Self‐assembled Helical Tetramer Stack of Terrylene Bisimide in Solution and Crystalline State
Simon Soldner, Kazutaka Shoyama, Matthias Stolte, Frank Würthner

TL;DR
A TBI derivative self-assembles into helical tetramers in solution and forms complex structures with guest molecules.
Contribution
The study reveals helical tetramer stacks of TBI and their interaction with aromatic guest molecules in both solution and crystalline states.
Findings
TBI forms helical tetramers via π–π interactions with a strength of ΔG = −40.1 kJ mol−1.
Addition of perylene leads to hexalayer stacks, while coronene forms 1:2 trilayer complexes.
X-ray analysis confirms the stacking arrangements in crystalline states.
Abstract
A terrylene bisimide (TBI) derivative 1 bearing bulky meta‐terphenyl imide substituents is shown to self‐assemble into helical [14] tetramer stacks in solution. Driven by strong dispersion and electrostatic forces, a direct transition from monomers into defined tetramers is already observed in the micromolar concentration range (∼10−6 M) in methylcyclohexane at room temperature, corresponding to a π–π‐interaction strength of ΔG = −40.1 kJ mol−1 for the respective π–π‐stacking interaction between two neighboring TBIs. Upon addition of perylene (P), a further growth of the stacks toward [P.14 .P] hexalayers is observed where the guest molecules are stacked at the peripheral positions. In contrast, upon addition of the larger coronene (C), the equilibrium shifts toward 1:2 complexes [C.1.C] where each TBI is surrounded by two coronene neighbors. Single crystal X‐ray analyses of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
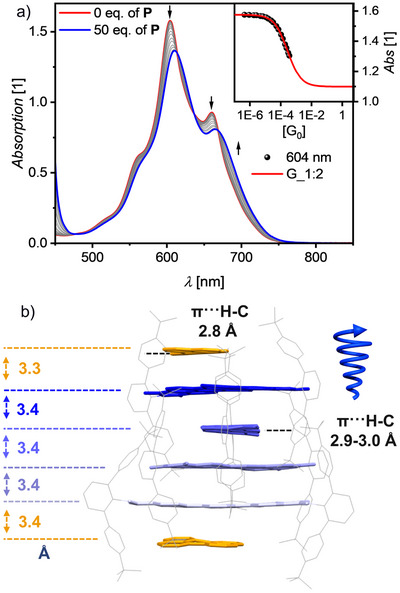 Figure 1
Figure 1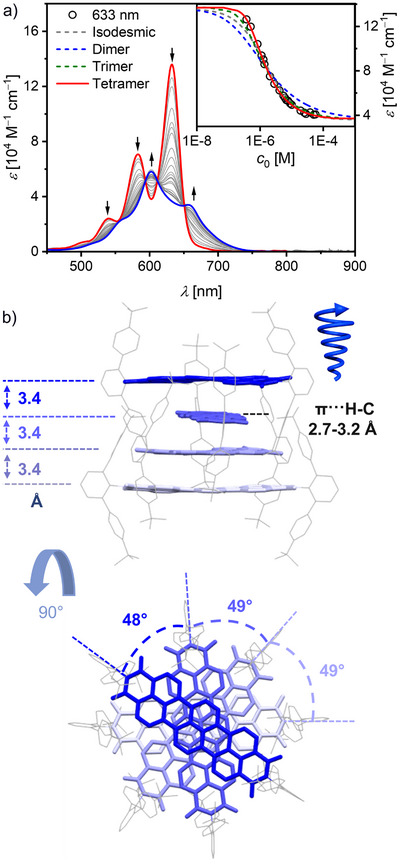 Figure 2
Figure 2 Figure 3
Figure 3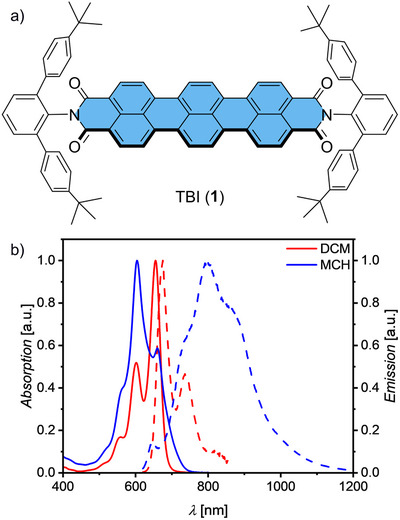 Figure 4
Figure 4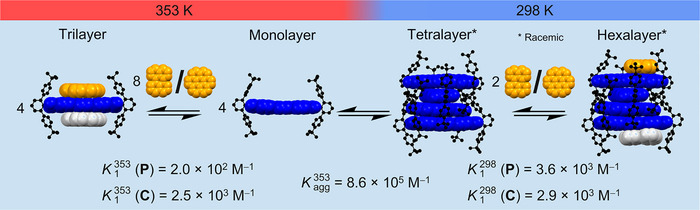 Figure 5
Figure 5- —Solar Technologies go Hybrid10.13039/100012027
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —Deutsches Elektronen‐Synchrotron
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Properties of Aromatic Compounds · Surface Chemistry and Catalysis · Supramolecular Self-Assembly in Materials
After about two decades of intensive research, the understanding of self‐assembly processes has advanced significantly, particularly for supramolecular polymers [1, 2] and metallosupramolecular polygonal structures [3, 4]. However, with the exception of dimers [5], the rationally controlled formation of larger oligomers of defined structure and size, and their use for a subsequent step of hierarchical self‐assembly toward larger nanoobjects [6, 7] remains as a formidable challenge. This is particularly true for flat aromatic π‐systems [8, 9] whose distinct organization on the one hand holds great promise for advanced photophysical functions, like those found in natural light harvesting systems [10], but whose organization is, on the other hand, hampered by the less directional dispersion forces that govern the formation of π‐stacks [11].
Our own interest has for many years been devoted to the organization of rylene dyes in π‐stacked supramolecular architectures with a particular emphasis on perylene bis(dicarboximides) (PBIs). Showing absorption in the visible range, these dyes entered the market as high‐grade color pigments already in the 1950s, which raised the interest in packing structure—color relationships [12], establishing a sound starting base for our supramolecular research in the late 1990s [13, 14]. Among the most striking features of these dyes is that solution self‐assembly of derivatives equipped with solubilizing chains shows a strong preference for rotationally displaced stacking arrangements [15, 16], affording macroscopic helicity [17, 18], whilst for pigments in crystalline systems such helical packing has never been observed. With the exception of a few examples reporting rotationally displaced PBI stacking in the solid state, in which stacks are always present in an alternating M‐/P‐ fashion [19], stacking structures with only translational offsets prevail across the entire range of red, maroon, or black PBI pigments [20]. Also, for the larger homologue terrylene bisimide (TBI), only alternating π‐stacks are reported in the crystalline solid state [21]. Here, we show that the highly desired rotationally stacked packing motif of rylene bis(dicarboximide) dyes can be accomplished with a TBI, leading to a preferential helical arrangement in both solution and crystalline solid state.
For our study we applied a cross‐coupling‐annulation protocol [22] for the synthesis of TBI 1 from peri‐dibromo‐naphthalene imide 2 and 1,4‐diborylated naphthalene 3 (Scheme S1, Supporting Information). Following our previous research on the self‐assembly of various polycyclic aromatic imides leading to a variety of interesting multilayer stacking motifs in solid state [23, 24], however, never rotationally displaced ones, we selected meta‐terphenyl groups equipped with tert‐butyl in para‐position as imide substituents (Figure 1a). The novel TBI 1 was characterized by high‐resolution mass spectrometry (HRMS) as well as ^1^H‐ and ^13^C‐NMR, UV/Vis and fluorescence spectroscopy and X‐ray analysis [25] (for details see Supporting Information). In dichloromethane (DCM) at 298 K the sterically shielded chromophore exhibits the TBI‐typical intense absorption band at λ abs = 655 nm (ε max = 129000 M^−1^ cm^−1^) and a fluorescence band at λ em = 675 nm (Φ f = 57%, τ f = 3.01 ns) with well‐resolved mirror‐image vibronic progressions (Figures 1b and S1,S2 and Table S1) [22]. In contrast, in methylcyclohexane (MCH) the absorption spectrum of TBI 1 significantly broadens and λ abs is strongly hypsochromically shifted to 604 nm even at elevated temperature of 353 K, indicating H‐type exciton coupling between self‐assembled chromophores (c 0 = 5.80 × 10^−5^ M; Figure 2a) [26]. Additionally, the broad and structureless excimer‐like emission spectrum is strongly bathochromically shifted to 797 nm with decreased fluorescence quantum yield (Φ f) of 2.8% and a longest lifetime component of 14.2 ns (Figure 1b and Table S1). To investigate this strong aggregation behavior of the well‐soluble TBI 1 in detail, a concentration (c 0)‐dependent study was performed in MCH at elevated temperature of 353 K where almost the entire transformation from the monomers to the aggregate (Figure 2a) could be covered, different from lower temperatures where even under highly dilute conditions a significant fraction of aggregates prevail (Figure S3).
Different analytical models like the isodesmic [27], dimer, trimer, and tetramer model [28] were applied to deduce the aggregation constant (K agg) as well as to gain insights over the size of the TBI aggregate in MCH. Both local (633 nm) as well as global (800–400 nm) fit analysis indicate that the tetramer model describes the experimental data best (Figures 2a and S4,S5). The resulting K agg per binding site [28] is unusually large at 8.6 × 10^5^ M^−1^ even at an elevated temperature of 353 K. This quantifies a significant increase in π–π‐stacking strength compared to the smaller PBI chromophore with the same meta‐terphenyl imide substituents, where only dimers with Greek cross‐like assembly [29] could be detected in MCH at 298 K with a K agg of 4.3 × 10^4^ M^−1^ [30]. For TBI 1, the larger π‐surface enables the formation of more extended, yet still defined monodisperse aggregates consisting of four π‐stacked chromophores. This tailored self‐assembly of TBI 1 in MCH toward defined tetrameric π‐stacks happens under thermodynamic control as verified by dilution as well as temperature (T)‐dependent UV/Vis‐absorption studies (Figures S6,S7). Accordingly, unlike the case with larger nanographene multiimides [31], no kinetic effects affect the self‐assembly of TBI 1 toward defined tetramers [1_4_].
Single crystals suitable for X‐ray analysis could be grown from solution, which provide proof for the self‐assembly of TBI 1 into discrete tetrameric stacks [1_4_] in accordance with the results from our studies in MCH (Figure 2b and Table S2). Here, surprisingly to us was the unique helical stacking arrangement of the four TBI units with either P‐ or M‐helicity (Figure S8). The neighboring TBI molecules stack on top of each other with about 49° rotational displacement and at π–π‐distances of 3.4 Å, establishing well‐defined P‐ or M‐helical tetralayers. This unique helical tetramer π‐stack of 1 originates from the meta‐terphenyl groups. These groups not only induce a 49° rotational displacement but also limit aggregate growth to four chromophores due to the steric congestion of the bulky imide substituents of the four π‐stacked TBI chromophores (Figure S8c). Interestingly, the two outer π‐surfaces of the tetrameric stack are occupied by π‐stacked chlorobenzene molecules in the single crystal, which demonstrate the accessibility of the chromophore π‐surface. The helical arrangement is also present in solution as circular dichroism (CD) measurements in chiral S‐ or R‐limonene solvent reveal a bisignated Cotton effect at about 615 nm (Figure S9). However, the CD signal remains rather weak, as the self‐assembly in this more polar solvent is not complete and the chiral bias induced by the chiral solvent is only moderate (g abs ∼ 1 × 10^−4^) [32, 33].
Inspired by the chlorobenzene molecules stacked to the peripheral positions of the tetramer stack [1_4_] in our single crystal [34] we envisioned that these binding sites could also host larger PAHs. Therefore, titration studies with perylene (P) and coronene (C) were performed in MCH at 298 K where 1 prevails in its tetramer state even for dilute conditions (c 0(1) ∼ 3 × 10^−5^ M; Figure S3). Spectral changes in the absorption spectra upon addition of these guest molecules indicated binding (Figures 3a and S10,S11 and Table S3). However, due to insufficient solubility of these guests in MCH saturation with two bound guests could not be seen. For the smaller and more soluble guest P a higher degree of complexation could be achieved, leading to values of K 1 = 7750 and K 2 = 1767 M^−1^ in 1:2 binding model (Figure 3a and Table S3).
Fortunately, for this guest we were also able to grow single crystals from chloroform solution at a host–guest ratio of 1:2, demonstrating the formation of the [P^.^1_4_ ^.^P] hexalayer complex by single crystal X‐ray analysis. In this hexalayer, the structure of the original [1_4_] tetramer is preserved (racemic mixture of P and M) with P bound at the two peripheral positions (Figures 3b and S12 and Table S4). Thus, despite containing different guest molecules (chlorobenzene, P), the supramolecular helix of [1_4_] tetramer remains almost unchanged as illustrated by the π–π as well as the C–H···π interactions between the imide substituent and the TBI core in Figure 3b and rotational angles of around 46° between the TBI molecules (Figure S13).
In addition to the π···π interaction between P molecules and TBI, also π···H–C interactions are recognized from P to the TBI imide substituents. Following the helical organization of the TBIs, the peripheral P molecules are arranged with rotational displacements of 54° and 13°, respectively (Figure S12). A geometry optimized structure of [P^.^1_4_ ^.^P] at the GFN2‐xTB level reveals only minor differences compared to the actual crystal structure (Figure S14).
From calculations, an optimized structure could also be obtained for [C^.^1_4_ ^.^C] (Figure S15). However, due to steric encumbrance originating from the tert‐butyl‐functionalized meta‐terphenyl substituents in the [1_4_] stack, the available peripheral cavities have problems in accommodating the two larger C guests, even though the π−π‐interaction of C with a single TBI 1 is increased compared to the smaller guest P. This leads to a situation where under more dilute conditions or at higher temperature a notable difference is observed upon addition of P or C guest molecules. Accordingly, whilst the [1_4_] stack prevails in the presence of P, in case of C the equilibrium shifts toward the 1:2 complex [C^.^1^.^C] (Figures S16–S19) according to Le Chatelier's principle as illustrated in Figure 4.[35] These 1:2 complexes between TBI 1 and C are also the preferred species in chlorinated solvents as demonstrated by a titration experiment (Figure S20) and by crystals grown in CHCl_3_ which show a polylayer assembly of [C^.^1^.^C] with close distances between cofacially stacked C units at 3.3 Å (Figure S21 and Table S5).
In summary, we show that a sterically shielded TBI self‐assembles into helically π‐stacked tetramers with rotational displacements of ∼49°. Due to the high binding affinity between the TBI units this stack prevails even at elevated temperatures in methylcyclohexane and showed the capability to complex polycyclic aromatic hydrocarbons such as perylene (P) and coronene (C) at its periphery, leading to chiral hexalayer π‐stacks. For the case of C, we noticed concentration‐ and temperature‐dependent changes of the equilibrium between the TBI tetralayer stack with bound C and 1:2 complexes of [C^.^1^.^C], enabling the disassembly of the tetramer at higher temperature and lower concentration of TBI 1 according to Le Chatelier's principle. Our future research is directed toward the formation of homochiral π‐stacks both in solution and in the solid state, and the use of the peripheral binding sites for enzyme‐like catalysis [36].
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting File 1: The authors have cited additional references within the Supporting Information.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1P. K. Hashim , J. Bergueiro , E. W. Meijer , and T. Aida , “Supramolecular Polymerization: A Conceptual Expansion for Innovative Materials,” Progress in Polymer Science 105 (2020): 101250, 10.1016/j.progpolymsci.2020.101250. · doi ↗
- 2J. Matern , Y. Dorca , L. Sánchez , and G. Fernández , “Revising Complex Supramolecular Polymerization Under Kinetic and Thermodynamic Control,” Angewandte Chemie International Edition 58 (2019): 16730–16740, 10.1002/anie.201905724.31271244 PMC 6900041 · doi ↗ · pubmed ↗
- 3S. Pullen , J. Tessarolo , and G. H. Clever , “Increasing Structural and Functional Complexity in Self‐assembled Coordination Cages,” Chemical Science 12 (2021): 7269–7293, 10.1039/d 1sc 01226 f.34163819 PMC 8171321 · doi ↗ · pubmed ↗
- 4C. T. Mc Ternan , J. A. Davies , and J. R. Nitschke , “Beyond Platonic: How to Build Metal–Organic Polyhedra Capable of Binding Low‐Symmetry, Information‐Rich Molecular Cargoes,” Chemical Reviews 122 (2022): 10393–10437, 10.1021/acs.chemrev.1c 00763.35436092 PMC 9185692 · doi ↗ · pubmed ↗
- 5G. Wu , F. Li , B. Tang , and X. Zhang , “Molecular Engineering of Noncovalent Dimerization,” Journal of the American Chemical Society 144 (2022): 14962–14975, 10.1021/jacs.2c 02434.35969112 · doi ↗ · pubmed ↗
- 6M. Yoshizawa , J. Nakagawa , K. Kumazawa , et al., “Discrete Stacking of Large Aromatic Molecules Within Organic‐Pillared Coordination Cages,” Angewandte Chemie International Edition 44 (2005): 1810–1813, 10.1002/anie.200462171.15714451 · doi ↗ · pubmed ↗
- 7S. Datta , H. Itabashi , T. Saito , and S. Yagai , “Secondary Nucleation as a Strategy towards Hierarchically Organized Mesoscale Topologies in Supramolecular Polymerization,” Nature Chemistry 17 (2025): 477–492, 10.1038/s 41557-025-01764-5.40164783 · doi ↗ · pubmed ↗
- 8D. Bialas , E. Kirchner , M. Röhr , and F. Würthner , “Perspectives in Dye Chemistry: A Rational Approach Toward Functional Materials by Understanding the Aggregate State,” Journal of the American Chemical Society 143 (2021): 4500–4518, 10.1021/jacs.0c 13245.33719435 · doi ↗ · pubmed ↗