Case Report: De novo USP9X missense mutation in a male fetus with pulmonary atresia and ventricular septal defect: expanding the genotype-phenotype spectrum of USP9X-related disorders
Tingting Man, Hairui Sun, Xiaoyan Hao, Xiaowei Liu, Yihua He

TL;DR
A new USP9X gene mutation in a male fetus is linked to severe heart defects, expanding the known effects of this gene beyond brain development issues.
Contribution
First report linking a specific USP9X missense mutation to pulmonary atresia with ventricular septal defect in a male fetus.
Findings
The p.His1729Arg variant in USP9X was identified in a male fetus with PA/VSD.
The variant affects a conserved residue in the zinc-finger domain of USP9X.
This finding expands the phenotypic spectrum of USP9X-related disorders to include CHD.
Abstract
Pathogenic variants in the X-linked USP9X gene, which evades X-chromosome inactivation, have been predominantly linked to neurodevelopmental disorders (NDDs). Accumulating evidence has linked USP9X dysfunction to congenital heart disease (CHD), yet the specific genotype-phenotype correlations in this context remain poorly characterized. Pulmonary atresia with ventricular septal defect (PA/VSD) represents a severe and complex form of congenital heart disease (CHD), characterized by heterogeneous etiological mechanisms. A 34-year-old G2P1L1 woman was referred at 22 weeks of gestation for prenatal echocardiography due to suspected fetal cardiac anomaly. Echocardiographic evaluation identified a male fetus with PA/VSD. Following detailed counseling, the couple elected to terminate the pregnancy due to the poor prognosis and opted for subsequent genetic testing. Trio whole-exome sequencing…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
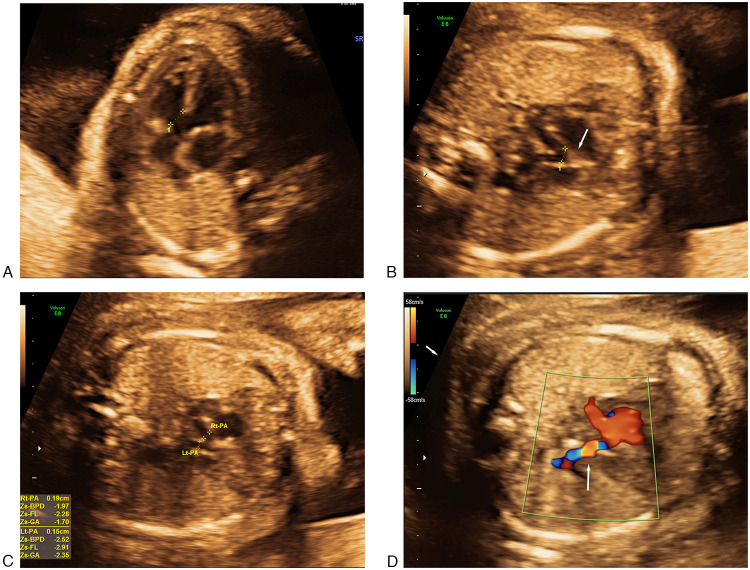 Figure 1
Figure 1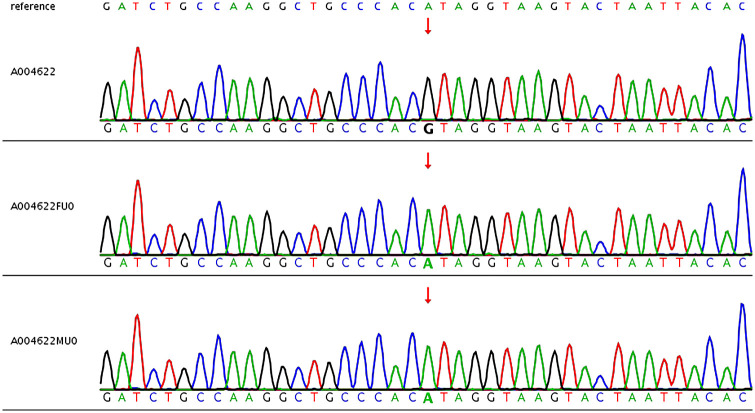 Figure 2
Figure 2| Evidence category | ACMG code | Strength (points) | Evidence description & application |
|---|---|---|---|
| PS2 | Strong (4) | The variant is confirmed as | |
| Functional domains | PM1 | Moderate (2) | The variant is located in the critical CHC2-type zinc-finger motif and affects a conserved zinc-coordinating histidine (His1729), which is essential for protein stability and catalytic integrity. |
| Functional data | PS3 | Supporting (1) | |
| Population data | PM2 | Supporting (1) | The variant is absent from large population databases (gnomAD), indicating extreme rarity (PM2 downgraded to Supporting level per ClinGen SVI V1.0 recommendations). |
| Computational data | PP3 | Supporting (1) | Multiple computational algorithms (SIFT, PolyPhen-2, CADD) predict a deleterious effect on the gene or gene product. |
| Total score | 9 points | Calculation: 4 (PS2) + 2 (PM1) + 1 (PS3) + 1 (PM2) + 1 (PP3) = 9 | |
| Final classification | Likely pathogenic | Posterior Probability > 90% (Consistent with Likely Pathogenic range: 6–9 points) |
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsUbiquitin and proteasome pathways · Congenital heart defects research · Genetics and Neurodevelopmental Disorders
Introduction
1
Pulmonary atresia with ventricular septal defect (PA/VSD) is among the most complex cyanotic congenital heart diseases (CHDs), defined by complete obstruction of the right ventricular outflow tract (RVOT) at the level of pulmonary valve, in conjunction with VSD (1). Its etiology is multifactorial, with established associations to 22q11.2 deletion syndrome (2), yet the majority of cases remain idiopathic. Identifying novel genetic etiologies is pivotal for unraveling disease pathogenesis and enhancing genetic counseling accuracy.
The USP9X gene, mapped to Xp11.4, encodes a deubiquitinase (DUB) that plays a critical role in regulating protein turnover and stability by removing ubiquitin chains (3). Crucially, USP9X evades X-chromosome inactivation, rendering its dosage critical for normal development (3). Pathogenic variants in USP9X are established causes of X-linked neurodevelopmental disorders (NDDs), including X-Linked Intellectual Developmental Disorder 99 (XLID99; OMIM #300919) (5) in males and a more complex syndromic form (MRXS99F; OMIM #300968) (4) in females, both attributed to haploinsufficiency. The recognized phenotypic spectrum encompasses intellectual disability (ID), global developmental delay (GDD), behavioral abnormalities, and structural brain anomalies (5).
While predominantly associated with NDDs, accumulating evidence indicates that USP9X dysfunction can also disrupt cardiac morphogenesis. Cardiac anomalies have been documented as part of the female-specific XLID99 phenotype (5). A recent report (6) further described a male individual with a de novo USP9X missense variant (p.Met1824Val) presenting with complex CHD, including VSD and aortic arch anomalies, in conjunction with NDD features. However, a definitive association between USP9X variants and the severe phenotype of PA/VSD, specifically in males, remains to be established.
Furthermore, the specific variant p.His1729Arg (rs2147230302), categorized as “Likely Pathogenic” for female-associated NDDs in the ClinVar database (VCV001338822) (9), has not been previously reported in conjunction with any CHD phenotype. Here, we report the case of a male fetus with a diagnosis of PA/VSD harboring a de novo hemizygous p.His1729Arg variant in USP9X. This report aims to broaden the known genotype-phenotype landscape of USP9X and furnish novel clinical evidence underpinning its role in molecular pathways governing cardiac outflow tract and septal morphogenesis.
Case presentation
2
A 34-year-old gravida 2, para 1, living child 1 (G2P1L1) woman underwent routine prenatal care. Her prior pregnancy resulted in a healthy full-term female infant, and her medical history was unremarkable. The early pregnancy course was uncomplicated, with the exception of a common cold. First-trimester screening, including Non-Invasive Prenatal Testing (NIPT), revealed low-risk results for common aneuploidies.
A detailed second-trimester routine screening at 22 weeks and 5 days of gestation uncovered severe cardiac anomalies in the male fetus. Fetal echocardiography confirmed the diagnosis of PA/VSD (Figures 1A–D). The main pulmonary artery (MPA) was not identified, and only the pulmonary confluence and right and left pulmonary branches were visualized. The left and right heart exhibited symmetrical proportions, with preserved left and right ventricular function. The atrioventricular valves were within normal limits with regard to both morphology and function. Ultrasound imaging detected no other major structural anomalies. Following multidisciplinary consultation on the severity of the CHD, the necessity for complex multi-stage surgical palliation, and the uncertain long-term prognosis, the parents elected to terminate the pregnancy. The family declined postmortem examination.
Cardiac abnormalities in fetus. (A) Subaortic ventricular septal defect(4.3 mm). (B) Absent main pulmonary artery with confluent central pulmonary arteries. (C) Dysplastic left and right pulmonary artery branches. (D) Reverse blood perfusion of ductus arteriosus.
Genetic analysis
3
Upon obtaining informed consent, fetal tissue collected post-termination and parental peripheral blood samples underwent trio whole-exome sequencing (WES). Library preparation, sequencing, read alignment, variant calling, and annotation were performed using established protocols (7). Candidate variants were validated by Sanger sequencing.
Chromosomal abnormalities were assessed by copy number variation sequencing, and the analysis revealed a normal karyotype without evidence of aneuploidy or pathogenic copy number variations—including 22q11.2 deletion syndrome and other common chromosomal etiologies of congenital heart defects. WES identified a hemizygous missense variant in exon 33 of the USP9X gene (NM_001039590.3): c.5186A > G (Figure 2), predicted to result in a histidine-to-arginine substitution at codon 1729 (p.His1729Arg). This variant (dbSNP ID: rs2147230302) was absent in both parental genomes, confirming its de novo origin in the fetus. The potential pathogenicity of the identified de novo hemizygous missense variant in USP9X, p.His1729Arg (NM_001039590.3: c.5186A > G), was assessed using multiple lines of evidence. This variant is exceedingly rare, as it was not observed in gnomAD, a large-scale population database (8). In clinical significance databases, this variant is annotated in ClinVar (VCV001338822) as “Likely Pathogenic” based on a submission from a single clinical laboratory, which associates it with a female-restricted syndromic neurodevelopmental disorder. In support of its potential pathogenicity, multiple in silico prediction algorithms, including SIFT (9), PolyPhen-2 (10), and CADD (11) (Phred-scaled score: 24.3), predicted a deleterious effect of the p.His1729Arg substitution on protein function (Supplementary Table S1). Furthermore, the His1729 residue is localized to a conserved CHC2-type zinc-finger (ZnF) motif within the catalytic domain of USP9X (15). Considering the critical role of ZnF motifs in maintaining protein tertiary structure and catalytic activity, substitution of the zinc-coordinating histidine residue at this position with arginine is highly predicted to disrupt the local zinc-binding network and compromise the functional integrity of the USP9X catalytic domain.
Sanger sequencing shows the de novo status of USP9X of c.5186A > G in the fetus.
The pathogenicity of the USP9X c.5186A > G variant was systematically evaluated following a Bayesian adaptation of the ACMG/AMP guidelines (12, 13). We integrated the confirmed de novo status (PS2), its localization to a critical region within the zinc-finger motif (PM1), absence from population frequency databases (PM2_Supporting), supportive in silico computational predictions (PP3), and emerging functional evidence indicative of splice-site alteration (PS3_Supporting). A cumulative score of 9 points was derived, leading to a Likely Pathogenic classification (Table 1). Collectively, these convergent lines of evidence strongly support this variant as the genetic etiology underlying the PA/VSD identified in the fetus.
Discussion
4
This case report describes for the first time a novel genotype-phenotype association between the de novo hemizygous USP9X missense variant p.His1729Arg and the severe CHD (PA/VSD) in a male fetus. While USP9X variants have been primarily associated with NDDs (7, 8), our finding substantially advances the understanding of USP9X's pleiotropic roles beyond neurodevelopment, particularly in cardiogenesis.
Previous studies have suggested cardiac involvement in USP9X-related disorders. Cardiac defects are recognized features of XLID99 in females, and Agazzi et al. described a male case with a distinct USP9X variant (p.Met1824Val) presenting with complex CHD, including VSD and aortic arch anomalies (8).
Our case extends this emerging evidence base while distinguishing itself in several critical dimensions. First, this study establishes a mechanistic link between USP9X functional impairment and the specific severe phenotype of PA/VSD, characterized by complete RVOT obstruction, a cardiac feature not previously documented in USP9X-related cases.
Furthermore, this study establishes a specific association between the severe cardiac phenotype and USP9X p.His1729Arg, a variant classified as “Likely Pathogenic” in ClinVar database that was previously exclusively linked to female NDDs. This finding expands the phenotypic spectrum associated with this specific USP9X allele.
The biological plausibility linking USP9X dysfunction to PA/VSD is strongly supported by its role as a deubiquitinase critical for early embryonic cardiogenesis (14); Complete loss-of-function of USP9X is presumably embryonic lethal in males. Surviving males typically carry hypomorphic alleles that cause partial functional impairment (4, 5) The reported p.His1729Arg variant localizes to a zinc-coordinating residue within a ZnF motif, strongly predicting disruption of the zinc-binding network and functional impairment. USP9X has been shown to physically interact with SMAD4 (15), a central transcriptional mediator of the TGF-β and BMP signaling pathways. These pathways are critical for multiple cardiogenic processes, including cardiac neural crest cell migration, outflow tract septation, and cardiac valve formation—developmental events that are fundamentally disrupted in PA/VSD (15). Dysregulation of TGF-β/BMP signaling is a well-documented genetic cause of various CHDs (15). Notably, USP9X evades X-chromosome inactivation, a feature that renders embryonic developmental processes exquisitely sensitive to gene dosage fluctuations. Therefore, partial loss of USP9X function resulting from the hemizygous p.His1729Arg mutation, likely disrupted critical signaling for cardiac development, consequently leading to the observed PA/VSD in this male fetus. Although NDDs could not be evaluated in utero, it is biologically plausible that neurodevelopmental sequelae would have emerged postnatally had the pregnancy proceeded, given the well-established role of USP9X.
Expanding on the functional evidence outlined in our genetic analysis, it is noteworthy that beyond the predicted disruption of the zinc-finger protein domain, emerging functional data suggests a potential impact on transcript processing. An entry in the ClinVar database (Accession: SCV007017646.1), based on RNA-seq analysis, indicates that the c.5186A > G (p.His1729Arg) variant may alter an exonic cryptic donor splice site, leading to significant intron inclusion (p = 0.0004). This finding suggests the variant may exert a deleterious effect via a dual mechanism: impairment of the protein's catalytic structure and induction of aberrant splicing. Both mechanisms converge to elicit a loss-of-function effect, which is consistent with the well-established haploinsufficiency mechanism underlying USP9X-related disorders.
This case underscores the critical need to include USP9X in the genetic differential diagnosis of severe, idiopathic CHD, particularly in male patients or those with syndromic features, even when NDDs are not the primary prenatal presentation. It underscores the critical utility of comprehensive genomic profiling, such as WES, in disentangling novel gene-disease associations within complex developmental disorders. The finding that a USP9X variant previously linked exclusively to female NDDs manifests as severe CHD in a male fetus underscores the gene's phenotypic pleiotropy and highlights potential sex-specific differences in variant expression.
Study limitations include its single-case design and the absence of post-mortem histological analysis or mechanistic validationto to directly assess the impact of p.His1729Arg variant on USP9X-mediated signaling and cardiac morphogenesis. However, while no specific animal models were generated for the present study, our systematic Bayesian analysis (presented in the Results and Table 1) yielded a cumulative score of 9, which firmly classifies this variant as Likely Pathogenic. This designation is supported by multiple independent lines of evidence, including confirmed de novo status, a critical structural impact on the zinc-finger domain, and emerging RNA-seq data indicative of splicing defects.
Conclusion
5
We herein report the first case establishing a de novo p.His1729Arg missense mutation in USP9X as a cause of PA/VSD in a male fetus. This discovery significantly expands the clinical spectrum of this “Likely Pathogenic” variant, and reinforces USP9X's critical role in human cardiogenesis, putatively via modulation of essential developmental pathways like TGF-β/BMP signaling. Our findings underscore the clinical imperative for comprehensive cardiac assessment in individuals harboring pathogenic USP9X variants. Concurrently, we advocate for the inclusion of USP9X in multi-gene diagnostic panels for severe CHD. Future investigations, including large-cohort studies and mechanistic analyses, are essential to fully characterize USP9X genotype-phenotype correlations and dissect the pathomechanisms underlying its role in cardiac development.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hock J Schwall L Pujol C Hager A Oberhoffer R Ewert P Tetralogy of fallot or pulmonary atresia with ventricular septal defect after the age of 40 years: a single center study. J Clin Med. (2020) 9(5):1533. 10.3390/jcm 905153332438748 PMC 7290291 · doi ↗ · pubmed ↗
- 2Skowyra A Allan LA Saurin AT Clarke PR. USP 9X limits mitotic checkpoint complex turnover to strengthen the spindle assembly checkpoint and guard against chromosomal instability. Cell Rep. (2018) 23(3):852–65. 10.1016/j.celrep.2018.03.10029669289 PMC 5917450 · doi ↗ · pubmed ↗
- 3Johnson BV Kumar R Oishi S Alexander S Kasherman M Vega MS Partial loss of USP 9X function leads to a male neurodevelopmental and behavioral disorder converging on transforming growth factor β signaling. Biol Psychiat. (2020) 87(2):100–12. 10.1016/j.biopsych.2019.05.02831443933 PMC 6925349 · doi ↗ · pubmed ↗
- 4Reijnders MR Zachariadis V Latour B Jolly L Mancini GM Pfundt R De Novo loss-of-function mutations in USP 9X cause a female-specific recognizable syndrome with developmental delay and congenital malformations. Am J Hum Genet. (2016) 98(2):373–81. 10.1016/j.ajhg.2015.12.01526833328 PMC 4746365 · doi ↗ · pubmed ↗
- 5Meira JGC Magalhães BS Ferreira IBB Tavares DF Kobayashi GS Leão EKEA. Novel USP 9X variant associated with syndromic intellectual disability in a female: a case study and review. Am J Med Genet A. (2021) 185(5):1569–74. 10.1002/ajmg.a.6214133638286 · doi ↗ · pubmed ↗
- 6Agazzi C Magliozzi M Iacoviello O Palladino S Delvecchio M Masciopinto M Novel variant in the USP 9X gene is associated with congenital heart disease in a male patient: a case report and literature review. Mol Syndromol. (2023) 14(2):158–63. 10.1159/00052742437064340 PMC 10090979 · doi ↗ · pubmed ↗
- 7Sun H Yi T Hao X Yan H Wang J Li Q Contribution of single-gene defects to congenital cardiac left-sided lesions in the prenatal setting. Ultrasound Obst Gyn. (2020) 56(2):225–32. 10.1002/uog.2188331633846 · doi ↗ · pubmed ↗
- 8Karczewski KJ Francioli LC Tiao G Cummings BB Alföldi J Wang Q The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581(7809):434–43. 10.1038/s 41586-020-2308-732461654 PMC 7334197 · doi ↗ · pubmed ↗