Viral genome editing methods and applications in the CRISPR era
Kihye Shin, Eui Tae Kim

TL;DR
This review explains how CRISPR technology can be used to edit large DNA viruses like herpesviruses, offering a more efficient and flexible alternative to traditional methods.
Contribution
The paper introduces a practical framework for optimizing CRISPR-based editing of large DNA viruses, including strategies for improving HDR efficiency and clone validation.
Findings
CRISPR enables scarless knock-ins and conditional gene manipulation in essential viral loci.
Optimized CRISPR designs work effectively without the need for complementing cell lines.
CRISPR outperforms traditional BAC recombination for editing clinical isolates of large DNA viruses.
Abstract
CRISPR-Cas systems have transformed viral genetics by enabling precise and efficient manipulation of large DNA virus genomes. This review provides a practical framework for applying CRISPR technology to herpesviruses and other large DNA viruses as an alternative and complement to traditional BAC recombination. Key considerations include nuclease choice; sgRNA design that minimizes cut-to-edit distance and prevents re-cutting; donor template configuration and homology arm length; and synchronized delivery of Cas complexes and donor DNA. Strategies to promote HDR efficiency, such as the use of small-molecule modulators, are also summarized. In addition, practical workflows for clone selection, genotypic validation, and phenotypic confirmation are summarized. Case studies in herpes simplex virus type 1 and human cytomegalovirus illustrate how optimized CRISPR designs achieve reproducible,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
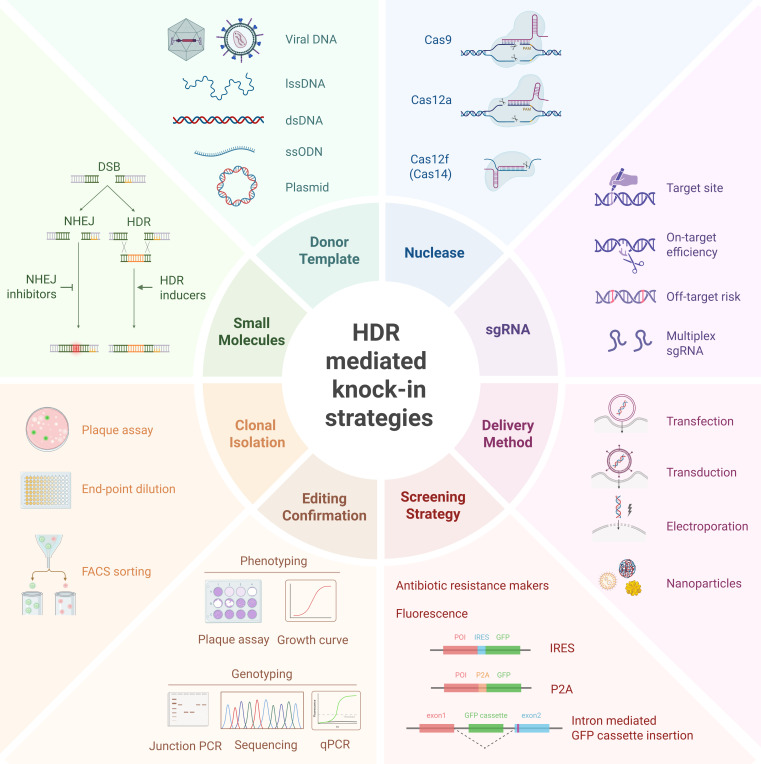 Fig 1
Fig 1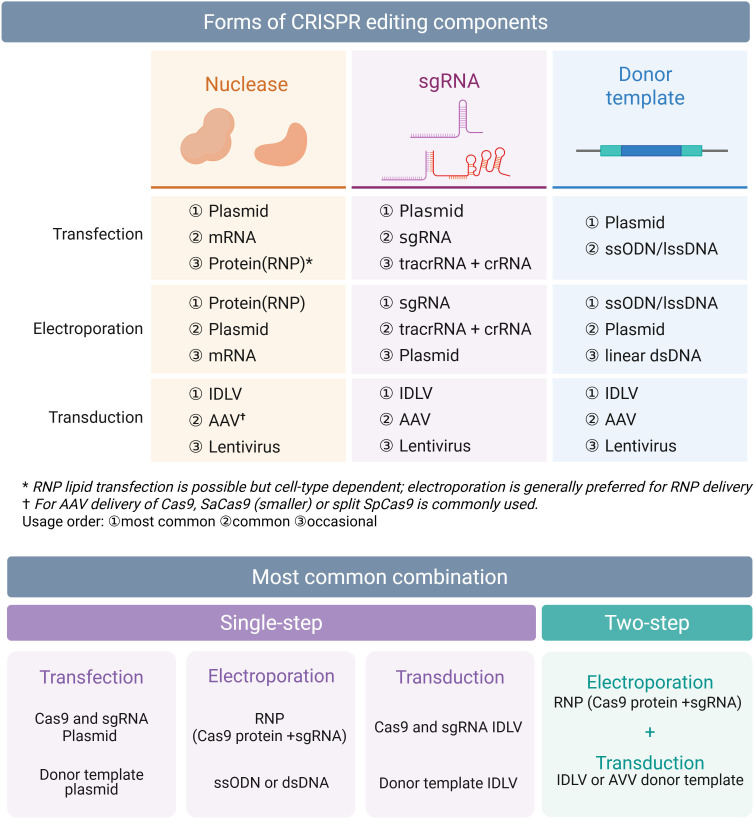 Fig 2
Fig 2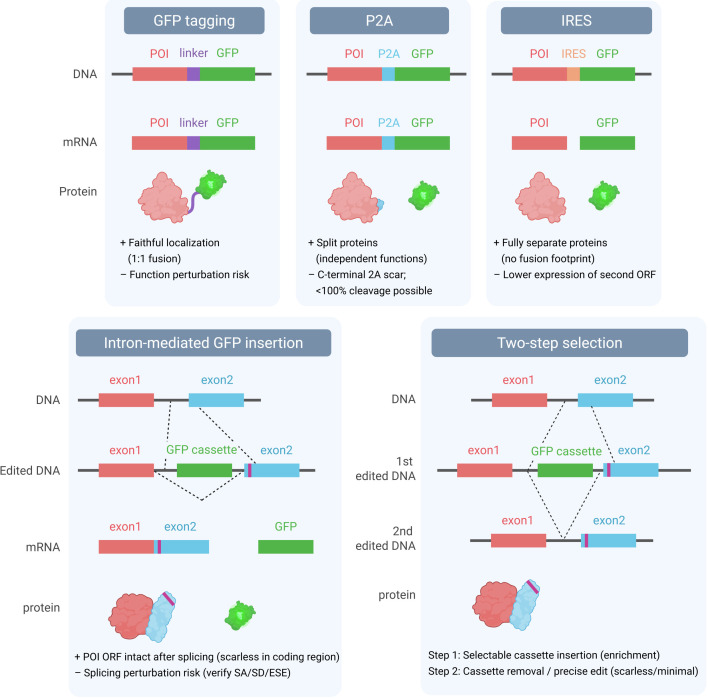 Fig 3
Fig 3| Virus | Target gene/modification | Editing strategy | Delivery of CRISPR editing components | Notes/outcome | Reference |
|---|---|---|---|---|---|
| HSV-1 | gE knockout and revertant | HDR | Co-transfection of Cas9/gRNA and ssODN DNA | First CRISPR editing report in HSV-1 | ( |
| HSV-1 | UL23 with EGFP insertion | HDR | Co-transfection of Cas9/gRNA and donor plasmids | Reporter knock-in | ( |
| HSV-1 | ICP0 knock-in | HDR | Stable Cas9/gRNA 293T cell; donor plasmid transfection | Precise insertion | ( |
| HSV-1 | ICP0 knockout | NHEJ | Transfection of Cas9/gRNA plasmid | Loss-of-function mutant | ( |
| HSV-1 | TdTomato insertion (UL26–UL27) | HDR | Co-transfection of Cas9/gRNA and donor plasmids | Reporter virus | ( |
| HSV-1 | Genome-wide KO (2D/3D) | NHEJ | Transduction of AAV expressing Cas9/gRNA | Editing validated in | ( |
| HCMV | US34 knock-in | HDR | Stable Cas9/gRNA MRC5 cell; donor DNA electroporation | Reporter insertion | ( |
| HCMV | IE1 knockout | NHEJ | Transduction of lentivirus expressing Cas9/gRNA | Functional dissection | ( |
| HCMV | MIEP knockout | NHEJ | Transduction of lentivirus expressing Cas9/gRNA | Promoter deletion | ( |
| VZV | ORF63 knockout | NHEJ | Transduction of lentivirus expressing Cas9/gRNA | Latency-associated gene study | ( |
| DEV | Vaccine strain modification | NHEJ/HDR | Co-transfection of Cas9/gRNA and donor plasmids | Attenuated vaccine | ( |
| Marek’s | V5/GFP tagging | HDR | Electroporation of Cas9/gRNA plasmid and donor DNA | Reporter vaccine strain | ( |
| PRV | Knock-in/dual reporter (GFP, Luc) | HDR | Co-transfection of Cas9/gRNA and donor plasmids | Dual-reporter virus | ( |
| Turkey herpesvirus | Knockout/tagging | NHEJ | Co-transfection of Cas9/gRNA and donor plasmids | Vaccine vector modification | ( |
| EBV | Knockout and knock-in episome | NHEJ/HDR | Co-transfection of Cas9/gRNA and donor plasmids | Functional analysis | ( |
| KSHV | ORF57 knockout (BCBL-1) | NHEJ | Transfection of Cas9/gRNA plasmid | Latency gene editing | ( |
| HBV | Episomal DNA knockout | NHEJ | Transfection of Cas9/gRNA plasmid | Replication inhibition | ( |
| JC polyomavirus | Knockout in 2D/3D models | NHEJ | Transduction of lentivirus expressing Cas9/gRNA | Functional analysis | ( |
| HIV-1 | Provirus knockout | NHEJ | Transduction of AAV expressing Cas9/gRNA | Targeting integrated genome | ( |
| HPV | E7 knockout (plasmid injection) | NHEJ | Nanoparticle-mediated transfection of Cas9/gRNA plasmid | Cervical cancer prevention | ( |
| HPV18 | E6 knockout (AAV-delivered Cas9) | HDR | Transduction of AAV expressing Cas9/gRNA | HeLa model | ( |
| Vaccinia | Genome editing | NHEJ/HDR | Electroporation of Cas9/gRNA and donor plasmids | Smallpox vaccine vector | ( |
| HSV-1 | UL36 tagging | HDR | Co-transfection of Cas9/gRNA and donor plasmids | UL36 essential gene two-step KI using IRES | ( |
| HCMV | IE1/IE2 tagging | HDR | Co-transduction of Cas9/gRNA and donor IDLVs | Inducible degron system knock-in | ( |
| Category | Compounds | Mechanism | Reported effects | Notes | References |
|---|---|---|---|---|---|
| Cell cycle modulators | Nocodazole, Vinblastine, RO-3306 | Arrest cells in G2/M phase, favoring HDR | Increased HDR frequency | May be less effective in viral editing due to asynchronous infection | ( |
| XL413 (CDC7 inhibitor) | Extends S phase duration | Provides longer time window for HDR | Effect is system-dependent | ( | |
| NHEJ inhibitors | NU7026 (DNA-PKcs inhibitor) | Blocks DNA-PKcs kinase activity | 2-3 fold increase in HDR (iPSCs, HEK293, K562) | Moderate enhancement, widely used | ( |
| M3814 (Peposertib) | Inhibits DNA-PKcs activity | Increased HDR in iPSCs and T cells | In clinical testing | ( | |
| AZD7648 | Potent DNA-PKcs inhibitor | >50 fold enhancement in screens | Risk of large deletions, translocations | ( | |
| SCR7 (Ligase IV inhibitor) | Prevents DNA end ligation by LIG4 | Reported suppression of | Variable results, possible cytotoxicity | ( | |
| HDR activators | RS-1 | Stabilizes RAD51 nucleoprotein filaments | Promotes strand invasion and HDR | Variable efficacy across cell types | ( |
| HDAC inhibitors (e.g., TSA) | Increase chromatin accessibility via histone acetylation | Up to 4-fold increase in HDR (iPSCs) | Cytotoxic at higher doses | ( | |
| ATR inhibitors (VE-822) | Block ATR-mediated DDR signaling | Shift repair balance toward HDR | Limited validation in viral editing | ( | |
| CHEK1 inhibitors (AZD-7762) | Inhibit CHK1 checkpoint kinase | Promote HDR by altering DNA damage response | Under investigation, potential toxicity | ( |
- —Korea Health Industry Development Institutehttp://dx.doi.org/10.13039/501100003710
- —Korea Health Industry Development Institutehttp://dx.doi.org/10.13039/501100003710
- —National Research Foundation of Koreahttp://dx.doi.org/10.13039/501100003725
- —National Research Foundation of Koreahttp://dx.doi.org/10.13039/501100003725
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCRISPR and Genetic Engineering · Cytomegalovirus and herpesvirus research · Herpesvirus Infections and Treatments
INTRODUCTION
Conventional viral gene editing
Large DNA viruses, including poxviruses, adenoviruses, and herpesviruses, have long served as important models and platforms for molecular genetics and vaccine development. Their genomes are typically between 100 and 300 kilobases in size and contain many regulatory regions, which make precise genetic manipulation technically challenging.
Before the advent of programmable nucleases, most viral genome engineering relied on in vitro recombination or bacterial artificial chromosome (BAC) cloning systems. These methods enabled targeted modification of several large DNA viruses, such as herpes simplex virus type 1 (HSV-1) and human cytomegalovirus (HCMV), by combining homologous recombination with counter-selection markers. Because of their efficiency and reproducibility, BAC-based strategies became the standard for functional studies across diverse herpesviruses and other large DNA viruses (1–3).
Nevertheless, these methods are labor-intensive, often require complementing cell lines and complex selection procedures and are restricted to strains for which BAC clones have been established. In addition, scarless editing requires extra steps, and residual BAC sequences frequently need to be eliminated to restore the authentic viral genome (4). By contrast, CRISPR-based approaches provide a more direct and versatile platform. Guide RNA and Cas nuclease are sufficient to introduce targeted DNA cleavage within infected cells, allowing precise modification of viral genomes without BAC intermediates. Recent studies have demonstrated that CRISPR can be used to edit the genomes of HSV-1 and HCMV or suppress their replication in infected cells, supporting its potential to meet the growing demands of modern virology and therapeutic development (5–7).
CRISPR-Cas-mediated genome editing
The development of CRISPR and associated endonucleases has transformed genome editing across cellular organisms and viruses alike (8–12). In CRISPR-Cas systems, a guide RNA hybridizes with the target sequence and directs nucleases such as Cas9, Cas12, or Cas13 to the specified locus, resulting in cleavage and subsequent modification of the target nucleic acid. Cas9 and Cas12 target DNA, whereas Cas13 acts on RNA (13, 14).
While this review focuses primarily on DNA virus genome editing, the RNA-targeting Cas13 platform extends CRISPR capabilities to the transcript level, offering complementary strategies for manipulating viral gene expression through RNA knockdown or base editing. In infection models, Cas13 systems have been applied for modification of both viral and host transcripts, particularly targeting RNA viruses such as influenza (15), SARS-CoV-2 (15), dengue virus (16), and HIV-1 (17). These RNA-level interventions may provide advantages in specific contexts, such as transient modulation of viral gene expression or targeting viral transcripts without permanently altering the genomic DNA template. For readers interested in detailed case studies of Cas13 applications, we recommend recent comprehensive overviews (18–20).
The most widely used nuclease is Streptococcus pyogenes Cas9 (SpCas9), which introduces a double-strand break (DSB) at a DNA site defined by a single-guide RNA (sgRNA). In eukaryotic cells, such breaks are mainly repaired by non-homologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ), typically leading to insertions or deletions (indels) and loss-of-function mutations. When a donor template is available, however, homology-directed repair (HDR) can enable precise sequence integration. This dual repair potential allows CRISPR to mediate both gene knockout and precise knock-in, including single-nucleotide substitutions and tag insertions, within the same platform (13, 21–23). Variants that avoid DSB formation, including Cas9 nickase (nCas9), which generates site-specific single-strand nicks, catalytically inactive Cas9 (dCas9) fused to transcriptional effectors for gene repression (CRISPRi) or activation (CRISPRa) (24), and ultracompact nucleases such as CasMINI that enable genome editing with minimal cargo size, further expand system versatility (25–28).
Beyond Cas9, several alternative nucleases have been developed for genome editing. Cas12 family members (Cas12a, Cas12b, and Cas12f) generally recognize T-rich protospacer adjacent motifs (PAMs) and introduce staggered cuts at PAM-distal sites, creating 5′ overhangs (29). These staggered breaks can be advantageous for HDR in certain designs. Because cleavage occurs at a distance from the PAM, early indels often leave the seed region of the protospacer intact, which allows repeated cleavage and progressive deletions through iterative NHEJ cycles until the PAM or protospacer sequence is lost (30). Cas12f, in particular, has been noted for its ability to induce large deletions when paired guides are used, making it suitable for targeted excision strategies (31, 32). The compact Cas12f offers advantages for vector delivery. Recent protein engineering has generated Cas12f variants with substantially improved editing activity in mammalian cells, broader PAM compatibility, and enhanced capacity for paired-guide designs that promote defined deletions (32).
Over the past decade, the application of CRISPR-Cas technology in virology has expanded rapidly. These systems have been applied not only to cellular genomes but also to DNA viruses and integrated DNA intermediates of RNA viruses, supporting diverse purposes including antiviral therapy, functional dissection of viral factors, and engineering of vaccine vectors. The programmable cleavage capacity of CRISPR enables precise modification at defined sites within viral genomes, facilitating gene knockouts, knock-ins, and point mutation corrections with speed and flexibility. Importantly, this approach allows scarless introduction of desired mutations without residual sequences, providing an attractive alternative to BAC-based recombination for the genetic manipulation of herpesviruses and other large DNA viruses.
DNA viral gene editing using CRISPR-Cas9
Over the past decade, the scope of CRISPR-Cas9 applications has expanded rapidly to include not only cellular genomes but also DNA viruses and even integrated DNA intermediates of RNA viruses (33). Major applications in virology include the development of antiviral strategies, functional studies of viral determinants, and engineering of vaccine vectors (34–37). While the underlying principles are similar to those in host genome editing, viral systems present unique opportunities and challenges. During the lytic phase, viral genomes are replicated in large numbers, increasing the likelihood of editing events. Replication compartments in the nucleus provide localized environments enriched in both viral proteins and host DNA repair factors, creating conditions favorable for CRISPR-mediated editing (38, 39). Moreover, virus-specific phenotypic readouts, such as plaque formation or growth curves, enable rapid functional validation, and clonal viral populations can be readily obtained through plaque purification or endpoint dilution.
At the same time, viral genome editing is sensitive to experimental timing. The infection stage, multiplicity of infection (MOI), and the timing of Cas9 and donor template delivery strongly influence editing outcomes. High copy numbers of viral genomes also increase the risk of repeated cleavage at the same locus, which may lead to progressive large deletions or unintended rearrangements. To minimize these events, donor constructs can be designed with silent mutations in the PAM or seed region to prevent re-cleavage. When essential genes are targeted, compensatory mutations or recombination-driven escape variants may arise rapidly; in such cases, conditional expression systems or cellular complementation provide important safeguards.
Previous reviews of CRISPR use in virology have largely focused on vaccine development, therapeutic applications, diagnostics, and host-virus interactions (34, 40–44). Here, we highlight the following practical considerations specific to viral genome editing.
Tool selection: choice of nuclease and sgRNA design based on gene essentiality, PAM availability, target site position (frameshift vs. precise edit), and prevention of re-cutting.Delivery strategy: optimization of RNPs, plasmids, or viral vectors depending on the infection model.Donor design*:* selection of single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA (dsDNA) templates depending on the intended modification and screening approach.Screening and validation: verification of on-target editing by junction PCR or amplicon sequencing; clonal virus isolation by plaque purification or endpoint dilution; genome-wide validation by whole-genome or long-read sequencing to assess deletions or rearrangements; and phenotypic validation by growth kinetics, plaque size, and viral yield. For essential genes, rescue or revertant experiments are recommended to confirm specificity.
CRISPR-based genome editing has now been demonstrated in a variety of DNA viruses, including herpesviruses, polyomaviruses, papillomaviruses, and poxviruses. Reported applications range from targeted knockouts and knock-ins to the generation of vaccine strains. Representative examples are summarized in Table 1.
CONSIDERATION FOR EFFICIENT GENOME EDITING
Efficient genome editing ultimately hinges on steering DNA repair toward the desired pathway. Because NHEJ is active across most cellular contexts, gene knockout is typically achievable with relatively simple design decisions, although the resulting indels are stochastic and limit precision. By contrast, HDR-based knock-in affords precise sequence changes but operates within a narrow window of conditions. Performance depends on nuclease properties (PAM, cleavage pattern, and cut position), donor format and length, strand bias, cut-to-edit distance, silent mutations to block re-cutting, delivery method and timing, and, in some settings, small-molecule modulation. In what follows, we emphasize determinants that most strongly influence knock-in outcomes (Fig. 1).
Overview of HDR-mediated knock-in strategies. Schematic summary of key components influencing HDR efficiency, including nuclease selection, sgRNA design, donor template configuration, delivery methods, screening strategies, and clonal validation approaches.
Selection of CRISPR-Cas proteins
The choice of nuclease should be guided by the editing goal (knockout or knock-in), the availability of PAM sequences near the target locus, and the characteristics of the cut site. SpCas9 is generally the first option, as it is the most widely used nuclease and supported by extensive protocols and validation tools. However, Cas12 family members, particularly compact nucleases such as Cas12f, can serve as useful alternatives when PAM sequences are limited, when multiplex editing is required, or when vector capacity is restricted.
Cas9
SpCas9 is a type II nuclease that recognizes its target through an sgRNA and cleaves dsDNA using its HNH and RuvC domains, typically three nucleotides upstream of the NGG PAM (70). Engineered variants such as SpCas9-NG (recognizing NG PAMs) and SpG/SpRY (capable of near-PAM-less targeting) expand the range of accessible target sites (71). To reduce off-target cleavage, high-fidelity variants (eSpCas9 and SpCas9-HF) and paired nicking strategies using nCas9 are effective for precise editing (72, 73). SaCas9 from Staphylococcus aureus is smaller and recognizes an alternative PAM (NNGRRT), making it particularly useful when vector packaging capacity is limited or PAM availability is constrained (74).
Cas12a (Cpf1)
Cas12a (type V) recognizes T-rich PAM sequences (commonly TTTV) and generates staggered 5′ overhangs at PAM-distal cut sites located approximately 18–23 nucleotides from the PAM. It operates with a crRNA alone and is capable of processing a single RNA transcript into multiple crRNAs, which enables multiplex editing using one promoter (75, 76). Cas12a is advantageous when NGG PAMs are scarce or when simultaneous disruption of multiple loci is required.
Cas12f (Cas14)
Cas12f is a small nuclease (~400–600 amino acids) that has shown improved activity in mammalian cells through protein engineering. Its compact size makes it compatible with packaging into adeno-associated virus (AAV) vectors, which is an important consideration for delivery in viral systems. However, PAM preferences differ between Cas12f variants; hence, locus-specific screening and optimization of guide design are usually required (25, 77).
Selection of sgRNAs
Designing sgRNAs requires balancing on-target activity, off-target risk, and cut placement relative to the desired edit. For knockout, the main goal is to ensure robust cleavage, since even random indels are sufficient to disrupt gene function if they cause frameshifts. For knock-in, however, cleavage must be carefully positioned close to the intended modification site, and donor design must include silent mutations that prevent re-cleavage after HDR.
sgRNAs for knockout
A practical workflow begins with in silico prediction followed by experimental validation. Web-based prediction tools can prioritize candidates based on expected cleavage efficiency and off-target probability (78–82). These scores are useful for initial filtering, but they are not fully reliable, as performance varies by cell type and locus. Final activity must therefore be confirmed experimentally, for example, by in vitro cleavage assays using Cas protein and sgRNA or by amplicon sequencing after transfection (70, 83–85).
When designing sgRNAs for knockout, it is best to target constitutive coding exons shared by all isoforms. Cuts placed within the central portion of such exons increase the likelihood of frameshifts and premature stop codons (70, 80, 86). Guides that cut too far upstream or downstream may allow production of partially functional proteins or the use of alternative start codons. In such cases, targeting near-essential protein domains or using paired sgRNAs to delete critical intervals can improve knockout efficiency.
In rapidly replicating viral systems, editing of essential genes can generate strong growth defects that enrich for escape variants. To avoid misinterpretation, complementation approaches, conditional expression or degradation systems, or the use of multiple sgRNAs to enforce large deletions should be considered.
sgRNAs for knock-in
For knock-in, the distance between the cleavage site and the desired edit is critical because HDR efficiency decreases as this distance increases (87). Therefore, the best strategy is to select sgRNAs that cut as close as possible to the intended insertion or substitution site. After HDR, re-cleavage of the edited allele can occur if the PAM or seed region is left intact. This can be avoided by introducing silent mutations into the donor sequence that disrupt the PAM or seed without altering protein coding (36, 75, 88, 89). If this is not possible, alternative guides or nucleases should be considered. Some studies report that using two sgRNAs can increase knock-in efficiency by promoting larger insertions, although this also increases the risk of off-target effects (90).
Donor templates
Precise knock-in or replacement requires a donor template to be present during the HDR window. Donor type, delivery method, and homology arm design depend on cell type, infection model, and insert size. For the best outcomes, the donor should be available at high nuclear concentration at the time of nuclease activity.
Donor formats
ssODN or long single-stranded DNA (lssDNA): best for small edits such as SNPs or short insertions. Synthesis is straightforward, risk of random integration is low, and asymmetric homology arms can improve HDR efficiency. The main limitation is length, typically ≤200 nt for ssODN in total and a few hundred nt for lssDNA (91–95).Linear dsDNA/plasmid dsDNA: suitable for larger insertions (hundreds of bp to several kb). Linear dsDNA integrates quickly but can undergo unwanted end-joining. Plasmid donors are stable but may persist in the nucleus, necessitating clonal validation. In vivo linearization, achieved by placing Cas target sites near homology arms of the donor template, can synchronize cleavage of both target and donor, improving recombination through HDR or HMEJ (96, 97).AAV: provides high knock-in efficiency across many cell types and can package up to ~4.5 kb. Particularly effective in primary cells and in vivo settings but constrained by packaging limits and production costs (98).IDLV (integrase-deficient lentivirus vector): support the delivery of larger cassettes with lower integration risk compared to standard lentivirus. However, low-level random integration still occurs, requiring plaque purification and sequencing validation (99).Silent mutations: regardless of donor type, silent PAM/seed mutations should be incorporated to prevent re-cutting after HDR.
Homology arm length
ssODN: each homology arm is typically ~30–60 nt (total donor length 100–200 nt), which aligns with the synthesis limit described above.lssDNA: ~150–400 nt per arm (total several hundred nt to ~1 kb).Linear dsDNA/plasmid: 300–800 bp per arm is common, with longer arms supporting more reliable large insertions.Cas9 blunt cuts: knock-in efficiency is highest when edits lie within ±5–10 bp of the cut site.Cas12a staggered cuts: the position of the staggered overhang relative to the edit can influence efficiency.
Delivery methods for CRISPR components
In viral genome editing, efficient delivery is often the limiting step. For HDR-based knock-in, Cas nuclease, sgRNA, donor template, and viral genome must coincide within the same cell and nucleus during the HDR window (100–102). Each delivery method also imposes restrictions on the forms of CRISPR components it can accommodate, and efficiency varies with format (Fig. 2).
Delivery formats and combinations for CRISPR-Cas-mediated genome editing. Summary of typical forms of Cas nucleases, sgRNAs, and donor templates used for transfection, electroporation, or transduction. Common single-step and two-step combinations are highlighted, illustrating optimal configurations for HDR-based viral genome editing.
Recombination is most active shortly after cleavage; hence, the donor should reach peak nuclear concentration at that time. Co-delivery of RNP plus donor by electroporation aligns cut initiation with donor availability and shortens nuclease exposure, which often reduces off-target effects and re-cutting. With plasmid transfection, expression delay can desynchronize cleavage; in this case, donors should be provided concurrently or within 6–12 hours, and in some systems, a second “booster” transfection is beneficial. AAV donors usually reach maximum nuclear levels within several hours to 1 day after infection, while IDLV donors peak later. Thus, MOI and infection timing should be optimized, taking into account the dynamics of viral replication compartments. When IDLV donors are combined with RNP, one effective approach is to perform IDLV transduction first, followed by RNP electroporation about 24 h later (103).
Transfection
Lipid-based transfection remains one of the simplest and most reproducible delivery methods. It is particularly effective in permissive cell lines such as HEK293, HeLa, or Vero, where high transfection efficiency allows straightforward proof-of-concept studies. It is also cost-effective and compatible with both plasmid DNA and donor oligonucleotides. However, its performance is strongly cell line-dependent, and efficiency drops in primary or suspension cells. For HDR, one limitation is that plasmid expression involves a lag period, which can cause misalignment between nuclease cleavage and donor availability. To address this, donors should be supplied at the same time or within 6–12 h, and booster transfections may improve overlap. Recently, commercial reagents have been optimized for direct RNP delivery, increasing editing efficiency and reducing off-target effects (104). Overall, transfection is best-suited for initial optimization, but careful attention to timing is required for knock-in applications.
Electroporation
Electroporation is the most versatile and efficient method for hard-to-transfect cells, including primary cells, monocytes, CD34+ hematopoietic progenitors, and suspension lines. It is especially effective for delivering Cas9 RNP, which allows immediate cleavage upon entry and rapid nuclease degradation, minimizing off-target accumulation (105–109). Electroporation can co-deliver a wide range of donor types (ssODN, lssDNA, linear dsDNA, and plasmid DNA), making it flexible for both small and large knock-in designs (107–109). However, optimization of pulse conditions, buffer composition, and cell density is essential, as excessive stress can reduce viability. The method also requires specialized instrumentation and can become costly in large-scale experiments. Despite these drawbacks, its combination of high efficiency and compatibility with diverse donor forms makes electroporation the preferred strategy for demanding viral editing contexts.
Viral vectors
Viral vectors offer unique advantages when stable or efficient delivery is required. Integrating lentivirus supports long-term Cas9/sgRNA expression, which is useful for screens or systems requiring sustained activity. However, random integration is a concern, especially in therapeutic contexts. IDLV provides transient expression and is widely used for donor delivery. IDLV accommodates larger inserts compared to AAV, and several studies have reported all-in-one IDLV systems where Cas9, sgRNA, and donor are packaged together (99, 110–112). This approach is particularly attractive for editing large viral genomes such as herpesviruses. However, low-level integration events can occur; hence, plaque purification and whole-genome sequencing are recommended for validation. Recombinant AAV (rAAV) has become a gold standard donor vector for precise knock-in. Its ability to deliver single-stranded DNA directly into the nucleus supports high HDR efficiency even in primary cells or in vivo (113–115). In herpesvirus genome editing, rAAV donors have been widely used to introduce tags or deletions that are difficult to achieve with plasmid donors. Recent innovations include capsid engineering (e.g., Y704T variants) to enhance nuclear trafficking and capsid-tethered HDR-promoting factors to improve recombination specificity. Nevertheless, rAAV is constrained by a packaging limit of ~4.7 kb, carries a low but detectable risk of random integration, and may provoke immune responses to viral proteins (116–119).
Other methods
Several additional methods provide niche advantages. Calcium phosphate transfection remains inexpensive and highly reproducible in HEK293 cells, although it is inefficient and often toxic in primary cells (120, 121). Lipid nanoparticles (LNPs) can encapsulate Cas9 mRNA with sgRNA or Cas9 RNP, offering transient expression with low immunogenicity (122, 123). This makes them particularly useful where electroporation is toxic. Cell-penetrating peptides conjugated to Cas9 RNP have enabled direct delivery into sensitive or neuronal cells and are also being tested for base editing and prime editing (124, 125). Magnetofection, which uses magnetic nanoparticles to concentrate editing components on the cell surface, has achieved strikingly high uptake (>99%) and notable knock-in efficiencies (~43%) in iPSC-derived neural progenitors (126–129). Although still experimental, such approaches highlight the diversity of delivery platforms under exploration.
Small molecules to enhance HDR
Cells primarily repair DSBs through NHEJ, whereas HDR is restricted to the S and G2 phases of the cell cycle. Upon DSB induction, DNA damage response pathways are rapidly activated (130). In G1 or M phase, end resection is suppressed, and NHEJ dominates, while in S/G2 phase, end resection is activated, promoting HDR (131).
In host genome editing, cell cycle modulators such as nocodazole, vinblastine, and RO-3306 increase HDR by synchronizing cells in G2/M, when homologous recombination is most active (132, 133). Similarly, XL413, a CDC7 inhibitor, prolongs S phase duration, providing an extended time window for HDR (134). However, these strategies may be less effective in viral genome editing, since infection proceeds asynchronously across the cell population, but they can still be tested empirically.
NHEJ is initiated by the Ku70/Ku80 heterodimer binding to DSB ends, preventing further degradation. This recruits DNA-PKcs, which together form the presynaptic complex. Long-range synapsis is first established, followed by XRCC4-XLF recruitment, which bridges the DNA ends and enables ligation by DNA ligase IV (135, 136). In contrast, HDR initiation depends on recruitment of the MRN complex (MRE11-RAD50-NBS1). MRE11 provides Mn²^+^-dependent endonuclease activity, RAD50 forms a homodimer that constitutes the structural core, and NBS1 coordinates downstream repair proteins. These mechanistic differences explain why blocking NHEJ or promoting HDR can substantially influence editing outcomes.
NHEJ inhibitors
Several small molecules that inhibit NHEJ have been used to enhance HDR. DNA-PKcs inhibitors such as NU7026 (an ATP-competitive inhibitor) and M3814 (peposertib) increase HDR efficiency by approximately 2-fold to 4-fold in systems such as iPSCs, HEK293, K562, and T cells (115, 137–139). High-throughput screens also identified AZD7648, which produced >50-fold enhancement in some assays (140–142). However, potent DNA-PK inhibition has been linked to large deletions, chromosomal arm loss, and translocations, raising concerns about genomic instability (143). Inhibition of DNA ligase IV by SCR7 prevents ligation of DNA ends, thereby blocking NHEJ, although its reported effects are inconsistent across cell types and target loci. Moreover, SCR7 can show dose-dependent cytotoxicity, necessitating careful titration (131, 138, 144–146).
HDR activators
Compounds that directly stimulate homologous recombination have also been explored. RS-1 enhances HDR by stabilizing RAD51 filaments on single-stranded DNA (ssDNA), thereby promoting strand invasion, although the results vary significantly between cell types (138, 147, 148). Chromatin remodeling approaches provide another avenue: histone acetylation relaxes chromatin into a euchromatic state, increasing DNA accessibility. Inhibitors of histone deacetylases (HDACs), such as trichostatin A (TSA), increased HDR up to 4-fold in iPSCs (149). However, HDAC inhibitors are associated with cytotoxicity and apoptosis at higher doses, limiting their general utility. In addition, inhibition of ATR (VE-822) and CHEK1 (AZD-7762) has been reported to alter DNA damage responses in ways that bias repair toward HDR, although their broader application in viral editing has yet to be fully validated (150).
Collectively, these studies demonstrate that HDR efficiency can be modulated by manipulating either the cell cycle state, blocking NHEJ, or directly stimulating HDR pathways. Representative compounds, their mechanisms, and reported outcomes are summarized in Table 2.
SELECTION AND SCREENING METHODS
Fluorescence-based selection markers
Fluorescent reporters are commonly integrated as markers to enrich or identify successfully edited viral genomes. Several strategies have been developed, each with distinct advantages and limitations (Fig. 3).
Representative strategies for fluorescence-based selection in viral genome editing. Comparison of commonly used reporter integration approaches, including direct GFP tagging, P2A or IRES bicistronic expression, intron-mediated GFP insertion, and two-step selection systems for scarless editing.
Internal ribosome entry site (IRES)
IRES elements allow independent translation of two ORFs from a single transcript, thereby preserving the amino acid sequence of the target protein (151, 152). However, translation efficiency is biased toward the upstream ORF, and expression from the downstream ORF is often substantially lower. Thus, a configuration such as GFP-IRES-target gene can compromise expression of the target protein, whereas target gene-IRES-GFP may produce a weak GFP signal and reduce the sensitivity of selection (151, 152).
2A self-cleaving peptides (P2A)
2A peptides, including P2A, enable the production of two nearly equimolar proteins from a single ORF via ribosomal skipping (153). This balanced expression favors reliable selection. The drawback is that 2A sequences leave short peptide scars: additional residues at the C-terminus of the upstream protein and an N-terminal proline on the downstream protein. These extra amino acids can, in some cases, affect protein stability or function (154).
Intron-based insertion strategies
Reporter integration within introns provides an alternative, scarless tagging approach. In the GEIS (gene editing through intron-mediated scarless integration) system, CRISPR induces a cut within an intron adjacent to the target exon, and a fluorescent reporter cassette with its own promoter is inserted by HDR (155). This allows visualization of target gene expression without modifying the coding sequence. Since dsDNA donors frequently lead to random integration or spurious expression, single-stranded DNA donors are preferred. These are often generated by heat-denaturing PCR products, and homology arms of at least ~500 nt are typically required for efficient integration.
Another method, CRISPIE (CRISPR-mediated intron-encoded tagging), inserts a synthetic exon module into an intron (155). Upon splicing, the artificial exon becomes part of the mature mRNA, placing the fluorescent tag in-frame with the endogenous coding sequence. This design enables functional tagging of proteins at either terminus while preserving the surrounding genomic context, offering a flexible and scarless way to monitor protein expression (155).
Two-step selection
When completely scarless editing is required, a two-step strategy can be applied. In the first step, a removable GFP or drug-resistance cassette is inserted by HDR near the target locus to allow the easy selection of positive clones. In the second step, the cassette is precisely removed, leaving only the intended mutation without any additional sequences (156). Although this approach requires two rounds of editing and increases the risk of off-target events, it ultimately provides the advantage of producing fully scarless genomes, which should be validated by whole-genome sequencing.
Beyond fluorescence-based selection, combined positive/negative systems have also been introduced to improve precision. One example is the CD/UPRT-T2A-NPTII cassette, which links neomycin phosphotransferase II (NPTII) for positive selection with cytosine deaminase/uracil phosphoribosyltransferase (CD/UPRT) for negative selection, separated by a T2A sequence to ensure co-expression. This design allows the first round of selection through antibiotic resistance and the subsequent removal of unwanted clones by drug-induced killing, thereby enriching for scarless edited genomes (157).
Clonal purification of edited viruses
After genome editing, the initial viral population is heterogeneous, with both edited and unedited genomes often co-existing in the same infected cell. Clonal purification is therefore essential. Moreover, plaque morphology (e.g., size, growth rate) can bias clone selection and carries a risk of inadvertently enriching recombinants or escape variants. Rigorous purification and quality control are therefore mandatory, typically involving two or more rounds of isolation.
Plaque purification
Plaque isolation on semisolid overlays (agarose or methylcellulose) remains the standard for DNA viruses (158). Fluorescent reporters can accelerate the process by enabling direct selection of positive plaques. Typically, at least two rounds of plaque purification are required to ensure clonality. Overlay composition influences plaque size and number; hence, the conditions must be optimized for each virus species (159).
Cell sorting
When a fluorescent reporter or other detectable marker is available, infected cells can be pre-enriched using flow cytometry. Positive cells are sorted and subsequently subjected to plaque assay or limiting dilution to obtain a clonal virus (160). Reporter-linked editing enables higher clonal recovery rates and shorter timelines. In viruses with weak plaque formation, combining FACS pre-enrichment with end-point dilution is advantageous.
End-point dilution
Sequential dilution ensures that individual wells receive, on average, fewer than one infectious unit. Wells showing cytopathic effect are then expanded. This approach is especially useful for viruses that do not form clear plaques or when overlays are not feasible. Viral titers are quantified in parallel by TCID₅₀, using methods such as Reed-Muench or Spearman-Kärber (161, 162).
Droplet microfluidics
Microfluidic droplet systems encapsulate single infected cells or virions in nanoliter compartments, enabling high-throughput isolation and analysis (163, 164). Although not yet widely applied to herpesviruses, this approach has been successfully demonstrated with other viruses and may provide future avenues for automated single-virus purification.
Selecting appropriate viral purification strategies
For viruses with robust plaque formation (e.g., HSV-1), plaque purification combined with fluorescent reporters is the most efficient. For viruses with weak plaque formation, overlay difficulties, or those requiring primary cells, FACS pre-enrichment followed by end-point dilution is preferred. For high-throughput or rapid processing, droplet microfluidics, potentially combined with AI-driven automation, represents a promising option.
Confirmation of genome editing
After obtaining isolated candidate clones, both genotypic and phenotypic verification steps are required. The main objectives are as follows: (i) to confirm whether the intended on-target modification has occurred, (ii) to exclude unwanted rearrangements, large deletions, or donor backbone retention (off-target events), and (iii) to assess whether the edited virus maintains expected growth and functional properties.
Genotypic assays
On-target editing is commonly verified by junction PCR and Sanger sequencing across the donor-genome junctions to ensure precise recombination. If the design is scarless, the absence of any residual selection cassette should also be confirmed. For quantitative assessment, amplicon-based next-generation sequencing (NGS) can be performed to profile mutation spectra, including HDR efficiency, indel frequency, and frameshift distribution. Mixed sequencing signals indicate heterogeneity within the population and typically warrant additional purification rounds (165).
To evaluate potential off-target effects, the top predicted off-target sites can be amplified and sequenced using targeted amplicon NGS. Depending on the project scale and risk level, long-read sequencing or even whole-genome sequencing may be implemented to identify large deletions, rearrangements, or cryptic integrations that may not be detected by short-read approaches (165).
Phenotypic assays
Phenotypic validation is essential to determine whether the edited virus retains expected replication kinetics and protein function. Standard assays include single-step and multi-step growth curves, plaque size comparison, and viral yield quantification relative to wild-type controls. Changes in viral replication may indicate functional alteration of the target gene. Protein expression and integrity can be analyzed by immunoblotting or immunofluorescence using antibodies specific to the target. When a fluorescent or reporter tag is used, its expression can serve as a direct visual confirmation of successful knock-in or inducible system performance.
APPLICATIONS AND REPRESENTATIVE STUDIES
CRISPR-based genome editing has expanded the experimental and translational scope of herpesvirus research by enabling precise manipulation of large DNA genomes. This technology facilitates functional genomics studies, such as mapping gene essentiality, dissecting protein domains, and probing virus-host interactions, while also advancing applications in vaccine development, antiviral screening, and oncolytic virotherapy. Because herpesviruses possess large genomes with complex regulatory elements, the programmable and modular nature of CRISPR tools makes them particularly suitable for targeted manipulations without global genomic disruption.
Auxin-inducible degron-mediated protein control in HCMV
Editing HCMV in hard-to-transfect cells such as human foreskin fibroblasts (HFFs) poses a major technical challenge. In this study, an IDLV was used to deliver Cas9/sgRNA together with the donor template into infected cells. The donor template was designed with additional Cas9 cut sites at both ends to promote in vivo linearization, thereby enhancing HDR efficiency through improved accessibility and co-localization of donor and target sequences.
An auxin-inducible degron (AID) tag was inserted immediately downstream of the IE1 gene together with a P2A-GFP module, creating a dual-function construct for both visual selection and inducible degradation. In HFF-TIR1 cells, treatment with indole-3-acetic acid (IAA) triggered rapid degradation of the AID-tagged IE1 protein, confirming the feasibility of conditional protein depletion within the viral context. This work established a practical pipeline for applying protein-targeted degradation systems to HCMV without the need for BAC-based recombination, providing a model for posttranslational control of essential viral proteins (69).
CRISPR-based editing strategy for essential genes in HSV-1
Editing essential viral genes requires strategies that maintain viral viability throughout the process. In this example, a two-step knock-in/knock-replacement method was employed to generate a point mutation in the essential UL36 gene of HSV-1.
First, a CMV promoter-eGFP-IRES cassette was inserted upstream of UL36 using HDR, enabling simultaneous expression of eGFP and the essential UL36 protein. This ensured viral propagation even during intermediate editing stages. Cas9 and sgRNA targeting the UL36 N-terminus were delivered along with a donor plasmid containing ~500 bp homology arms. Following transfection into HEK293 cells and infection at MOI 0.5, progeny viruses were passaged with re-targeting sgRNAs to selectively enrich edited genomes.
From this intermediate CMVp-IRES-GFP-UL36 virus, a second round of HDR was performed to remove the GFP cassette and introduce the C40S point mutation, which abolishes the N-terminal DUB activity of UL36. GFP-negative plaques were selected and validated by sequencing.
This strategy offers several advantages. It preserves essential gene expression during editing, eliminates the need for complementing cell lines, reduces time compared with BAC recombination, and enables scarless editing through cassette replacement. The dual-sgRNA design minimizes NHEJ-mediated artifacts, increasing HDR precision. Moreover, the intermediate GFP-UL36 construct can serve as a versatile platform for generating additional UL36 N-terminal modifications (68).
CONCLUSION
This review provides a practical framework for applying CRISPR-Cas systems to DNA viral genome editing, demonstrating their potential as both an alternative and a complementary approach to conventional BAC recombination. The focus was to offer an experiment-oriented guide for improving the precision and efficiency of knockout and knock-in strategies in large DNA viruses. We summarized key design considerations, including the choice of Cas nucleases, optimization of sgRNA design to minimize cut-to-edit distance and prevent re-cutting through silent mutations, donor template configuration and homology arm length, co-delivery timing of Cas complexes and donor DNA, and the use of small molecules to promote HDR. In addition, this review outlined practical approaches for clone selection and validation, encompassing both genotypic and phenotypic assays, to ensure accuracy and reproducibility in viral genome editing.
Two recent case studies illustrate how these design principles can be successfully implemented. In one example, an IRES-based two-step editing strategy was employed to introduce a precise C40S point mutation into the essential HSV-1 UL36 gene while maintaining its expression throughout the editing process. This approach enabled accurate modification of an essential gene without the need for a complementing cell line (68). In another case, an IDLV delivery system was used to introduce Cas9, sgRNA, and donor templates into hard-to-transfect human fibroblasts, resulting in efficient knock-in of an AID-P2A-GFP tag downstream of the HCMV IE1 and IE2 loci. Incorporating Cas9 cut sites at both donor ends and optimizing the Cas-to-donor ratio significantly improved HDR efficiency and reproducibility (69). Together, these examples demonstrate how systematically optimized CRISPR workflows can achieve rapid, precise, and scarless genome editing in large DNA viruses without relying on BAC-based systems. Such developments are expected to accelerate functional genetic studies of clinical strains, elucidate mechanisms of viral pathogenesis, and identify novel therapeutic targets.
PERSPECTIVES
Practical challenges in viral genome editing arise from the intricate relationship between biological constraints and experimental limitations. Editing essential genes often results in nonviable or growth-defective viruses or leads to the rapid selection of adaptive escape mutants. To address this, strategies such as conditional expression or degradation systems, complementing cell lines, and two-step editing designs should be considered at the planning stage to maintain replication competence during genome manipulation.
In the lytic phase, viral genomes replicate multiple copies within nuclear replication compartments, which not only increases opportunities for editing but also raises the risk of cumulative repair events, including large deletions and structural rearrangements. Therefore, careful validation at the junction level is indispensable, and the use of long-read or whole-genome sequencing is strongly recommended before confirming clonal candidates. Editing efficiency is further influenced by cell type, infection stage, MOI, and the timing of Cas nuclease and donor introduction. Because efficient co-delivery of both viral genomes and CRISPR components is technically demanding, optimization must be tailored to each virus-host system.
Although inhibition of the NHEJ pathway can promote HDR, it also introduces the risk of extensive deletions or chromosomal instability. Thus, the use of DNA repair inhibitors requires precise dose optimization and post-editing genome integrity checks. Furthermore, predictive models for sgRNA activity and PAM distribution remain limited for viral genomes, underscoring the need for improved computational tools specialized for viral editing contexts.
Despite these challenges, the unique advantage of CRISPR technology lies in its ability to directly edit clinical isolates without the need for BAC cloning. This capability dramatically enhances experimental speed, accessibility, and adaptability, paving the way for a new era of functional virology. However, CRISPR editing also introduces its own sources of variability, including off-target cleavage, random integration, and background mutations accumulated during viral passaging. Even comprehensive whole-genome sequencing cannot always distinguish these events from naturally occurring mutations.
The most reliable solution is to adopt a revertant-based validation framework that rigorously establishes causality between genotype and phenotype. This includes confirming phenotypic reproducibility across independently edited clones, restoring wild-type function through reversion or complementation, validating on-target junctions and copy numbers by long-read or droplet digital PCR analysis, and employing appropriate negative controls such as scrambled sgRNAs, catalytically inactive Cas variants, or mock-passaged samples. Transparent disclosure of viral passage history and sequencing data will further enhance reproducibility and community confidence.
By integrating these validation standards with the flexibility of CRISPR-based tools, researchers can safely leverage the strengths of the technology, including direct editing of clinical strains, rapid functional interrogation, and scalable design, while maintaining scientific rigor. The continued refinement of CRISPR systems, combined with advances in delivery, bioinformatics, and synthetic virology, will ultimately transform how we manipulate and understand large DNA viruses at the genomic level.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Post LE, Roizman B. 1981. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25:227–232. doi:10.1016/0092-8674(81)90247-66268303 · doi ↗ · pubmed ↗
- 2Domi A, Moss B. 2002. Cloning the vaccinia virus genome as a bacterial artificial chromosome in Escherichia coli and recovery of infectious virus in mammalian cells. Proc Natl Acad Sci USA 99:12415–12420. doi:10.1073/pnas.19242059912196634 PMC 129459 · doi ↗ · pubmed ↗
- 3Messerle M, Crnkovic I, Hammerschmidt W, Ziegler H, Koszinowski UH. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci USA 94:14759–14763. doi:10.1073/pnas.94.26.147599405686 PMC 25110 · doi ↗ · pubmed ↗
- 4Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system, p 421–430. In Braman J (ed), In vitro mutagenesis protocols. Vol. 634. Humana Press, Totowa, NJ.10.1007/978-1-60761-652-8_3020677001 · doi ↗ · pubmed ↗
- 5King MW, Munger J. 2019. Editing the human cytomegalovirus genome with the CRISPR/Cas 9 system. Virology (Auckl) 529:186–194. doi:10.1016/j.virol.2019.01.021PMC 638255130716580 · doi ↗ · pubmed ↗
- 6Amrani N, Luk K, Singh P, Shipley M, Isik M, Donadoni M, Bellizzi A, Khalili K, Sariyer IK, Neumann D, Gordon J, Ruan G-X. 2024. CRISPR-Cas 9-mediated genome editing delivered by a single AAV 9 vector inhibits HSV-1 reactivation in a latent rabbit keratitis model. Mol Ther Methods Clin Dev 32:101303. doi:10.1016/j.omtm.2024.10130339610766 PMC 11602521 · doi ↗ · pubmed ↗
- 7Ying M, Wang H, Liu T, Han Z, Lin K, Shi Q, Zheng N, Ye T, Gong H, Xu F. 2023. CLEAR strategy inhibited HSV proliferation using viral vectors delivered CRISPR-Cas 9. Pathogens 12:814. doi:10.3390/pathogens 1206081437375504 PMC 10302303 · doi ↗ · pubmed ↗
- 8Gwon LW, Badon IW, Lee Y, Kim H-J, Lee SH. 2025. Advances in large-scale DNA engineering with the CRISPR system. Exp Mol Med 57:1902–1912. doi:10.1038/s 12276-025-01530-040887498 PMC 12508223 · doi ↗ · pubmed ↗