A novel compound heterozygous mutation in CYP11B2 (p.A153P and p.Q337*) associated with primary hypoaldosteronism
Jianmei Yang, Lingyu Li, Yan Sun, Jie Jiang, Xiangbo Xie, Weiwei Xu, Chen Chen

TL;DR
A new CYP11B2 gene mutation is linked to primary hypoaldosteronism in a newborn, offering insights into the genetic causes of this rare condition.
Contribution
The study identifies and confirms a novel CYP11B2 mutation (p.A153P) associated with primary hypoaldosteronism, expanding the known genetic spectrum.
Findings
A novel compound heterozygous mutation in CYP11B2 (p.A153P and p.Q337*) was found in a newborn with primary hypoaldosteronism.
The mutation was confirmed to affect protein structure and function, contributing to the disease phenotype.
Treatment with 9α-fluorohydrocortisone normalized biochemical and plasma renin levels within a month.
Abstract
Congenital aldosterone synthase deficiency (ASD), a subset of primary hypoaldosteronism caused by CYP11B2 mutations, is characterized by hyponatremia, hyperkalemia, and elevated plasma renin (with normal cortisol production). This article focused on the clinical and genetic analysis of aldosterone synthase deficiency type II to achieve a deep understanding of the ASD pathophysiology. The clinical biochemical data and whole exome genetic data of a newborn ASD patient were analyzed. The pathogenicity of the novel mutations was predicted using the Result Prediction Software (REVEL). Three-dimensional structures of the mutated gene coded protein was calculated. Steroid hormone level was measured by mass spectrometry. Literatures of all related reports were collected using the HGMD database and Pubmed to analyze the similarity and difference of the novel mutations. The male neonate, born…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
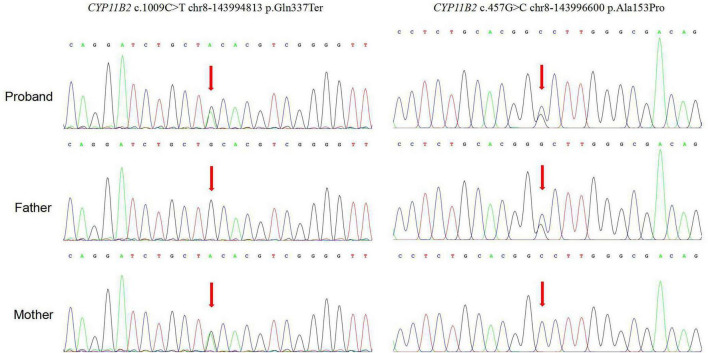 FIGURE 1
FIGURE 1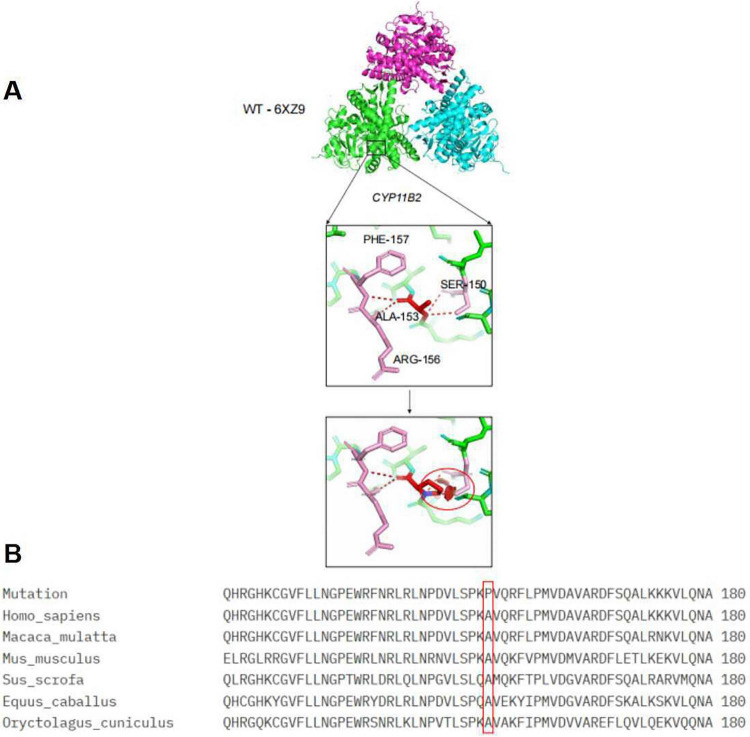 FIGURE 2
FIGURE 2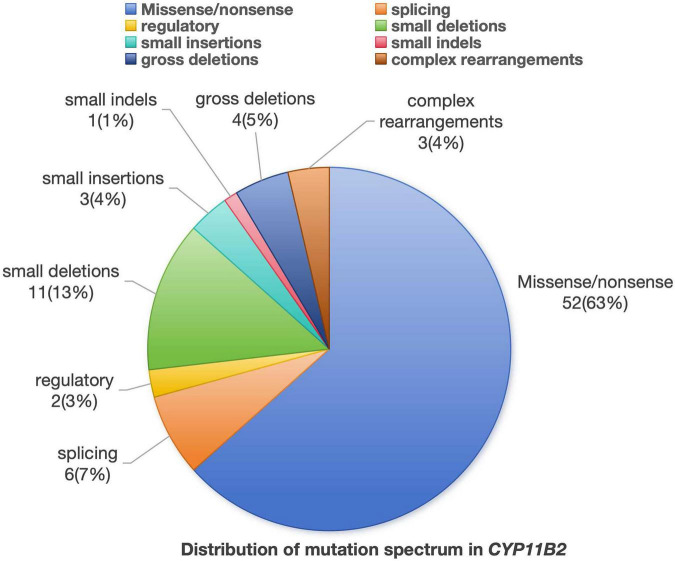 FIGURE 3
FIGURE 3| Patients | Gender | Age of onset | Symptoms | COR (nmol/L) | Renin (pg/mL) | Aldosterone | K (mmol/l) | Na (mmol/l) | Cl (mmol/l) | Gene mutation | Prognosis | PMID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 5 months | Poor feeding, failure to thrive, repeated vomiting, hyponatremia | 274.3 | - | 0.166nmol/l | - | - | - | c.977C > A (p.T326K) | A good long-term prognosis | 24,694,176 |
| 2 | Female | 41 weeks | Poor feeding, failure to thrive | - | - | - | - | – | - | - | 12,788,848 | |
| 3 | Female | 23 days | Poor feeding, failure to thrive, | 18.46 μg/dL | 31 ng/mL/h | 235.4 pg/mL | - | - | - | p.T185I (ACC-ATC) | Good clinical effect on | 32,857,717 |
| 4 | Male | 39 weeks | Vomiting, failure to thrive | 62ng/ml | 3760 ng/L | < 0.03 ng/mL | - | - | - | Homozygous missense mutation c.554C > T(p.T185I) | - | 22,565,077 |
| 5 | Male | 4 weeks | Failure to thrive, vomiting, hypotonia, hyperkalemia, and hyponatremia | - | > 75 ng/mL/h | 3.7 ng/dL | - | - | - | - | - | 6,991,942 |
| 6 | Male | 8 weeks | Failure to thrive, spitting up | 9151 ng/dL | 15.2 ng/mL/h | 19ng/dl | 5.2 | 121 | 93 | - | Poor | 7,485,152 |
| 7 | Male | 1 year 10 months | Failure to thrive, dehydrated | - | - | - | 114 | - | - | Good clinical effect | 2,044,581 | |
| 8 | Male | 3 months | Failure to thrive, infection, hypovolaemic shock | Normal | - | - | 5.4 | 122 | - | - | Good clinical effect | 2,044,581 |
| 9 | Female | 2 weeks | Vomiting, diarrhea, and severe dehydration | 23 μg/dL | - | 1.8 ng/dL | 5.0 | 136 | – | – | – | 3,510,001 |
| 10 | Male | 6 weeks | Failure to thrive, dehydrated | 5.4 μg/dL | - | 6.9 ng/dL | 7.3 | 125 | – | – | – | 3,510,001 |
| 11 | Male | 1 month | Failure to thrive, repeated vomiting | - | 45.5 ng/mL/h | - | 6.0 | 122 | – | – | – | 1,709,913 |
| 12 | Male | 3 months | Infection, hypovolaemic shock | - | - | 130 pmol/L | – | – | – | – | – | 9,625,333 |
| 13 | Male | 10 days | Poor sucking, repeated vomiting | 5.7 μg/dL | 50 ng/mL/h | 6 ng/dL | 5.9 | 127 | – | – | – | 1,601,005 |
| Variant locus | Protein change | HGMDpro database report status | Family origin | GnomAD Frequency (East Asian) | ACMG Evidence | Interpretation conclusion |
|---|---|---|---|---|---|---|
| c.1009C > T | p.Q337 | Reported as pathogenic variant (DM), associated with Aldosterone synthase deficiency, PubMed ID:26936515 | Mother | 0.000163 | PVS1 + PM2_Supporting + PM3 | Pathogenic |
| c.457G > C | p.A153P | No report available | father | 0 | PM2_Supporting + PM3 | Variant of Uncertain Significance (VUS) |
| Steroid profile | Numerical value | Unit | Reference range |
|---|---|---|---|
| Pregnenolone | 372.5 | pg/mL | Not applicable |
| Progesterone | 0.03 | ng/mL | ≤ 0.5 |
| 11-deoxycorticosterone | 298.7 | pg/mL | < 300.0 |
| Corticosterone | 5.06 | ng/mL | 0.18–19.70 |
| 18-Hydroxycorticosterone | 657.49 | pg/mL | 100–6,700 |
| Plasma Aldosterone | 62.2 | pg/ml | ≤ 400.0 |
| 17a-Hydroxypregnenolone | 0.20 | ng/mL | 0.35–7.12 |
| 17-Hydroxyprogesterone | 0.07 | ng/mL | >6.30 |
| 11-Deoxycortisol | 169.5 | pg/mL | <3,440 |
| 21-Deoxycortisol | 0.7 | pg/mL | <50 |
| Cortisol | 35.6 | ng/mL | 50–250 |
| Cortisone | 12.9 | ng/mL | 5.0–39.0 |
| 18-hydroxycortisol | 241.1 | pg/mL | 318–1,666 |
| 18-oxocortisol | 0.3 | pg/mL | Not applicable |
| Dehydroepiandrosterone | 0.03 | ng/mL | < 2.3 |
| Dehydroepiandrosteron e sulfate | 4.1 | ng/mL | 110-1200 |
| Androstenedione | 9.3 | pg/mL | < 690.0 |
| Testosterone | 8.5 | pg/ml | 50.0–4,000.0 |
| Dihydrotestosterone | 3.6 | pg/mL | ≤ 1,200.0 |
| 11-hydroxyandrostenedione | 85.3 | pg/mL | 370–3,508 |
| 11-ketoandrostenedione | 2.7 | pg/mL | Not applicable |
| 11-hydroxytesterone | 4.0 | pg/mL | Not applicable |
| 11-ketoTestoterone | 43.9 | pg/mL | Not applicable |
| Epi-Testoterone | 0.82 | pg/mL | Not applicable |
| Androsterone | 9.61 | pg/mL | Not applicable |
| Epi-Androsterone | 1.47 | pg/mL | Not applicable |
| T/DHT | 2.36 | Not applicable | Not applicable |
| 11OHAD/AD | 9.17 | Not applicable | Not applicable |
| 11KAD/AD | 0.29 | Not applicable | Not applicable |
| 11OHT/T | 0.47 | Not applicable | Not applicable |
| 11KT/T | 5.16 | Not applicable | Not applicable |
| Estrone | 0.0 | pg/mL | ≤ 16.0 |
| β-Estradiol | 3.1 | pg/mL | ≤ 13.0 |
| Estriol | 0.00 | pg/mL | ≤ 180.0 |
| Date | Height (cm) | Weight (kg) | Age (days) | ACTH (pg/ml) | COR (nmol/l) | Renin (pg/ml) | Aldosterone | Creatinine | K (mmol/L) | Na (mmol/L) | Cl (mmol/L) | Therapy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 29/04/2023 | - | - | 0 | - | - | - | 7.43 | 125.2 | 112.2 | - | ||
| 31/05/2023 | - | - | 32 | 18.4 | 124 | - | - | - | 4.6 | 114 | 83.2 | Oral 20% NaCl, ivdrip0. 9%NaCl |
| 02/07/2023 | - | - | 64 | 7.73 | 29 | - | - | - | - | - | - | Hydrocortisone1 + 0.5 mg |
| 08/09/2023 | - | 3.4 | 132 | 13.4 | 68.4 | >500 | 224.52 | 250 | 5.5 | 134 | 103 | Hydrocortisone 1.5+1 +1 mg, 5 mL 0. 9%NaCl q3h |
| 20/10/2023 | - | 3.4 | 174 | 11.3 | 159 | 56.24 | 68.92 | 8.7 | 4.81 | 138 | 106 | Hydrocortisone 1.5 mg, Fludrocortisone 0.033 +0.033 mg |
| 04/01/2024 | 63 | 6.8 | 250 | 30.2 | 251 | 92.73 | 81.76 | 18.7 | 4.83 | 139.4 | 106.8 | Hydrocortisone 1.5 mg, Fludrocortisone 0.033 +0.033 mg |
| 01/03/2024 | 67 | - | 306 | 12 | 271 | 65.72 | 74.87 | 18.2 | 4.76 | 137 | 104 | Hydrocortisone 1 mg, Fludrocortisone 0.033 + 0.033 mg, Oral 0.9% NaCl 10 mL |
| 21/05/2024 | 70 | - | 387 | 12.2 | 319 | 96.65 | 107.32 | 18.9 | 5.08 | 137.1 | 104.7 | Fludrocortisone 0.033 + 0.033 mg |
| 30/07/2024 | 71 | 7.5 | 457 | 78.3 | 155 | 126.7 | 95.37 | 26 | 5.1 | 137.5 | 106.2 | Fludrocortisone 0.033 + 0.033 mg |
| 13/09/2024 | 71 | 7.5 | 502 | 29.3 | 216 | 64.63 | 81.24 | 31.8 | 4.78 | 138.3 | 106.6 | Fludrocortisone 0.033 + 0.033 mg |
| 14/01/2025 | 78 | 9 | 625 | 31.8 | 208 | 3.46 | 68.54 | 21.2 | 4.76 | 141.5 | 108 | Fludrocortisone 0.033 + 0.033 mg |
| 24/06/2025 | 82.5 | 9.3 | 786 | 30.9 | 540 | 140.54 | 176.39 | 28.6 | 4.05 | 137.5 | 102.3 | Fludrocortisone 0.033 + 0.033 mg |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHormonal Regulation and Hypertension · Ion Transport and Channel Regulation · Renin-Angiotensin System Studies
Introduction
1
Congenital aldosterone deficiency (ASD) is a rare genetic endocrine disorder caused by CYP11B2 mutations, which impair aldosterone biosynthesis (1). The incidence rate is less than 1/1,000,000 (2). It was first linked to CYP11B2 defects by Russell in 1963, presenting with neonatal hyponatremia, hyperkalemia, metabolic acidosis, and failure to thrive (1). ASD is classified into two subtypes based on steroid profiles: Type I features undetectable aldosterone, elevated 18-OHDOC, and low 18-OHB; while Type II presents with low aldosterone, normal/high 18-OHB, and a reduced 18-OHB/aldosterone ratio. The current standard treatment for ASD involves lifelong fludrocortisone replacement, dietary sodium supplementation, and regularly monitoring blood electrolytes and pressure to prevent hyperkalemia and hypertension. Consistent with established practice, this case study applied the standard protocol. Notably, the efficacy of standard protocol in a patient with the rare p.A153P CYP11B2 mutation was validated, reinforcing the applicability of current guidelines to this specific population. The diagnosis of ASD is often challenging due to its nonspecific symptoms similar to other pathological conditions, and requires a combination of biochemical tests, imaging studies, and genetic analysis (3, 4). To date, eighty-two CYP11B2 mutation variants have been reported. This study reported a patient with hypoaldosteronism carrying a CYP11B2 compound heterozygous mutations (including a new mutation p.A153P) and explored the association between genotype and phenotype of the case.
Materials and methods
2
Patient and ethical approval
2.1
Ethical approval
2.1.1
The Institutional Human Ethics Review Board at Shandong Provincial Hospital affiliated to Shandong First Medical University approved this study (SWYX:NO.2023-532). The participant or legal guardians of the participant were given a written information to obtain the signed consent to participate in the study. This study conformed to the provisions of the Declaration of Helsinki.
Patient
2.1.2
In 2023, a newborn boy, delivered by emergency cesarean section in Shandong Provincial Hospital. He was admitted due to “low vitality 21 min after birth.” G1P1, gestational age 26^+2^ weeks, extremely preterm, birth weight 800g. After birth, he exhibited weak respiration, with Apgar scores of 4 at 1 min, 6 at 5 min, and 8 at 10 min. The patient experienced severe respiratory distress and required intubation and mechanical ventilation. The parents of this patient were physically healthy and had a nonconsanguineous. The clinical evaluation, baseline and dynamic hormonal levels, and genetic analyses were performed and obtained with signed consent form by the parents.
Clinical observations
2.2
Initial and sequential plasma levels (ionogram and hormonal profile) in patient were measured. Adrenocorticotropic hormone (ACTH), Cortisol (COR), Renin, aldosterone, creatinine, and ionogram (K^+^, Na^+^, Cl^–^) were measured. Laboratory reference ranges were as follows: ACTH: 7.2–63.3 pg/ml, COR: 166–507 nmol/L, Renin: 3.8–38.8 pg/ml, aldosterone: 40–310 pg/ml, creatinine: 19–44 μmol/L, K: 3.5–5.5 mmol/L, Na: 135–145 mmol/L, Cl: 98–110 mmol/L. All hormones were measured by chemiluminescent methods (Roche, Basel, Switzerland) following the manufacturer’s instructions. Renin, aldosterone, creatinine, ionogram (K Na Cl) were measured in the hospital laboratory.
Steroid hormone determination
2.3
Steroid hormone determination was measured using mass spectrometry. Plasma samples were subjected to protein precipitation using methanol, and then the supernatant was purified and enriched for the analytes through solid-phase extraction. Impurities were eluted with n-hexane and 10% acetonitrile, and finally, the analytes were eluted with 90% acetonitrile and pure water, resulting in samples containing the analytes and their internal standards. The processed samples were analyzed for signal collection and quantification using high-performance liquid chromatography-tandem mass spectrometry.
Genome sequencing
2.4
A volume of 3–5 mL of peripheral venous blood was obtained from the proband as well as his parents. The DNA extracted from the peripheral blood underwent whole exome sequencing (WES). Following the manufacturer’s instructions, the exons derived from the genomic DNA of the patient were fragmented, ligated, amplified, and purified. Subsequently, the SeqCap EZ Med Exome Enrichment Kit (Roche NimbleGen) was utilized to capture exons and their adjacent regions from all known genes. Post-capture amplification and purification were performed, after which the DNA library was constructed using the Illumina HiSeq system.
The sequence data were aligned to the human genome reference version 19 (hg19) using NextGene V2.3.4 to ensure adequate coverage and an appropriate depth of mean reads within the targeted regions. Information regarding conserved nucleotide bases and amino acids, as well as the frequencies within normal populations (sourced from the 1,000 Genomes Project, ExAC, dbSNP DNA, and other locus-specific databases), predictions of biological functions, and data from The Human Gene Mutation Database (HGMD), ClinVar, and Online Mendelian Inheritance in Man (OMIM) were gathered through NextGene V2.3.4. Variants were screened in accordance with established guidelines. The pathogenicity of variants was assessed following the criteria set forth by the American College of Medical Genetics (ACMG) for the interpretation of sequence variants, as published in 2015, using the nomenclature established by the Human Genome Variation Society (HGVS) (5).Sanger sequencing was used to verify the variants in the proband revealed by WES, and to test the co-segregation of variants in the family. Genome sequencing was completed in collaboration with GrandOmics.
Bioinformatic analysis
2.5
The spatial structure of the CYP11B2 protein and the affected protein regions after mutations were demonstrated. Prediction of three-dimensional protein structures based on the three-dimensional structure of mutant CYP11B2 was achieved using I-TASSER software (6)^1^ The PyMOL Viewer software was used to visualize the effects of altered residues on the protein structure models. Additionally, we used the following result prediction softwares (REVEL), including SIFT,^2^ PolyPhen2 HVAR,^3^ Mutation Taster,^3^ MutationAssess,^4^ and FATHMM^5^ were used.
Literature comparison
2.6
To comprehensively review the mutations in patients with ASD type II, searches for primary studies of ASD were conducted on PubMed and WANFANG MED ONLINEMEDLINE on June 25, 2025. Mesh terms “aldosterone synthase deficiency type II or aldosterone synthase deficiency type 2” and “CYP11B2 mutation” were applied for the literature search. A total of 13 patients were included, and their general information, biochemistry, genetics, and prognosis were compiled into a table and analyzed (Table 1).
Results
3
Clinical features
3.1
A male neonate, born at 26^+2^ weeks (birth weight 800 g) of gestation via emergency cesarean section, presented with poor respiratory effort and low Apgar scores (scored 4, 6, or 8 at 1, 5, and 10 min after delivery in infant). Initial assessments revealed severe respiratory distress requiring intubation and mechanical ventilation. The infant appeared as a preterm neonate with poor responsiveness, and perioral cyanosis. Under intubation, breath sounds were consistent, with no dry or moist rales noted. Heart beats were strong and regular, with no detected murmurs detected in the valve auscultation areas. The abdomen was soft, with the umbilical cord intact and free of bleeding or discharge. Reduced muscle tone was noted in the limbs, with diminished primitive reflexes.
Laboratory tests and treatment
3.2
The first symptom of the child after birth was hyponatremia, with blood sodium levels of 114 mmol/L, blood potassium levels of 7.43 mmol/L, and blood chlorine levels of 84.4 mmol/L. The child was treated with oral concentrated sodium and intravenous sodium supplementation. After treatment, the effect was poor. At the same time, it was found that cortisol decreased from 124 to 29 nmol/L and ACTH decreased from 18.4 to 7.73 pg/ml. Oral sodium salt was discontinued, and 30 ml of intravenous physiological saline and 1.2 mg of hydrocortisone were administered intravenously once a day. After treatment, the blood sodium level increased to 132 mmol/L, blood potassium level was 5.67 mmol/L, blood chlorine level was 102 mmol/L, cortisol level was 703 nmol/L, and ACTH level was 7.95 pg/ml. After the condition improved, the patient switched to oral physiological saline and acetic acid chloride cortisone tablets. Examination showed that cortisol level decreased to 46.7 nmol/L after 193 nmol/L, and ACTH level increased to 13.4 pg/ml, but the blood sodium level remained stable. After the diagnosis of CYP11B2 mutation through genetic examination, oral saline was stopped and 9a fluoro-hydrocortisone was added. Afterwards, cortisol levels steadily increased to 159 nmol/L, and blood sodium remained stable at 134–139 mmol/L. The original treatment plan was maintained and continued.
Bioinformatic analysis
3.3
The patient had compound heterozygous pathogenic variants in the CYP11B2 gene. The mother was heterozygous for the c.1009C > T chr8-143994813 p.Q337*, which had been previously reported (7) and the father was heterozygous for the c.457G > C chr8-143996600 p.A153P, which was a new pathogenic variant (Figure 1). The pathogenicity of novel mutation c.457G > C was predicted using multiple software tools, with a combined score of pathogenicity (Table 2). A conservation analysis discovered that the 153 rd alanine (A) of the CYP11B2 gene (NP_000489.3) was conserved across different species. The p.A153P (exon3) mutation site affects its polar interaction with serine at position 150, which may affect protein function (Figure 2).
Genetic testing results of the patient’s family.
(A) WT-6XZ9 represents a three-dimensional structural model of the wildtype CYP11B2 protein, designated 6XZ9 in the Protein Data Bank (PDB). CYP11B2 represents the gene. PHE-157, SER-150, ALA-153, ARG-156,the numbers represent amino acid residues of PHE (phenylalanine), SER (serine), ALA (alanine), ARG (arginine). (B) Mutation: indicates that this section displays sequence information related to the ALA-153 mutation. Homo_sapiens: indicates that this sequence originates from humans. Macaca_mulatta: indicates that this sequence originates from macaques (a non-human primate species). Mus_musculus: indicates that this sequence originates from mice (a commonly used model organism). Sus_scrofa: indicates that this sequence originates from wild boars/domestic pigs. Equus_caballus: indicates the sequence originates from horses. Oryctolagus_cuniculus: indicates the sequence originates from European rabbits/domestic rabbits. QHRGHKCGVFLNLNG…. (long string): this is an amino acid sequence (represented by single-letter abbreviations), displaying the primary structure of the CYP11B2 protein fragment corresponding to this species. 180: indicates the amino acid residue number corresponding to the right-aligned end of the amino acid sequence (i.e., residue 180), used for localization.
The c.1009C > T variant carried by the proband has been clearly identified as a pathogenic variant, with strong evidence chains of PVS1 (nonsense variant triggering nonsense mediated mRNA degradation), PM3 (trans distribution with another pathogenic variant), and PM2 (extremely low population frequency). The other variant c.457G > C, although separately classified as a clinically unknown variant (VUS), is trans distributed with pathogenic variants and has supporting evidence for PM2 (population frequency of 0) and PM3 (presence of pathogenic variants in the trans position), which conforms to the rule of “one definite pathogenic and one supporting pathogenic in a compound heterozygous pair.” Final classification conclusion: According to the ACMG guidelines, this complex heterozygous variant combination should be classified as a pathogenic complex heterozygous variant, which can clearly explain the pathogenesis of aldosterone synthase deficiency in the proband (Table 2).
Steroid profiling analyzes
3.4
Plasma steroid profiling was conducted after a 1-week cessation of therapy when the subject was 1 year old (Table 3). The mass spectrometry results indicated that 11-deoxycorticosterone, corticosterone, and 18-hydroxycorticosterone were all within normal or slightly above normal concentration levels for children of the same age group, but the aldosterone level is very low. During the administration of fludrocortisone at a daily dosage of 66 μg, the subject exhibited normal developmental milestones, electrolyte levels, and blood pressure.
Management and follow-up
3.5
This patient was followed up closely since his diagnosis in endocrinology clinics and has demonstrated a favorable clinical and laboratory response to mineralocorticoid therapy. Regular monitoring of ACTH, cortisol, renin, aldosterone, creatinine, and biochemical parameters was implemented to adjust medication dosages accordingly. On May 21, 2024, after cortisol levels returned to normal (319 nmol/L), hydrocortisone acetate was discontinued and only treatment with fluoxetine was maintained. The most recent follow-up on January 14, 2025, showed normal parameters with the infant measuring 78 cm in height and weighing 9 kg. The patient will be followed up in the future to maintain a healthy physiological condition (Table 4). In summary, the patient was treated with 9α-fluorohydrocortisone and sodium supplementation for 1 month, to achieve biochemical maker and plasma renin levels returned to normal. After 3 months, cortisol levels were normalized, and the patient is currently under long-term follow-up schedule.
Literature review
3.6
To review the clinical information and genetic data in patients with ASD type II, reports of primary studies were thoroughly searched in PubMed and WANFANG MED Onlinemedline databases. A total of 13 papers were recorded (8–17), and available clinical information and genetic data were tabulated and analyzed (Table 1).
A total of 82 mutation sites related to the CYP11B2 gene have been reported, including 52 missense/nonsense mutations (63%), 11 small deletions (13%), 6 splicing mutations (7%), 4 gross deletions (5%), 3 complex rearrangements (4%), 3 small insertions (4%), 2 regulatory (3%), and 1 small indels (1%) (Figure 3 and Supplementary Table 1).
All mutation sites related to the CYP11B2 gene.
Discussion
4
In this study, a novel variant in the CYP11B2 gene was identified in a boy of ASD type II. This patient was the second ever reported CYP11B2 gene mutation-ASD type II case in China and the fourteen reported CYP11B2 gene mutation-ASD type II case internationally (5–14). This discovered variant has never been reported in literature. The significant deteriorating property of this novel mutation was verified by bioinformatic analysis. The prognosis of the patient after 9α-fluorohydrocortisone and sodium supplementation treatment was determined in a follow-up regular check showing an excellent recovery in clinic observation and blood hormone and biochemical assay. Such outcome was particularly rare, and this case highlighted the importance of considering Congenital Hypoaldosteronism in the differential diagnosis of infants presenting with such electrolyte imbalances and failure to thrive. Early recognition and appropriate long-term follow-up management were crucial to prevent life-threatening complications associated with ASD, emphasizing the need for heightened awareness among clinicians when evaluating similar cases (8, 15). Current literature indicates that congenital hypoaldosteronism, particularly with CYP11B2 mutations, presents a unique clinical symptoms characterized by electrolyte imbalances, growth failure, and potentially life-threatening adrenal crises (3). The genetic underpinnings of this condition reveal a variety of mutations within the CYP11B2 gene, which is crucial for aldosterone synthesis (18).
ASD is caused by mutations in the CYP11B2 gene located on chromosome 8q.24.3, encompassing nine exons and coding for 503 amino acids. The CYP11B2 gene has evolved from the CYP11B1 gene, with both genes exhibiting high homology within a 40 kb range in the DNA sequence (19). To date, approximately 82 mutations in the CYP11B2 gene have been reported, including missense mutations, nonsense mutations, deletions, insertions, splice mutations, large deletions, and complex rearrangements, with missense and nonsense mutations being the most common (19). CYP11B2 mutations exhibit ethnic and geographic heterogeneity, with p.V386A and p.R181W being prevalent in Iranian Jews, while p.T185I being a hotspot variant among Greeks and Albanians. Reports of ASD cases in Asia are relatively scarce, and no hotspot variants have been identified. Additionally, the CYP11B2 gene shows significant genetic polymorphism (20). This current case presented a novel mutation, highlighting the complexities associated with ASD, specifically congenital aldosterone deficiency due to mutations in the CYP11B2 gene. This condition was of significant interest due to its rarity and the critical implications it holds for neonatal health. The patient’s diagnosis and subsequent management in this report underline the importance of early identification and tailored therapeutic strategies in improving outcomes for affected individuals. Two mutations in CYP11B2 gene in this patient were validated, including: c.1009C > T chr8-143994813 p.Q337*, and c.457G > C chr8-143996600 p.A153P (novel mutation). The A153P variant had not been reported in the Clinvar/ HGMD databases for diseases, and there were also no reports of this variant in the normal population databases, indicating that this variant was rare. Although there is no evidence to support its pathogenicity rating, and it is currently classified as a variant of uncertain clinical significance, the clinical presentation is very consistent in this report. ASD type II (aldosterone synthase deficiency type II) is an autosomal recessive genetic disorder caused by mutations in the CYP11B2 gene leading to aldosterone synthesis disorders. Its onset is directly related to genetic defects and has no causal relationship with preterm birth (21). The disturbed renin angiotensin aldosterone system (RAAS) in premature infants may prevent the mature process and aldosterone levels may be reduced and fluctuated, ASD type II patients have the electrolyte imbalances (such as low sodium and high potassium) as the “symptom overlap.” Aldosterone replacement therapy with dexamethasone as the core, supplemented by oral sodium chloride to correct electrolyte imbalance, emergency management of hyperkalemia complications, and adjustment of treatment plan through long-term biochemical monitoring and imaging evaluation to maintain internal environment stability. This was the same as the treatment for children with this disease. Although the gene test for this patient revealed a paternal-derived A153P variant currently defined as of uncertain significance, this mutation may not be excluded from a pathogenic mutation in conjunction with the patient’s medical history of this case. The identification of novel mutations further emphasized the heterogeneous nature of ASD and the necessity for genetic testing to confirm diagnosis and to guide treatment.
In the process of aldosterone synthesis, 11β-hydroxylase (CYP11B1) catalyzes the conversion of 11-deoxycorticosterone to corticosterone. It provides the necessary precursor for aldosterone synthesis and is an important upstream enzyme in the aldosterone synthesis pathway. Its deficiency may lead to reduced aldosterone synthesis, resulting in related endocrine disorder diseases. Aldosterone synthase (CYP11B2) is the key enzyme in aldosterone synthesis, primarily catalyzing the conversion of corticosterone to aldosterone, and is expressed only in the adrenal zona glomerulosa. The patient’s levels of 11-deoxycorticosterone, corticosterone, and 18-hydroxycorticosterone were all within normal or slightly above normal levels in this patient, but the aldosterone level was very low at 62.2 pg/ml (normal range around 1240 pg/ml). This reflects the improvement in some indicators after treatment with fludrocortisone. The aldosterone synthesis process is influenced by two enzymes, 11β-hydroxylase and aldosterone synthase. The normal concentrations of corticosterone and 18-hydroxycorticosterone in this case indicate that the precursors for aldosterone synthesis were sufficient, but the significantly low aldosterone concentration suggested a deficiency related to a mutation in the CYP11B2 gene that causes a lack of aldosterone synthase.
In managing the patient, the treatment regimen was adjusted based on the genetic findings, transitioning from hydrocortisone therapy to a combination of hydrocortisone and fludrocortisone. This adjustment was particularly notable as it reflected the importance of personalized medicine in the treatment of endocrine disorders, where genetic insights can significantly influenced therapeutic strategies (22). The timely initiation of appropriate steroid replacement therapy was proven effective in alleviating the symptoms associated with adrenal insufficiency and improving the overall quality of patient life. As the patient continues to be monitored, the long-term implications of congenital aldosterone deficiency are becoming clearer. Regular follow-up assessments of hormone levels, growth parameters, and overall health were essential in managing this case and preventing complications. This case illustrated the critical role of continuous monitoring and the potential for positive outcomes with appropriate medical intervention (23).
Future research should focus on expanding our understanding of the genetic variations associated with congenital hypoaldosteronism and their clinical implications. Additionally, studies exploring the psychosocial aspects of living with such a condition, as well as the long-term management strategies, will be invaluable in enhancing clinical practice and patient education. By integrating genetic insights into routine clinical assessments, diagnosis and treatment of neonates suffering from ASD may be improved (24).
Conclusion
5
Aldosterone synthase deficiency (ASD) type II shows an important disease related with hereditary hyperkalemia and hyponatremia. Genetic diagnosis achieves an early confirmation, and continuous treatment follow-up is crucial for optimizing treatment and ensuring long-term health. This current study identifies a novel variant in the CYP11B2 gene, enriching the genetic spectrum of CYP11B2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Turan H Dağdeviren Çakır A Özer Y Tarçın G Özcabi B Ceylaner Set al. Clinical and genetic characteristics of patients with corticosterone methyloxidase deficiency type 2: novel mutations in CYP 11B 2. J Clin Res Pediatr Endocrinol. (2021) 13:232–8. 10.4274/jcrpe.galenos.2020.2019.0216 32539318 PMC 8186340 · doi ↗ · pubmed ↗
- 2Liu X Xie Y Tang J Zhong J Zeng D Lan D. Aldosterone defects in infants and young children with hyperkalemia: a single center retrospective study. Front Pediatr. (2023) 11:1092388. 10.3389/fped.2023.1092388 36726778 PMC 9885047 · doi ↗ · pubmed ↗
- 3Lee S Baranowski E Sakremath R Saraff V Mohamed Z. Hypoglycaemia in adrenal insufficiency. Front Endocrinol. (2023) 14:1198519. 10.3389/fendo.2023.1198519 38053731 PMC 10694272 · doi ↗ · pubmed ↗
- 4Shaikh S Nagendra L Shaikh S Pappachan J. Adrenal failure: an evidence-based diagnostic approach. Diagnostics. (2023) 13:1812. 10.3390/diagnostics 13101812 37238296 PMC 10217071 · doi ↗ · pubmed ↗
- 5Richards S Aziz N Bale S Bick D Das S Gastier-Foster Jet al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. 10.1038/gim.2015.30 25741868 PMC 4544753 · doi ↗ · pubmed ↗
- 6Yang J Yan R Roy A Xu D Poisson J Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. (2015) 12:7–8. 10.1038/nmeth.3213 25549265 PMC 4428668 · doi ↗ · pubmed ↗
- 7Li N Li J Ding Y Yu T Shen Y Fu Qet al. Novel mutations in the CYP 11B 2 gene causing aldosterone synthase deficiency. Mol Med Rep. (2016) 13:3127–32. 10.3892/mmr.2016.4906 26936515 · doi ↗ · pubmed ↗
- 8Gucev Z Tasic V Pop-Jordanova N Riepe F. Aldosterone synthase deficiency type II with hypospadias. Indian Pediatr. (2012) 49:318–20. 10.1007/s 00431-011-1638-822565077 · doi ↗ · pubmed ↗