solid Tumors, Prostate Cancer, Metastatic RCC* | Monoclonal antibody targeting IL-6. | Reduces CRP* levels; inhibits IL-6 signaling pathway; stabilizes disease in some patients. | Clinical Phase II ( | ( |
| Tocilizumab | Lung Cancer, TNBC*, Recurrent Epithelial Ovarian Cancer | Monoclonal antibody targeting IL-6 receptor; blocks the IL-6/STAT3/C/EBP-IL-6 feed-forward loop. | Reduces STAT3 activation and IL-6 production in TAMs. Safe and effective when combined with interferon-α2b chemotherapy. | Clinical application (Cachexia treatment); Preclinical studies (TNBC, Lung adenocarcinoma); Clinical Phase I (Ovarian cancer). | ( |
| IL-1R Antagonist | Pancreatic Cancer | Antagonizes IL-1 receptor; enhances chemotherapeutic efficacy. | Primarily acts by blocking IL-1 signaling (specific immune subsets not specified). | Preclinical studies. | ( |
| Infliximab | Colon Cancer | Monoclonal antibody targeting TNF-α; blocks TNF-α/TNFR2 signaling. Enhances chemotherapy efficacy. | Reduces CCR8+ regulatory T cells, decreasing TME immunosuppression. | Clinical studies; Clinical Phase I/II (well-tolerated); Preclinical studies show enhanced anti-PD-1 efficacy. | ( |
| Carlumab (CNTO 888) | Metastatic CRPC* | Monoclonal antibody targeting CCL2. | Transiently inhibits CCL2. | Clinical Phase I/II trials (did not show significant single-agent anticancer activity). | ( |
| Pattern recognition receptors and agonists/inhibitors of inflammasomes | |||||
| TLR3 Agonist (ARNAX) | TLR3 agonist; promotes interferon transcriptional induction; enhances immunotherapy efficacy. | Promotes dendritic cell maturation; enhances T cell activation and proliferation. | Promotes immunotherapy efficacy; Clinical studies. | ( | |
| TLR7/8 Agonist Nanoparticles | Activates TLR7/8; promotes polarization of tumor-associated macrophages (TAMs) toward an antitumor phenotype. | Promotes TAM polarization to M1 (antitumor) phenotype. | Enhances cancer immunotherapy efficacy; Preclinical studies. | ( | |
| STING Agonists (DMXAA, cGAMP, ADU-S100, etc.) | Pancreatic, Breast, Melanoma, and other cancers | Activates cGAS-STING pathway; induces Type I interferon production; promotes antitumor immunity. | Promotes dendritic cell maturation; enhances activation and infiltration of T cells and NK cells; reprograms macrophages. | Enhances antitumor effects when combined with radiotherapy, chemotherapy, CAR-T*, or CTLA-4/PD-1 antibodies. Preclinical and early clinical studies. | ( |
| Nigericin | Breast Cancer, Neuroblastoma | Activates NLRP3 inflammasome; controls tumor growth. | Regulates immune response via NLRP3 activation, likely influencing IL-1β and IL-18 secretion. | ( | |
| OLT1177 | Melanoma | Inhibits NLRP3 inflammasome; inhibits tumor growth. | Limits expansion of myeloid-derived suppressor cells, likely by affecting IL-1β. | Potential synergy with ICIs*. Preclinical studies. | ( |
| Aging Clearance Agents (Senolytics) and Targeted Delivery Systems | |||||
| Quercetin | Osteosarcoma | Inhibits SASP secretion (IL-6, IL-8, MMPs, IL-1α, CXCL12, VEGF). Induces senolysis in doxorubicin-induced senescent fibroblasts. | Attenuates protumorigenic effects of senescent fibroblasts. | Preclinical studies. | ( |
| ABT-263 (Navitoclax) | Lung Cancer, Breast Cancer, Murine Colon Cancer (MC38), Lymphoma (EL4) | Inhibits anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-w) to clear senescent cells. Specifically eliminates p16+/p21+ senescent cells; reverses senescence-induced immunosuppressive myeloid phenotypes; reduces p16/p21 expression in spleen and TME. | Restores CD8+ T cell proportion, accumulation, and activation (IFNγ+) within tumors; increases CD8+ T cell/CD11b+ myeloid cell ratio; abrogates myeloid cell-mediated suppression of CD8+ T cell proliferation. | Reverses immunotherapy resistance: Restores anti-PD-L1 efficacy in senescent mice; restores abscopal effects of “radiotherapy + anti-CTLA-4” therapy; significantly prolongs survival when combined with ICIs*. Clinical Stage: Preclinical studies (validated senolytic; entered human clinical trials). | ( |
| ABT-737/Navitoclax (Bcl-2 inhibitor) | NSCLC* (Kras G12V model) | Eliminates senescent cells; reduces SA-β-gal activity; decreases proliferation markers (Ki67, pRb); increases tumor cell apoptosis. | Immune cell changes not detailed. | Combined with cisplatin: significantly reduces tumor burden; decreases senescence and proliferation markers; improves therapeutic response. | ( |
| Ra/Ba SAzyme (²²³Ra/Ba SAzyme) | Lung Cancer | Induces senescence and ROS generation, followed by senescent cell clearance via anti-PD-L1. | Induces senescence to enhance antitumor immunity; effectively clears senescent cells upon combination with anti-PD-L1, reducing recurrence risk. | Preclinical studies. | ( |
| Targeting chemokine receptors and downstream signaling pathways | |||||
| PF-04136309 | PDAC* | Small molecule CCR2 antagonist. | Reduces CCR2+ monocytes and TAMs; alleviates immunosuppression; stimulates T cell infiltration into the TME. | Clinical Phase Ib trial (combined with chemotherapy; well-tolerated; showed early antitumor activity). | ( |
| BMS-813,160 | Pancreatic, colorectal cancer(combination with chemotherapy or nivolumab) | CCR2 inhibitor. | Disrupts prosurvival microenvironment driven by CCL2-mediated monocyte and macrophage migration. | Preclinical studies | ( |
| Galunisertib (TGFBR1 Inhibitor) | NSCLC*, HGSOC* | Inhibits TGFβ/TGFBR1 axis; blocks AKT/mTOR pathway activation; reduces p-AKT/p-p70S6K levels; attenuates tumor cell proliferation, migration, and sphere-forming capacity. | No significant changes in immune cell proportions (except normalization of alveolar macrophages to pre-cisplatin levels); exhibits no significant immunosuppressive activity. | Combined with cisplatin: significantly reduces tumor burden; prolongs survival (median survival increased by 33–42.7%); improves weight maintenance and tolerability. | ( |
| JAK-STAT signaling pathway inhibitor | |||||
| Bazedoxifene | Pancreatic Cancer, Breast Cancer | SERM*; effectively blocks IL-6/GP130-mediated STAT3 activation. | Preclinical studies (demonstrates repurposing potential in malignancies driven by aberrant IL-6 signaling). | ( | |
| Stattic | Prostate Cancer | STAT3 inhibitor. | Preclinical studies ( | ( | |
| Ruxolitinib | aCML*, CNL*, Inflammation-related tumors | JAK1/JAK2 inhibitor; blocks JAK/STAT3 signaling; inhibits CRP* expression. | Significantly reduces inflammation-associated symptoms (e.g., fever). | Clinical studies (effective in patients with CSF3R mutations); Preclinical studies (demonstrates anti-inflammatory effects). | ( |
| AZD1480 | HPV-related HNSCC* | JAK1/JAK2 inhibitor; targets JAK/STAT signaling to overcome cisplatin resistance. | Reduces protumor inflammation and immune evasion. | Preclinical studies (animal models). | ( |
| Momelotinib | NSCLC* | JAK1/JAK2 inhibitor; combined with EGFR inhibitor Erlotinib. | Targets JAK/STAT-mediated inflammation and resistance pathways to delay or overcome EGFR inhibitor resistance. | Clinical Phase Ib study (safe and tolerable). | ( |
| Tofacitinib | Inflammatory diseases (in context of cancer risk) | JAK1/JAK3 inhibitor; inhibits proinflammatory cytokines (IL-2, IL-6, IL-12, IL-23). | Inhibits immune system activation; may hinder immune surveillance of nascent tumor cells. | Systematic review and Meta-analysis (associated with slightly increased overall cancer risk). | ( |
| Jaktinib | Myelofibrosis | Novel JAK inhibitor. | Clinical Phase II study (shows promise in patients relapsed or refractory to Ruxolitinib). | ( | |
| Pacritinib | Metastatic Colorectal Cancer, AML* | JAK2/FLT3 inhibitor. | Inhibits STAT3 signaling and proinflammatory cytokine levels. | Clinical studies (limited clinical activity in heavily pretreated patients); Preclinical studies (synergistic with HDAC inhibitors). | ( |
| COX inhibitors and non-steroidal anti-inflammatory drugs | |||||
| Aspirin | Colorectal, Breast, Pancreatic, Liver, and other cancers | Irreversibly acetylates and inactivates COX; inhibits multiple pathways (PIK3CA/AKT/PTEN, Wnt-β catenin, NF-κB); suppresses TGF-β, IL-6, MCP-1; regulates angiogenic/inflammatory cytokines. | Modulates T cell and macrophage responses; activates resolution pathways; promotes M1 macrophage polarization and inhibits M2 polarization, thereby limiting tumor progression. | Efficacy combined with ICIs* (e.g., pembrolizumab/ipilimumab) is comparable to ICIs* monotherapy. Epidemiological studies, clinical prevention (USPSTF recommended), and Clinical Phase II trials. | ( |
| Sulindac | Colon, Laryngeal Cancer, etc. | Downregulates Sp proteins; induces apoptosis; inhibits Wnt/β-catenin pathway. | Reduces Fas ligand expression; may affect lymphocyte apoptosis. | ( | |
| Celecoxib | Colorectal, Breast, Pancreatic Cancer, etc. | Selective COX-2 inhibitor; inhibits Wnt/β-catenin signaling; induces apoptosis; inhibits EMT and metastasis. Suppresses tumor-derived PGE2; enhances chemotherapy (paclitaxel)-induced immunogenic cell death. | Inhibits IFN-γ-induced surface PD-L1 expression. | Enhances antitumor effects when combined with p65miRNA, statins, or TRAIL. Preclinical and clinical studies. | ( |
| Ibuprofen | Colorectal, NSCLC*, Prostate, Liver Cancer, etc. | Inhibits COX-2; reduces expression of HDACs and histone demethylases; regulates β-catenin signaling. | May regulate the inflammatory microenvironment by inhibiting COX-2-mediated PGE2 production. | Preclinical studies. | ( |
| Other drugs with anti-inflammatory or immune-regulating effects | |||||
| Silybum marianum (Silymarin) Flower Extract | Skin Cells | Inhibits IL-6 and MMP-1 expression. | Demonstrates anti-aging activity in human skin cells. | Preclinical studies. | ( |
| CD38 Blockers | Blocks CD38; reduces intratumoral adenosine accumulation; restores CD8+ T cell activity. | Relieves CD38-mediated suppression of CD8+ T cells. | Sensitizes tumors to anti-PD-L1 therapy. Preclinical studies. | ( | |
| A2A Adenosine Receptor Inhibitors | Breast Cancer, Colon Cancer | Inhibits A2A adenosine receptor. | Promotes T cell proliferation; generates T cell-dependent antitumor activity. | Enhances anti-PD-1 efficacy. Preclinical studies. | ( |
| Small Molecule OmoMYC | AML* | MYC-inhibiting peptide/miniprotein. | Increases immune cell infiltration into the tumor. | Sensitizes tumors to anti-PD-1 immunotherapy; currently in clinical development. Preclinical/Clinical trial development. | ( |
| Dexamethasone | Lymphoma, Breast, Pancreatic Cancer | Glucocorticoid (adjuvant for lymphoma); may promote progression in pancreatic/breast cancer. Combined with Tamoxifen: targets E2F3/SOX2/Wnt pathways; induces apoptosis; inhibits proliferation and migration. | Inhibits broad immune cell functions; induces lymphocyte apoptosis; may systemically suppress immune responses. | May induce resistance to immunotherapy and chemotherapy. Clinical application (as adjuvant), though protumorigenic risks reported. | ( |
| Simvastatin | Murine Colon Cancer (C26) | Liposome-encapsulated; enhances anticancer effect of 5-fluorouracil. | Preclinical studies ( | ( | |
| TAPI-1 | ESCC* | Metalloproteinase inhibitor; inhibits NF-κB pathway to reduce inflammatory signaling; suppresses tumor growth, migration, and invasion. | Preclinical studies ( | ( | |
| Trametinib + Palbociclib | Pancreatic Cancer | MEK + CDK4/6 inhibition induces tumor senescence and SASP (containing VEGF), remodeling tumor vasculature. | Promotes CD8+ T cell infiltration via VCAM-1, though T cells exhibit exhaustion. | Enhances PD-1 blockade efficacy (Preclinical): Combination therapy converts “cold” tumors to “hot” tumors, significantly improving anti-PD-1 efficacy. | ( |
- —National Natural Science Foundation of China10.13039/501100001809
- —China Postdoctoral Science Foundation10.13039/501100002858
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTelomeres, Telomerase, and Senescence · Cancer Immunotherapy and Biomarkers · Cancer Research and Treatments
Introduction
1
The progression of aging induces not only normal physiological aging, but is also accompanied by a considerable array of pathological alterations following the decline of systemic functions. Factors such as the accumulation of intracellular DNA damage, diminished efficiency of cellular repair mechanisms, recurrent exposure to environmental triggers, metabolic syndrome, and a state of chronic inflammation, collectively contribute to an elevated risk of carcinogenesis (1). Among these alterations, immunosenescence stands out as a hallmark feature shaping the clinical trajectory of elderly cancer patients (2, 3). The majority of elderly patients are diagnosed at advanced stages of the disease (4), which results in poor clinical prognoses and low treatment tolerance (5). Concurrently, their distinct physiological characteristics, including multi-system functional decline and reduced drug metabolism, necessitate a multifaceted assessment prior to the selection of a clinical regimen, thereby increasing the therapeutic complexity (6, 7). In response to this phenomenon, this review explores the correlation between aging and tumors, revealing a bidirectional interaction between the two. In particular, it highlights how immunosenescence and chronic inflammation promote tumor initiation, progression, and metastasis in elderly patients, while also exerting a significant impact on their treatment outcomes and prognosis. These effects are mediated by multiple senescence-associated signaling pathways, involving factors such as senescent immune cells and dynamic cellular interactions within the aging tumor microenvironment (TME).
In recent years, the advent of cancer immunotherapy, exemplified by immune checkpoint inhibitors (ICIs) and chimeric antigen receptor T cell therapy (CAR-T), has revolutionized the clinical treatment paradigm. However, evidence indicates significant heterogeneity in the response rates of elderly patients to ICIs, with the underlying mechanisms remaining unelucidated. The underrepresentation of older patients in some clinical trials has resulted in divergent data regarding efficacy and safety in this cohort (7). Based on this, this review synthesizes aging-related immunotherapy data and reveals that the impact of age on immunotherapy efficacy varies across different cancer types. Additionally, it summarizes senescence-modulating drugs, evaluating their therapeutic benefits and potential limitations in the context of immunotherapy. Through this analysis, the review explores tailored immunotherapeutic strategies for the elderly population.
Aging and immunosenescence
2
With advancing organismal age, the functionality of the immune system declines—a process termed immunosenescence—which is characterized by an impaired capacity to clear deleterious substances and a concomitant increase in excessive inflammatory responses. This process can perpetuate a vicious cycle of aging and predispose individuals to autoinflammatory and autoimmune disorders (2, 3). Immunosenescence dysregulates both the innate and adaptive arms of the immune system. In the innate immunity system, the components exhibit functional deficits, including reduced formation, phagocytic activity, and chemotaxis of neutrophils, alongside their increased susceptibility to apoptosis (8). In the adaptive immune system, thymic involution leads to a reduction in the naive T-cell pool, decreased T-cell receptor (TCR) diversity, and an imbalance in memory T-cell populations, which collectively impair antigen recognition capabilities (9). B-cell antibody affinity and the durability of the adaptive immune response are also reduced (10). The expansion of memory cells results in immunological resource depletion, and different antigens cause the upregulation of pro-inflammatory molecules (11, 12). This culminates in a comprehensive decline in immune function, which not only weakens host defense against pathogens but also intensifies chronic inflammation, impairs immune surveillance, and results in an elevated risk of infection and malignancy (3, 13).
Immunosenescence leads to a dysregulation of inflammatory homeostasis within the immune system, preventing the effective clearance of senescent cells and pro-inflammatory factors. This culminates in a systemic, low-grade inflammatory state known as “inflammaging” (14). In the absence of overt infection, this condition is also termed “sterile inflammation”. It is distinct from acute inflammation and is characterized as a low-grade, systemic, and persistent inflammatory state associated with elevated levels of inflammatory biomarkers such as interleukin-6 (IL-6), interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-α), and C-reactive protein (CRP). These age-related immunological impairments and the resultant inflammatory state shift the body towards a catabolic metabolism and are correlated with a spectrum of diseases, frailty, and mortality (15, 16).
Immunosenescence: immune cell-specific features and functional impacts
2.1
As shown in Figure 1, all immune cell lineages are impacted by aging to varying degrees, which compromises the capacity of the immune system to respond to novel antigens. Within the innate immune system, the phagocytic function of neutrophils and macrophages is attenuated, the antigen-presenting capacity of cells such as dendritic cells (DCs) is diminished, and the function of natural killer (NK) cells becomes dysregulated (8). Neutrophil Senescence is characterized by increased mitochondrial reactive oxygen species (ROS) and significantly elevated C-X-C motif chemokine ligand 4 (CXCR4) levels; the latter mediates neutrophil homing to the bone marrow for clearance and serves as a distinct marker of neutrophil senescence (17). Macrophages Senescence is characterized by elevated levels of p16 and SA-β-Gal, concurrent with mitochondrial dysfunction and increased ROS production; this subsequently leads to increased expression of NOX4 NADPH oxidase, resulting in polarization imbalance (18).Macrophages display attenuated phagocytic capacity, impaired debris clearance and polarize towards a CD80^+^ pro-inflammatory M1 phenotype, contributing to a chronic inflammatory state (19, 20). Senescent DCs exhibit reduced mitochondrial membrane potential, decreased ATP generation, and increased production of ROS. Mitochondrial dysfunction not only impairs phagocytic capacity, but the accumulation of ROS also further interferes with the process of antigen cross-presentation (18). Senescence in NK cells manifests as alterations in surface markers and functional impairment, characterized by the downregulated expression of activating receptors (NK group 2 member D (NKG2D), NKp30), the upregulated expression of inhibitory receptors (KIRs, NKG2A), and the activation of the killer cell lectin-like receptor subfamily G member 1 (KLRG1) -AMP-activated protein kinase (AMPK) signaling pathway (21). Furthermore, NK cells function is compromised, manifesting as impaired immune surveillance, a shift towards a more mature cytotoxic phenotype (CD56dim NK cells), and reduced cytokine secretion (CD56Bright NK cells). This collectively leads to attenuated immune surveillance and the occurrence of immune escape (the ability of tumor cells to avoid recognition and elimination by the immune system) (22).
Changes related to immune cell senescence. Aging is associated with alterations in both innate and adaptive immune cells, resulting in increased levels of senescence-associated secretory phenotype (SASP). Neutrophil senescence is marked by elevated mitochondrial reactive oxygen species (ROS) and a significant upregulation of CXCR4 (C-X-C motif chemokine receptor 4), which directs bone marrow homing for clearance and serves as a key senescent marker. Macrophage senescence is defined by upregulated p16, mitochondrial dysfunction/ROS, and increased NADPH oxidase 4 (NOX4) expression, leading to M1-skewed polarization. This results in impaired phagocytosis, defective debris clearance, and chronic inflammation. Senescent DCs show reduced mitochondrial membrane potential, ATP, and increased ROS. NK cell senescence features a shifted receptor profile and activation of the killer cell lectin-like receptor subfamily G member 1 (KLRG1) -AMP-activated protein kinase (AMPK) pathway, driving functional decline. Senescent T cells are defined by the CD28-KLRG1+CD45RA+ phenotype (cluster of differentiation 28 negative, KLRG1 positive, cluster of differentiation 45 RA isoform positive); this is accompanied by mitochondrial dysfunction and consequent ROS accumulation. B cell aging is marked by a metabolic shift toward glycolysis and elevated mitochondrial oxidative stress. These immunological changes contribute to the development of chronic low-grade inflammation, often referred to as “inflammaging”, which is linked to multi-system dysfunction and the onset of age-related disease.
In the adaptive immune system, B cells from elderly individuals demonstrate an enhanced ability to shift from oxidative phosphorylation to anaerobic glycolysis, accompanied by increased mitochondrial oxidative stress. This leads to a state of oxidative damage and cellular senescence, a metabolic reprogramming that parallels features observed in tumor cells (23). Romero et al. demonstrated that co-culturing B cells from elderly donors with normal CD4^+^ T cells induced the expression of inflammation-associated markers and increased lactate production in the T cells (23).
Senescent T cells are characterized by the loss of CD28, the upregulation of KLRG1, and the re-expression of CD45RA, accompanied by mitochondrial dysfunction that leads to the accumulation of ROS (24).T cells participate in inflammaging through functional dysregulation—including an expansion of pro-inflammatory subsets, a reduction in anti-inflammatory functions, and the secretion of pro-inflammatory cytokines (13, 25)—and further exacerbate the chronic inflammatory milieu through interactions with the Senescence-Associated Secretory Phenotype (SASP). SASP is composed of pro-inflammatory factors such as IL-6, IL-1, and TNF-α secreted by senescent cells, which can induce senescence in adjacent cells through paracrine signaling, thus establishing and intensifying a chronic inflammatory environment and accelerating systemic aging and age-related pathologies (26, 27). Research indicates that activated T cells in the elderly exhibit lysosomal dysfunction, which prevents the effective degradation of damaged mitochondria. Instead, these cells release necrotic mitochondria and their DNA via exocytosis, a process that contributes to inflammaging (28). Desdín-Micó et al. identified T cells deficient in mitochondrial transcription factor A as accelerators of immune system aging (29), while Ovadya et al. found that perforin gene knockout resulted in a more rapid accumulation of senescent cells across multiple murine organs, thereby accelerating aging (30). Elevated expression of the E3 ubiquitin ligase bifunctional apoptosis regulator (BFAR) in aged individuals inhibits the generation of tissue-resident memory T cells (TRM), thereby impairing the differentiation of CD8^+^ T cells into the TRM lineage (31). This impairs the capacity of T cells to recognize and eliminate novel tumor antigens.
Aging and cancer
3
In China, the age-specific incidence of all cancers is relatively low in the 0–34 age group, increases markedly from the 35–39 age group, and culminates in a peak in the 80–84 age group (32). Aging and cancer share numerous characteristics and exhibit a strong degree of equivalence (33). Concurrently, the state of chronic, low-grade inflammation inherent in elderly individuals exerts a pro-tumoral effect (34). Elevated levels of various inflammatory cytokines are known to have a detrimental impact on anti-tumor immunity (27).
Aging and the tumor microenvironment
3.1
With advancing age, the tissue microenvironment—comprising the extracellular matrix (ECM), immune cells, fibroblasts, and the vascular system—undergoes functional alterations.
ECM remodeling is a recognized hallmark of cancer, driven predominantly by the interplay between immune cells and cancer-associated fibroblasts (CAFs) (35). This process leads to the accumulation and increased cross-linking of non-cellular components such as collagen and elastin, which in turn alters tissue stiffness and mechanical stress (36). These changes promote tumorigenesis and establish a physical barrier that impedes drug delivery and immune cell infiltration, thereby fostering an immunosuppressive state (37). Tumor tissue often exhibits increased stiffness. Cells sense these physical properties of the ECM via integrin-conjugated focal adhesions, which influences cell migration, differentiation, and survival. Meanwhile, the remodeled ECM directly promotes tumor cell invasion and metastasis (38).
As shown in Figure 2, the TME in older individuals is characterized by immune cell dysregulation. Aging impairs the infiltration and function of CD8^+^ T cells within the tumor. Recent studies indicate that tumor-derived extracellular vesicles (tEVs) carrying Programmed death-ligand 1 (PD-L1) can induce T-cell senescence and immunosuppression via alterations in lipid metabolism and cAMP response element-binding protein (CREB) signaling (39). And the Programmed cell death protein 1 (PD-1)/PD-L1 interaction facilitates immune evasion by directly inducing T cell apoptosis and promoting the expansion of immunosuppressive regulatory T cells (Tregs) (40). The TME of older patients exhibits alterations in the NK cell-DC-CD8^+^ T cell signaling axis. These alterations encompass compromised NK cell immune surveillance, attenuated antigen presentation by DCs, and intrinsic changes in T cells themselves (8, 41, 42), which collectively diminish the priming of T cells by conventional type 1 DCs (cDC1s). This specifically impacts the differentiation of CD8^+^ T cells, driving them into a state of tumor-infiltrating age-associated dysfunction that is functionally, transcriptionally, and epigenetically distinct from canonical exhaustion and persists within the TME (42). The age-associated decline in the migratory capacity of DCs further exacerbates T-cell functional deficiencies, promoting tumor development, immune escape, and a suboptimal response to ICIs. Improving cDC1s function through bone marrow-targeted immunotherapy can enhance CD8^+^ T-cell immunity in aged mice (42). Similarly, inducing a hyperactivated state in DCs with agonists such as PGPC can rectify their age-related defects and enable Type 1 T helper cells (Th1) to acquire cytotoxic potential, thereby exerting anti-tumor activity (43).
The changes in the tumor microenvironment caused by aging. In aging tumor microenvironment (TME), senescent cells, M2-type macrophages, T cells, myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) mainly secrete the senescence-associated secretory phenotype (SASP), which activates fibroblasts and promotes the epithelial-mesenchymal transition of tumor cells. Simultaneously, SASP inhibits T cell function. Through interactions with SASP, T cells induce further cellular senescence and cytokine release, resulting in fibroblast dysfunction and pathological fibrosis of the matrix. SASP causes the weakening of vascular walls, which increasing tumor vessel density but compromises barrier function. Meanwhile, SASP recruits MDSCs and Tregs, suppresses dendritic cell (DC) function, and exerts immunosuppressive effects. However, SASP can also promote M1 macrophage differentiation and natural killer (NK) cell function, thereby exerting anti-tumor effects. Aging cells and fibroblasts influence tumor-associated macrophages (TAMs), promoting their differentiation into the M2 phenotype. M2-type macrophages, in turn, activate Tregs and secrete immunosuppressive cytokines such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which impair DCs and NK cells function. M2-type macrophages also upregulate PD-L1, thereby suppressing T cells. NK cells and DCs exhibit functional abnormalities due to alterations in signal transduction pathways, impairing their ability to activate CD8+ T cells and leading to suppressed T cell function. Additionally, tumor cells and MDSCs release extracellular vesicles containing PD-L1. PD-1/PD-L1 signaling drives immune evasion by inducing T cell apoptosis and expanding immunosuppressive Tregs. Senescent cells also secrete Bifunctional Apoptosis Regulator (BFAR), which suppresses the activity of CD8+ T cells. Some key items are circled in red.
Senescent fibroblasts can modulate tumor-associated macrophages (TAMs) to express pro-tumorigenic growth factors (M2-type), which in turn suppress the function of tumor-related DCs via the secretion of immunosuppressive factors such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), induce a Th2-polarized immune response, and promote the generation of Tregs, thereby attenuating anti-tumor immune response (27, 44). In the elderly, age-related macrophage dysfunction leads to a diminished capacity for clearing damaged ECM components as well as promoting tissue repair, and an upregulation of KEGG gene sets associated with fibrotic signaling (8, 45, 46). Furthermore, a decline in T-cell function results in aberrant, cytokine-mediated regulation of fibroblast activity, leading to the abnormal secretion of non-cellular matrix components (47).In aged individuals, persistent low-grade inflammation gives rise to a fibrotic SASP (fibrosis-related components of the SASP), activates fibroblasts, impairs tissue regeneration and repair, and facilitates tumor progression by modulating cancer cell stemness and the epithelial-mesenchymal transition (EMT) through various signaling pathways and inflammatory mediators such as TGF-β (48). Tumors harbor distinct populations of CAFs, which can be activated by fibrotic SASP derived from aged individuals. Stromal CAFs are pivotal in tumor angiogenesis, whereas inflammatory CAFs (iCAFs) are implicated in the formation of an immunosuppressive microenvironment (37). CAFs release higher levels of SDF-1, which recruits endothelial progenitor cells into carcinomas, enhancing angiogenesis and thus tumor growth (49). Stromal fibrosis is associated with diminished therapeutic efficacy: tumors characterized by a high degree of fibrosis, such as pancreatic, hepatocellular, and breast cancers, exhibit poor responses to chemotherapy and immunotherapy (50, 51). In breast cancer patients undergoing anti-PD-1 immunotherapy, iCAFs demonstrate an enhanced propensity to promote cancer cell proliferation, EMT, and the establishment of an immunosuppressive microenvironment. Conversely, in melanoma, patients with high iCAF scores exhibit significantly prolonged overall survival (OS) and progression-free survival (PFS), coupled with higher objective response rates to both anti-PD-1 and anti- Cytotoxic T-lymphocyte-associated protein 4 (CTLA4) therapies (52).
SASP facilitates angiogenesis and tumor invasion by secreting matrix metalloproteinases (MMPs) to degrade ECM and releasing growth factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) (53). Specifically, MMPs degrade elastin, causing the weakening of vascular walls (54). Myeloid-derived suppressor cells (MDSCs) promote EMT and angiogenesis by secreting IL-6, TGF-β, and EGF, and by producing large amounts of MMP-9 (55). In melanoma, the secretion of the ECM protein hyaluronan and proteoglycan link protein 1 (HAPLN1) by fibroblasts decreases with age. This indirectly upregulates intercellular adhesion molecule 1 (ICAM1) expression in vascular endothelial cells, leading to the phosphorylation and internalization of VE-cadherin. This process increases tumor vessel density but compromises barrier function, creating conditions for tumor cell entry into the bloodstream and distant metastasis, thereby promoting tumor progression (56).
Regulation of the tumor microenvironment by the SASP
3.2
SASP
3.2.1
SASP is a secretory phenotype unique to senescent cells, characterized by the secretion of various proinflammatory cytokines (interleukins), chemokines (C-X-C motif chemokine ligand 1 (CXCL1), C-C motif chemokine ligand 8 (CCL8)), growth factors, ECM remodeling factors (matrix metalloproteinases), and non-protein components such as ROS and nitric oxide (NO) into the surrounding microenvironment, despite cell cycle arrest (57). SASP plays a dual role in tumor progression. In the early stages, it enhances immunosurveillance and suppresses tumorigenesis by recruiting M1 macrophages, Th1, and NK cells (mediated by factors like IL-1α, IL-6, and interleukin-8 (IL-8)). Conversely, in advanced stages and under conditions of chronic inflammation, it drives the formation of an immunosuppressive TME and facilitates the recruitment of MDSCs and Tregs. By releasing immunosuppressive cytokines (some of which overlap with SASP factors, such as TGF-β and IL-10), it inhibits T cell and NK cell functions, ultimately enhancing tumor invasion, metastasis, and therapeutic resistance (53, 58, 59).
A state of tumor-promoting inflammation (TPI), a chronic inflammatory state that facilitates tumor progression, is a recognized hallmark of cancer (60). Meanwhile, the chronic inflammatory state of elderly can exacerbate TPI and facilitates tumor progression. Senescent cells secrete transcription factors and inflammatory cytokines that play a critical role in promoting tumor proliferation, angiogenesis, and invasion/metastasis. For instance, chronic inflammation can exert a pro-tumoral effect by inhibiting the clearance of senescent and neoplastic cells by myeloid cells (61–63). Cytokines play dual roles in cancer, with interferons like Interferon-gamma (IFN-γ) exerting core anti-tumor effects, while sustained signaling can induce resistance. TGF-β is stage-dependent, suppressing early tumors but promoting progression later. Pro-inflammatory cytokines (IL-1, IL-6) drive tumorigenesis, whereas immunosuppressive IL-10 dampens immunity. Even potentially therapeutic cytokines like interleukin-18 (IL-18) are neutralized by endogenous inhibitors in the TME (63, 64). This section focuses on several representative SASP factors for discussion.
IL-6
3.2.2
Elevated levels of IL-6 are considered a manifestation of primary aging and possess dual (65), context-dependent anti-tumor and pro-tumor functions. IL-6 has been observed to manifest in inflammatory sites, the TME, and during the process of aging. It has been demonstrated that IL-6 typically exerts a pro-carcinogenic effect by fostering chronic inflammation and activating pro-tumoral signaling cascades (66). High serum concentrations of IL-6 are an adverse prognostic indicator for a multitude of cancers (67).In the context of chronic inflammation and cancer, the signaling pathways associated with IL-6 are often constitutively activated (68). With respect to its anti-tumor activities, IL-6 released from skeletal muscle during physical exercise enhances insulin sensitivity in glycogen-storing tissues, stimulates the circulation of anti-inflammatory cytokines, mobilizes cytotoxic immune cells, and reduces DNA damage in cancer cells (69).
Supraphysiological levels of IL-6 are linked to T-cell immune dysregulation. Excess IL-6 can inhibit the de novo generation of induced Tregs from naive T cells—without affecting the development or function of natural Tregs—thereby diminishing anti-inflammatory capacity and promoting a pro-/anti-inflammatory imbalance that perpetuates a chronic inflammatory state (70, 71). The IL-6- janus kinase (JAK)- signal transducer and activator of transcription (STAT) 3 signaling pathway can promote tumor cell survival by inducing the expression of anti-apoptotic proteins (including Bcl-2, Bcl-xL, and survivin). It also enhances the expression of MMP-2, VEGF), and basic FGF (bFGF), thereby promoting tumor angiogenesis and metastasis (34, 72, 73). Furthermore, IL-6-dependent STAT3 signaling can drive the methylation and subsequent inactivation of the key tumor suppressor gene p53, which allows cancer cells to bypass critical cell cycle checkpoints and evade apoptosis induced by DNA damage (74, 75).
IL-1, IL-17, and IL-22
3.2.3
IL-1α and IL-1β, known as alarmins, activate and amplify local inflammation via the IL-1 receptor (IL-1R). In aged individuals, the expression of SASP-associated factors, including IL-6 and IL-1β, is upregulated in renal tissue, with the quantity of IL-1β^+^ macrophages being substantially higher than in younger cohorts (46). Furthermore, immunosenescence enhances the tumor-induced myelopoietic response, leading to the local accumulation of myeloid progenitor-like cells within the TME. These cells produce copious amounts of IL-1α, which promotes tumor development (62).
IL-1 has long been implicated in inflammation-induced carcinogenesis. In a chronic inflammatory state, IL-1α and IL-1β can directly promote the generation of carcinogenic mediators such as nitric oxide and ROS (76, 77). IL-1 also acts on epithelial cells via IL-1R to directly facilitate malignant transformation mediated by the nuclear accumulation of NF-κB (78). Studies have shown that IL-1α-expressing monocyte-derived macrophages correlate with poorer survival and higher recurrence rates; disruption of IL-1R1 signaling early in tumorigenesis can normalize myelopoiesis and decelerate the growth of lung, colon, and pancreatic tumors (62). In addition to the intrinsic pro-tumoral effects of IL-1, the activation of IL-1 signaling in T cells promotes the secretion of interleukin-17 (IL-17) and interleukin-22 (IL-22) (78). IL-17 is involved in the pathogenesis of numerous cancers, including breast, liver, lung, and colon cancer, through multiple pathways (79–84). And the IL-17/IL-17RA signaling axis is also implicated in pancreatic cancer progression (85–87). IL-22, via the phosphorylation of STAT3, provides proliferative and migratory signals to transformed malignant cells and/or cells harboring oncogenic mutations (88).
IL-18
3.2.4
In the context of cancer, IL-18 exhibits dual anti-tumor and pro-tumor activities. Its anti-tumor functions include enhancing NK cell activity, promoting the production of anti-tumorigenic IFN-γ, and modulating immune responses (89). Furthermore, when IL-18 is cleaved by caspase-3 within cancer cells, a 15 kDa truncated form, termed short IL-18, is generated. It translocates to the nucleus, where it facilitates STAT1 phosphorylation at serine 727 via CDK8 and enhances the expression and secretion of ISG15, and thereby mediate the clearance of various syngeneic tumors and colitis-associated colorectal cancer in murine models (90). Conversely, IL-18 can exert pro-tumor effects. The decoy receptor IL-18BP (IL-18 binding protein), which is abundantly secreted by cancer cells, binds competitively to IL-18, thereby limiting its anti-tumor activity in mice (91). Additionally, IL-18 can induce the phosphorylation of long-chain acyl-CoA synthetase 6 (ACSL6) at serine 674 (ACSL6 pS674) and its translocation to the cell membrane. ACSL6 pS674 can promote the stable formation of the IL-18 receptor (IL18R1-IL18 receptor accessory protein (RAP)) heterodimer, activating the IL-18R1/NF-κB signaling pathway and leading to the upregulation of various pro-cancer genes, ultimately promoting liver cancer growth and metastasis. The combination of knocking down ACSL6 or ACSL6 S674A with anti-PD-1 therapy can inhibit the recruitment of TAMs and neutrophils, significantly promote the recruitment and activation of CD8^+^ T cells, and enhance immunotherapy efficacy (92).In liver cancer and hematogenous metastases to the liver and lungs, NK cells have been found to highly express IL-1R8, which functions as a negative regulator of the IL-18 signaling pathway. By inhibiting the interaction between IL-18Rα and downstream signaling molecules (such as MyD88 and IRAK4), it attenuates NK cell activation, thereby exerting a pro-tumor effect (93).
TNF-α and IFN-γ
3.2.5
IFN-γ and TNF-α released by the senescent microenvironment activate immunosuppressive networks while simultaneously promoting tumor proliferation through chronic inflammation (94); conversely, TNF-α and IFN-γ signaling can induce tumor cell senescence, apoptosis, and ferroptosis, aiding in tumor growth control (95). TNF-α exhibits dual antitumor and protumor effects. High doses can induce tumor necrosis (96) and are often used in recombinant form combined with chemotherapy for isolated limb perfusion (97); however, long-term, low-dose TNF-α promotes tumor development by inducing DNA damage and angiogenesis (96). In the senescent microenvironment, TNF-α secretion by TAMs, combined with oxidative stress in the TME, leads to reduced nuclear translocation of PPARγ and increased TNF-α secretion, further driving tumor progression (98). TNF-α upregulates PD-L1 expression via the NF-κB pathway, promoting immune escape (99), and enhances the invasive and metastatic capabilities of multiple cancers, including melanoma (98). TNF-α also participates in EMT via NF-κB, inducing tumor cell dedifferentiation and promoting tumorigenesis (100). In vivo experiments demonstrate that blocking the TNF/TNFR1 signaling pathway with targeted antibodies increases the proportion of melanoma-specific CD8+ T cells in the microenvironment and delays tumor growth (101).
IFN-γ is generally not classified as a typical SASP factor. However, within the senescent TME, it acts as a critical immunomodulator, serving as both an amplifier of SASP effects and an effector molecule in the immune system’s response to senescent cells (53). As a pivotal component in CAR-T therapy, IFN-γ activates macrophages, promotes their polarization toward the M1 phenotype, enhances T cell cytotoxic function, and improves the organism’s immune response to tumors (102). Conversely, IFN-γ can upregulate PD-L1 via the STAT1/IRF-1 axis, promoting immune escape (53). Upon stimulation with IFN-γ, fibroblasts are induced to express high levels of major histocompatibility complex (MHC) class II and costimulatory molecules (e.g., CD40, CD80/86). This enables them to directly present antigens to T cells and trigger their reactivation, thereby establishing a positive feedback loop that amplifies and sustains inflammatory and fibrotic responses (103).
Aging and immune surveillance of tumors
3.3
In tumor immunology, the immune system performs immune surveillance to recognize and eliminate nascent transformed cells. The concept of tumor immune surveillance is based on three principles (1): cancer cells possess antigenicity (2); cancer cells can be destroyed by the host’s immune response (through mechanisms analogous to those in tissue or organ transplant rejection); and (3) immunosuppression is correlated with a higher incidence of tumors (104). The relevant antigens are typically tumor-associated antigens and neoantigens, which are presented to T cells via MHC/human leukocyte antigen (HLA) molecules (105). Immune surveillance imposes a selective pressure on tumors, driving continuous mutation and a process known as immunoediting. This is characterized by a dynamic equilibrium where tumor cells generate new mutations, selecting for those with lower immunogenicity, while a competent immune system continuously recognizes and eliminates these mutated cells. If mutated cells successfully evade immune-mediated clearance, immune escape occurs. This refers specifically to the acquisition by tumor cells, through genetic or epigenetic alterations, of insensitivity to immune recognition and/or elimination, allowing them to gain a clonal advantage and form a clinically detectable tumor (106). Immunosenescence and chronic inflammatory states are mutually influential with cancer. Key senescence-associated signaling pathways in tumors include the following: Constitutive activation of the NF-κB pathway drives the transcription of proinflammatory genes while inhibiting autophagy and apoptosis; this upregulation of anti-apoptotic proteins leads to the accumulation of senescent cells (65, 107, 108). Hyperactive nutrient sensing via the mTORC1 pathway increases ULK1 phosphorylation and blocks autophagy, resulting in anabolic imbalance and T cell exhaustion (109, 110). Chronic activation of the JAK-STAT pathway drives cytokine storms, causes hematopoietic stem cell (HSC) dysfunction, and leads to T cell reduction (111, 112). The cGAS-STING pathway, activated by damaged cytosolic DNA, induces NF-κB-mediated inflammation but exhibits impaired type I interferon (IFN-I) production, resulting in weakened antiviral responses (113–115). Blunted energy sensing in the AMPK pathway results in a failure of mTOR inhibition and obstructed autophagy, causing the uncontrolled expansion of MDSCs and impaired T cell memory formation (116–118). Decreased melatonin secretion diminishes its triple protective effects: inhibition of NF-κB, scavenging of ROS, and optimization of mitophagy (119, 120). Downregulation of Sirtuin family members (SIRT1/3/6) elevates mitochondrial oxidative stress and reduces histone deacetylation, resulting in epigenetic dysregulation (121–123). Concurrently, the synergistic interaction between the NF-κB and STAT pathways sustains a chronic inflammatory state within the TME, thereby enhancing tumor malignancy (124). For example, the chronic inflammation associated with inflammaging can promote cancer initiation and progression by activating STAT5-related pathways, which enhance cell proliferation, expand the cancer stem cell population, and confer resistance to chemotherapy and EMT (48, 125). TNF-α stimulation induces the expression of MMP-9, which, upon activation, acts on numerous inflammatory substrates (126), leading to ECM degradation and remodeling and promoting tumor metastasis (127). Concurrently, various oncogenes are involved in coordinating inflammatory transcriptional programs—including RAS, RAF, protein tyrosine kinases, tumor suppressor proteins, and transcription factors—and the inflammatory responses they mediate are linked to angiogenesis and the recruitment of myeloid-derived monocytic cells (63).
The senescence process partially overlaps with tumorigenesis, as immunosenescence involves the continuous deposition of inflammatory factors and senescent cells (128). Senescent cells activate chronic inflammation via SASP, creating an inflammatory microenvironment (27) that recruits Tregs and MDSCs. This further induces effector T cell senescence and suppresses CD8^+^ T cell function (129, 130). NKG2D ligands released by senescent cells inactivate NK cells, forming a malignant “immune clearance-escape” cycle (131). These processes ultimately lead to the accumulation of senescent cells within the TME, establishing an immunosuppressive microenvironment that accelerates cancer progression (132).
Immunosenescence leads to a dual escape from immunosurveillance: reduced CD8+ T cell cytotoxicity, failure in senescent cell clearance, decreased Tregs activity, and dysregulated immune tolerance (133–135). Increased release of soluble NKG2D ligands by senescent cells causes persistent receptor activation, ultimately leading to NK cell exhaustion and failure of immunosurveillance (131). Immunosenescence also drives resistance to immunotherapy; DCs senescence results in reduced IL-10: interleukin-15 (IL-15) and IFN-α levels, hindering NK cells activation (136). Elevated PD-1/T cell immunoglobulin and mucin domain-containing protein 3 (Tim-3) levels in senescent T cells attenuate immunotherapy responses. This ultimately leads to defective antigen presentation and a T cell exhaustion phenotype (137).
Aging and Immunotherapy
4
Immunotherapy has demonstrated substantial potential in clinical oncology research. However, its feasibility and efficacy in older patients remain critical clinical questions. The capacity of the older population to benefit from immunotherapy may be contingent upon factors such as tumor type.
As shown in Table 1, numerous studies have analyzed the efficacy of immunotherapy in the older population, but their conclusions are partially discrepant due to limitations such as sample or subgroup size, tumor heterogeneity, variable criteria for defining older cohorts, and differing observational endpoints. In 2019, a meta-analysis from the First Affiliated Hospital of China Medical University, encompassing 34 clinical trials on advanced cancer and over 20,000 elderly patients, evaluated data on ICI monotherapy and combination therapy. The findings indicated that while patients in the <65 years and ≥65 to <75 years age brackets derived an OS benefit compared to control groups, the OS benefit for patients ≥75 years was of a shorter duration than that of the control group. A PFS benefit was observed in the <65 and ≥65 age groups, but not in the <75 and ≥75 age groups (161). A meta-analysis by Kasherman et al., including 38 clinical trials on advanced cancer and 10,669 elderly patients over 65, concluded that following ICI treatment, both younger and older patients across different subgroups could achieve similar, age-independent OS benefits (162). A retrospective study by Corbaux et al. on cancer patients receiving ICI monotherapy reported no significant difference in OS or PFS between 150 patients ≥70 years and 185 patients <70 years across various metastatic tumor types (163).
Differential efficacy of immunotherapy in various cancers with aging
4.1
Non-small cell lung cancer and small cell lung cancer
4.1.1
In non-small cell lung cancer (NSCLC), multiple clinical studies have demonstrated that, in both first-line and subsequent-line settings, ICI monotherapy improves PFS (138), objective response rate (ORR) (142), median OS (mOS) (142), and OS duration compared to standard paclitaxel-based or platinum-based chemotherapy (141, 143–146, 164, 165).
However, neither a study by Nokihara of 1,208 patients with locally advanced or metastatic NSCLC using a 75-year age cutoff (775 on standard chemotherapy, 463 on ICI monotherapy) nor other clinical studies using a 65-year cutoff found a significant difference in the efficacy of ICI monotherapy between elderly and non-elderly cohorts (138, 141, 143–145, 165). But a study by Lichtenstein et al. involving 245 NSCLC patients treated with PD-1 and PD-L1 inhibitors suggested a trend toward prolonged PFS with increasing patient age. The risk of disease progression or death was significantly lower for patients aged 70–79 compared to younger patients, but a decline in PFS was observed in patients over 80. Patients aged 60–69 exhibited the longest survival, followed by younger patients. The mortality risk for patients aged 80 or older was significantly higher than for their younger counterparts (139). Similarly, another study of 53,719 patients with advanced NSCLC revealed that despite similar rates of ICI utilization across age groups, survival benefits differed markedly: younger patients with advanced disease derived a clear benefit, whereas a significant benefit was not observed in patients aged 75 and older (140).
Notably, a meta-analysis of randomized controlled trials (RCTs) confirmed that the OS benefit from ICI monotherapy in elderly NSCLC patients (≥75 years) was comparable to that of the general population, and the degree of clinical benefit correlated with PD-L1 expression levels, with patients having a PD-L1 tumor proportion score ≥50% experiencing longer OS (147). However, the use of ICI combinations in NSCLC has yielded slightly different outcomes. One clinical trial indicated that although the proportion of middle-aged and elderly patients in the combination therapy arms was relatively small, the results did not suggest a diminished benefit from combination treatment (164). The IMpower150 study evaluated the efficacy of adding bevacizumab to an ICI-chemotherapy backbone in NSCLC. The results showed that, irrespective of PD-L1 expression, both the three-drug (atezolizumab, carboplatin, paclitaxel) and four-drug (plus bevacizumab) regimens conferred superior OS compared to the bevacizumab-carboplatin-paclitaxel regimen. However, patients ≥75 years did not derive a benefit from the four-drug regimen, a finding presumed to be related to poorer tolerability (146). For small cell lung cancer, Shiono et al. retrospectively analyzed the efficacy of atezolizumab combined with carboplatin and etoposide in 65 patients with extensive-stage disease. The cohort included 36 patients in the elderly group (70–89 years; median age: 74) and 29 in the non-elderly group (43–69 years; median age: 67). The results showed an ORR of 73.8%, with 80.5% in the elderly group, and 65.5% in the non-elderly group. No significant differences were observed in mPFS (5.5 vs. 4.9 months, p = 0.18) or mOS (15.4 vs. 15.9 months, p = 0.24) between the two groups (148).
Digestive tract tumors
4.1.2
In colorectal cancer, older patients, particularly those over 75, exhibit a higher prevalence of tumors with a CpG island methylator phenotype (CIMP-high), microsatellite instability (MSI) or mismatch repair deficiency (dMMR), and v-Raf murine sarcoma viral oncogene homolog B (BRAF) mutations, which suggests a greater potential for benefit from immunotherapy (166). The KEYNOTE-177 study enrolled 307 patients with dMMR/MSI-H metastatic colorectal cancer (mCRC) for first-line therapy. After a median follow-up of 44.5 months, pembrolizumab demonstrated a significant improvement in ORR (43.8% vs. 33.1%) and PFS (16.5 vs. 8.2 months) compared to 5-FU-based chemotherapy. The median OS in the pembrolizumab arm exceeded 4 years, with a significantly reduced risk of death. A subgroup analysis stratified by age 70 (ratio of patients <70 to ≥70 was approximately 2.4:1) revealed no significant difference in clinical benefit between the two age groups (158, 167). Building on this, Saberzadeh-Ardestani et al. retrospectively analyzed the outcomes of ICI monotherapy in 41 similar patients aged >75: the ORR was 49%, disease control rate (DCR) 56%, complete response (CR) rate 32% (7 patients), median PFS 21 months, and OS 36 months, with the overall efficacy trend being consistent with KEYNOTE-177. However, subgroup analysis identified liver metastasis as a negative prognostic factor in this older cohort, with a median PFS of only 6 months (HR = 3.4); whether this phenomenon is also observed in non-older patients requires further investigation (159).
For gastric cancer, in the first-line treatment of human epidermal growth factor receptor 2 (HER2)-negative advanced disease, the ATTRACTION-4 study (N = 724 Asian patients) showed that nivolumab plus chemotherapy significantly reduced the risk of disease progression and prolonged PFS compared to placebo plus chemotherapy, although a statistically significant difference in OS was not achieved (149). The CheckMate 649 study (a global, multicenter trial with >1500 patients with HER2-negative gastric cancer, gastroesophageal junction cancer, or esophageal adenocarcinoma) confirmed that a nivolumab plus chemotherapy regimen conferred benefits in both PFS and OS (150). The discordance in OS results between these two RCTs remains incompletely understood but may be attributable to factors such as population heterogeneity and sample size. Subgroup analyses from both studies suggested that the clinical efficacy in the ≥65 years elderly subgroup was consistent with that of non-elderly patients. However, the subgroup with concomitant peritoneal metastases exhibited a poorer response to ICI-chemotherapy combinations (149). In the realm of HER2-positive advanced gastric cancer, the addition of pembrolizumab to the standard regimen of trastuzumab plus chemotherapy can further and significantly enhance the ORR (168). In the later-line setting for advanced gastric cancer, nivolumab significantly prolonged OS compared to placebo, irrespective of PD-L1 expression status, with no statistically significant difference in clinical benefit observed between elderly and non-elderly patients (151). A post-hoc analysis of the ATTRACTION-2 study data revealed that patients <60 years of age with peritoneal metastases or hyponatremia had a suboptimal clinical response to nivolumab. In contrast, patients ≥60 years of age without peritoneal metastases and with normal serum sodium levels still derived a high degree of clinical benefit (169). These findings, combined with the subgroup analysis from ATTRACTION-4, suggest that peritoneal metastasis in gastric cancer may be an independent negative prognostic factor for immunotherapy, while patient age itself is not the primary determinant of treatment efficacy.
Other malignancies
4.1.3
For head and neck squamous cell carcinoma, older patients, particularly those with recurrent or metastatic disease, can derive benefit from immunotherapy. The Check-Mate 141 trial demonstrated that nivolumab significantly prolonged OS compared to conventional chemotherapy; however, immunosenescence may diminish ICI efficacy, and the survival benefit from PD-1 inhibitors may be attenuated in patients over 75 years of age (170).
In breast cancer, the KEYNOTE-522 study, which included an 11.8% cohort of elderly patients (≥65 years) with triple-negative breast cancer (TNBC), showed that the pathological complete response (pCR) rate was superior with neoadjuvant pembrolizumab plus chemotherapy compared to chemotherapy alone, but the study did not include a dedicated analysis for the older subgroup (152). Preclinical evidence suggests that high levels of tumor-infiltrating lymphocytes (TILs) may correlate with better outcomes from neoadjuvant immunotherapy in TNBC (171). However, the percentage of TILs is known to decrease with age in breast cancer patients, particularly those with TNBC (166). This may be a contributing factor to the diminished immunotherapy efficacy observed in some elderly patients, and targeting this mechanism could potentially improve therapeutic outcomes (172).
For tumors with limited responsiveness to chemotherapy, such as advanced hepatocellular carcinoma and metastatic clear cell renal carcinoma, ICI monotherapy or ICIs in combination with anti-angiogenic agents targeting VEGF/VEGFR has been established as a first-line recommendation (Level I) in national and international guidelines. Age-stratified data from pivotal studies indicate that the treatment response in elderly patients (≥75 years) is comparable to that of the overall population, with a favorable safety profile (153, 154).
In the context of metastatic melanoma, clinical observations suggest that older patients (>60 years) exhibit a more favorable response to PD-1 inhibitors than their younger counterparts, and even nonagenarians can achieve a clinical response to ICIs (155–157). The underlying mechanism may be attributed to the presence of an aging-enriched enterotype (E/AE) in older individuals, which is associated with improved ICIs outcomes. Microbiota derived from the E/AE enterotype have been shown to enhance sensitivity to anti-PD-1 therapy in mice and remodel the TME (173).
Prostate cancer, typically managed surgically, is characterized by a low tumor mutational burden and an immunosuppressive TME, which constrains the efficacy of ICIs and necessitates the development of novel therapeutic strategies to augment the immune response (174). For instance, in metastatic castration-resistant prostate cancer, docetaxel remains the standard of care, and studies have shown that the addition of pembrolizumab did not increase efficacy in patients who had previously received docetaxel (160).
Strategies to sensitize older individuals to immunotherapy
4.2
A significant body of current research is focused on addressing immunosenescence in the context of cancer.
The formation of tertiary lymphoid structures (TLS) is an antigen-dependent process driven by chronic inflammatory signals within non-lymphoid organs. Single-cell analyses have revealed significant heterogeneity in their cellular composition and phenotypic states, which underlies their seemingly contradictory roles in disease. While the presence of TLS is generally associated with favorable clinical outcomes in cancer and infections, it conversely constitutes a hallmark of more severe disease in autoimmune and chronic age-related inflammatory disorders, such as chronic kidney disease (175). Based on this, a key therapeutic strategy for elderly cancer patients is to harness TLS that form within the context of immune aging. This approach aims to counteract the adverse effects of SASP on immune cells, thereby enhancing TLS function and ultimately unleashing their anti-tumor potential. Pharmacologically blocking PD-L1 while simultaneously downregulating the expression of inflammatory cytokines (including TNF-α, IL-1β, and IL-6) can disrupt the immunosuppressive network within the TPI, enhance cytotoxic T-lymphocyte infiltration, and thereby amplify therapeutic efficacy (176). As summarized in Table 2, various pharmacological strategies targeting the senescence-associated microenvironment and tumor-related inflammation have been identified. These agents exert antitumor effects primarily through two core mechanisms. The first involves acting as senolytics, such as ABT-263/Navitoclax (211, 212). These agents eliminate senescent cells within the TME, thereby reversing immunosuppressive traits and restoring the effector function of CD8^+^ T cells (211) to overcome resistance to immunotherapy (211, 213). Second, these agents function as SASP modulators, such as metformin (177), rapamycin (178–180), and biologics targeting critical inflammatory cytokines, particularly IL-6/IL-1β, exemplified by tocilizumab (190–193) and IL-1R antagonists (194).By intervening in signaling pathways including AMPK/mTOR (177), p38 MAPK (182, 183), and NF-κB (184), they inhibit the secretion of protumorigenic inflammatory factors such as IL-6 (177–180, 182, 210) and TNF-α (178–181, 183) by senescent cells, effectively remodeling the immune microenvironment. Furthermore, inhibitors targeting inflammation-associated pathways, including the NLRP3 inflammasome (206–209), COX-2 (245–251), and JAK/STAT (219–223), as well as certain clinically established drugs such as aspirin (229–238), have demonstrated significant potential in modulating both innate and adaptive immunity and enhancing the efficacy of ICIs (200, 211, 212, 214, 260). Currently, while the vast majority of these studies remain in the preclinical phase, select agents such as Navitoclax (211, 212)and tocilizumab (190–193) have already exhibited promising clinical utility in clinical trial.
At the cellular level, aging induces lysosomal dysfunction in activated T cells, which impairs the effective degradation of damaged mitochondria. Instead, these cells release necrotic mitochondria and mitochondrial DNA into the extracellular space via exocytosis, perpetuating the immunosenescence cycle (28). A potential strategy to counter this involves the intercellular transfer of healthy, stem-cell-derived mitochondria to cells with mitochondrial dysfunction, particularly T cells, to restore aerobic respiration, prevent cell death, and recover cellular function (273). Elevated expression of the E3 ubiquitin ligase BFAR in aged individuals inhibits the generation of TRM, thereby suppressing the differentiation of CD8^+^ T cells into the TRM lineage. Targeting this pathway with a small molecule inhibitor of BFAR, iBFAR2, can enhance sensitivity to PD-1 antibody-mediated immunotherapy while mitigating the side effect of systemic inflammation associated with high-dose PD-1 antibodies (31). Inhibiting the formation of PD-L1-containing tEVs, or targeting lipid metabolism or CREB signaling, can reverse T-cell senescence and sensitize tumors to PD-L1 inhibitor therapy (39). Sensitization of aged individuals to immunotherapy can be achieved by targeting the delivery of rejuvenating agents, such as rapamycin, to tumor-draining lymph nodes via a bioorthogonal click chemistry approach, thereby reversing T cell senescence and restoring anti-tumor immunity (274).
Discussion
5
Aging remodels the TME through multiple dimensions—fostering an immunosuppressive, pro-fibrotic, and metabolically abnormal niche—which is a fundamental reason for the high incidence, aggressive biology, and therapeutic resistance of cancer in the elderly. However, this does not preclude older cancer patients from deriving benefit from immunotherapy. Across various tumor types, including NSCLC (141, 143–146, 164, 165) and colorectal cancer (158, 167), older cohorts have demonstrated outcomes equivalent or even superior to those of non-older populations, although some results remain contentious due to limitations in sample size and subgroup characteristics. By contrast, elderly patients with cancers like prostate cancer (160, 174) drive less benefit, highlighting marked inter-tumoral heterogeneity in immunotherapy efficacy. Moreover, the poorer tolerance of combination regimens in the elderly population underscores the application and strategic use of immunotherapy in this demographic remain pressing issues requiring resolution (7).
The partial overlap between aging and tumorigenesis provides a rationale for targeting aging-related mechanisms to enhance anticancer immunity (128). For instance, suppressing chronic inflammation via SASP modulation may delay tumor progression, while improving mitochondrial function in aged T cells could restore their antitumor activity (273). Strategies targeting the senescence-associated microenvironment and SASP have demonstrated significant potential in preclinical studies to remodel the tumor immune landscape (177, 211–213). Nevertheless, their clinical translation, particularly for elderly cancer patients, remains constrained by multiple challenges.
Foremost, the complexity and pleiotropic effects of these mechanisms require rigorous clarification. Senescence is a double-edged sword: while SASP promotes tumors in early stages, it may activate immunosurveillance in specific contexts (211, 212). Currently, the effects of most agents, such as rapamycin (178–180) and dexamethasone (263–269), on immune cells are dose- and context-dependent. Consequently, systemic inhibition of SASP or indiscriminate clearance of senescent cells may disrupt normal tissue repair and immune function (210), thereby introducing unpredictable long-term risks.
Second, clinical translation is limited by methodological constraints. Most evidence derives from mouse models or in vitro assays (181, 213) that cannot recapitulate the physiological decline, inflammaging, and multimorbidity characteristic of elderly hosts (211, 212). The pharmacokinetic and pharmacodynamic profiles of elderly patients differ significantly from the general population (5); thus, combination therapies, such as senolytics combined with ICIs inhibitors (211, 212, 214) or chemotherapy (213) may exacerbate cumulative toxicity, negatively impacting the performance status and quality of life. In the context of current clinical regimens, such as multi-drug combinations of ICIs and chemotherapy for gastric cancer, the tolerability of elderly patients is a critical factor for consideration in clinical practice.
Furthermore, the severe underrepresentation of elderly population in clinical trials (185–193) introduces inherent bias when extrapolating existing evidence to this demographic. Future research should prioritize patient stratification based on senescence biomarkers, such as SASP factor profiling (210), and the design of adaptive clinical trials specifically tailored for elderly cancer patients that incorporate frailty assessments and polypharmacy management (211–213). Future investigations may fruitfully focus on these areas to elucidate the underlying mechanisms, enabling more targeted immunotherapeutic interventions for older cancer patients to improve efficacy and clinical outcomes. Only through the deep integration of senescence biology mechanisms with geriatric clinical practice can targeting senescence be transformed from a laboratory concept into a precision immunotherapy paradigm that benefits elderly cancer patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stead ER Bjedov I . Balancing DNA repair to prevent ageing and cancer. Exp Cell Res. (2021) 405:112679. doi: 10.1016/j.yexcr.2021.112679, PMID: 34102225 PMC 8361780 · doi ↗ · pubmed ↗
- 2Hakim FT Flomerfelt FA Boyiadzis M Gress RE . Aging, immunity and cancer. Curr Opin Immunol. (2004) 16:151–6. doi: 10.1016/j.coi.2004.01.009, PMID: 15023406 · doi ↗ · pubmed ↗
- 3Nikolich-Žugich J . The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. (2018) 19:10–9. doi: 10.1038/s 41590-017-0006-x, PMID: 29242543 · doi ↗ · pubmed ↗
- 4Shan T Ran X Li H Feng G Zhang S Zhang X . Disparities in stage at diagnosis for liver cancer in China. J Natl Cancer Cent. (2023) 3:7–13. doi: 10.1016/j.jncc.2022.12.002, PMID: 39036312 PMC 11256694 · doi ↗ · pubmed ↗
- 5Kadambi S Loh KP Dunne R Magnuson A Maggiore R Zittel J . Older adults with cancer and their caregivers — current landscape and future directions for clinical care. Nat Rev Clin Oncol. (2020) 17:742–55. doi: 10.1038/s 41571-020-0421-z, PMID: 32879429 PMC 7851836 · doi ↗ · pubmed ↗
- 6Wildiers H Heeren P Puts M Topinkova E Janssen-Heijnen MLG Extermann M . International society of geriatric oncology consensus on geriatric assessment in older patients with cancer. J Clin Oncol. (2014) 32:2595–603. doi: 10.1200/JCO.2013.54.8347, PMID: 25071125 PMC 4876338 · doi ↗ · pubmed ↗
- 7Marosi C Köller M . Challenge of cancer in the elderly. ESMO Open. (2016) 1:e 000020. doi: 10.1136/esmoopen-2015-000020, PMID: 27843603 PMC 5070391 · doi ↗ · pubmed ↗
- 8Mogilenko DA Shchukina I Artyomov MN . Immune ageing at single-cell resolution. Nat Rev Immunol. (2022) 22:484–98. doi: 10.1038/s 41577-021-00646-4, PMID: 34815556 PMC 8609266 · doi ↗ · pubmed ↗