Organophosphides: A New Class of Luminophore Ligands for Copper(I) Carbene Based TADF Emitters and Photocatalysts
Paul C. Ruer, Julian J. Holstein, Andreas Steffen

TL;DR
A new class of copper(I) carbene complexes using dimesityl phosphide ligands shows orange emission and photocatalytic potential for hydrophosphination.
Contribution
Dimesityl phosphide ligands are introduced as a new class of luminophores for copper(I) carbene complexes with enhanced photonic and catalytic properties.
Findings
Dimesityl phosphide ligands cause a bathochromic shift in emission to orange-red and near-infrared wavelengths.
The complexes exhibit thermally activated delayed fluorescence with high quantum yields and dissymmetry factors.
They act as efficient blue light photocatalysts for hydrophosphination of alkynes in solution.
Abstract
Luminescent carbene copper(I) charge transfer complexes are promising candidates as molecular materials for photonic applications. Apart from steric and electronic modification of the acceptor carbene, most of the research has been dedicated to amide donor ligands to control the luminescence properties, while the remaining pnictogen group as anionic electron donating ligands is photophysically underrepresented. Herein, we demonstrate that dimesityl phosphide (Mes2P–) as a heavier homologue in [Cu(cAAC)(PMes2)] (cAAC = cyclic amino(alkyl) carbene) gives rise to orange emission with quantum yields of up to ϕ max = 0.52 in the solid state that is bathochromically shifted by ∼3000 cm−1 in comparison to related amide complexes due to the lower electronegativity of phosphorus. Time‐resolved variable temperature studies reveals that the µs‐lifetimes and radiative rate constants of ca. k r =…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 SCHEME 1
SCHEME 1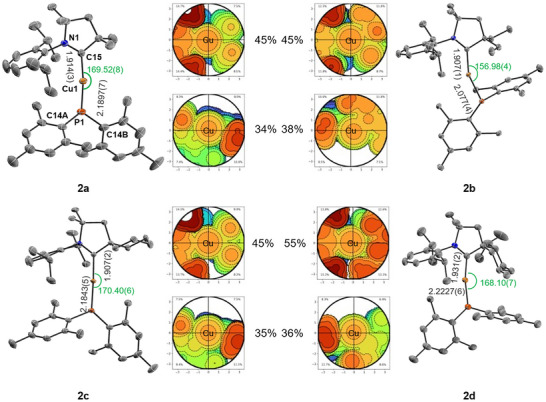 FIGURE 1
FIGURE 1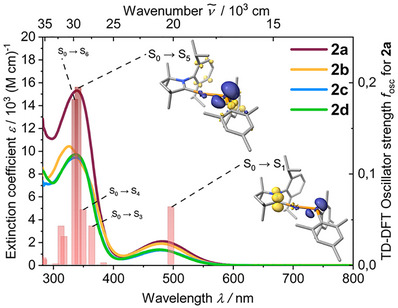 FIGURE 2
FIGURE 2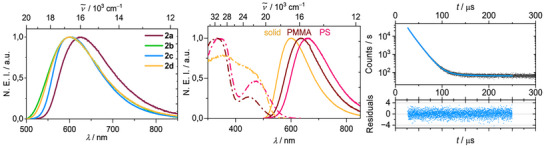 FIGURE 3
FIGURE 3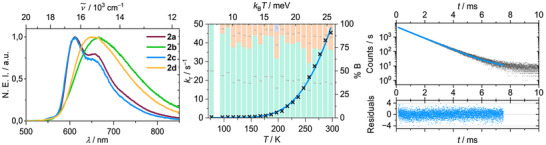 FIGURE 4
FIGURE 4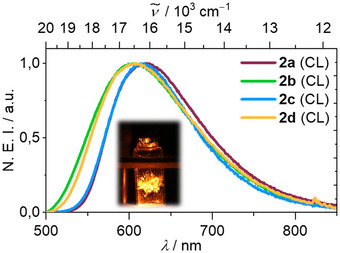 FIGURE 5
FIGURE 5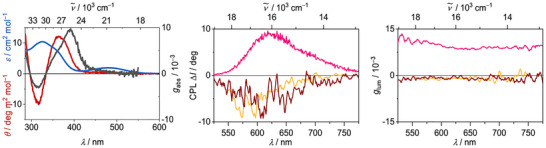 FIGURE 6
FIGURE 6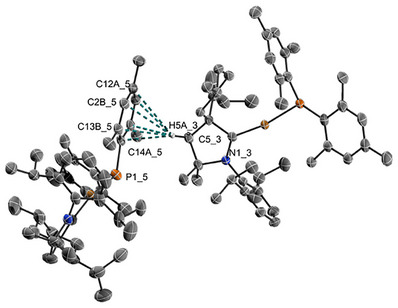 FIGURE 7
FIGURE 7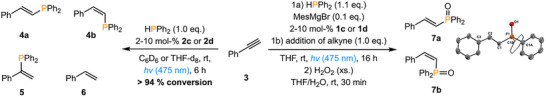 SCHEME 2
SCHEME 2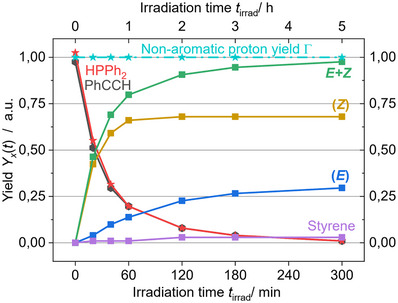 FIGURE 8
FIGURE 8| Cpd. | Medium |
|
|
| < | < | Cpd. | Medium |
|
|
| < | < |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| solid state | 297 | 626 | 0.47 | 7.91 | 59.0 |
| solid state | 297 | 600 | 0.52 | 12.0 | 43.3 |
| 77 | 612 | 0.35 | 1085 | 0.32 | 77 | 610 | 0.39 | 1079 | 0.36 | ||||
| PS | 297 | 682 | 0.04 | 1.51 | 26.5 | PS | 297 | 677 | 0.05 | 2.55 | 19.6 | ||
| PMMA | 297 | 635 | 0.07 | 2.56 | 27.3 | PMMA | 297 | 655 | 0.07 | 2.79 | 25.1 | ||
| toluene | 297 | 785 | n/d | n/d | n/d | toluene | 297 | 804 | 0.001 | n/d | n/d | ||
|
| solid state | 297 | 601 | 0.30 | 16.5 | 18.8 |
| solid state | 297 | 602 | 0.45 | 13.5 | 31.1 |
| 77 | 652 | 0.14 | 264 | 0.53 | 77 | 650 | 0.31 | 433 | 0.72 | ||||
| PS | 297 | 679 | 0.01 | 1.62 | 6.17 | PS | 297 | 660 | 0.05 | 5.99 | 8.3 | ||
| PMMA | 297 | 655 | 0.07 | 1.63 | 42.9 | PMMA | 297 | 636 | 0.07 | 8.15 | 8.5 | ||
| toluene | 297 | n/d | n/d | n/d | Toluene | 297 | 789 | 0.001 | n/d | n/d |
| Cat. | Loading | Solvent | Conversion (HPPh2) / % | NMR‐Yield / % | |||
|---|---|---|---|---|---|---|---|
| 4a ( | 4b ( | 5 ( | 6 | ||||
|
| 10 mol‐% | THF‐d8 | 98 |
| 10 | 0 | 5 |
| 10 mol‐% | C6D6 | 98 |
| 2 | 0 | 3 | |
| 5 mol‐% | THF‐d8 | 94 |
| 31 | 0 | 4 | |
| 2 mol‐% | THF‐d8 | 99 | 28 |
| 0 | 3 | |
| 2 mol‐% | C6D6 | 98 | 28 |
| 0 | 3 | |
|
| 2 mol‐% | THF‐d8 | 97 | 11 |
| 1 | 2 |
|
|
| ||||||
|
| 10 mol‐% | THF |
| 21 | |||
| 5 mol‐% | THF |
| 25 | ||||
| 2 mol‐% | THF | 43 | 43 | ||||
|
| 10 mol‐% | THF | 21 |
| |||
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —Deutsche Bundesstiftung Umwelt10.13039/100007636
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic Cross-Coupling Reactions · Organometallic Complex Synthesis and Catalysis · Organophosphorus compounds synthesis
Introduction
1
Current research on luminescent transition metal complexes (TMC) focuses on the use of highly abundant 3d transition metals as potential substitutes for photoactive compounds based on precious 4d and 5d elements [1, 2, 3, 4, 5, 6, 7, 8]. The latter can mediate beneficial spin‐orbit coupling (SOC) [9, 10, 11] to populate triplet excited states for photocatalysis, singlet oxygen sensitization and photodynamic and photoactivated therapy (PDT/PAT) [12, 13, 14], but also for formal spin‐forbidden phosphorescence T_1_→S_0_. One of the challenges is that 3d compounds are often prone to potential premature deactivation via the metal‐centered (MC) dd* transitions, which activate the M–L bond vibrational modes, or they exhibit only insufficient SOC, leading to relatively slow and inefficient spin‐flip processes. However, great efforts of creative ligand design have allowed to access emissive complexes of V, [15, 16] Cr, [17, 18, 19, 20, 21] Mn, [21, 22, 23, 24, 25] Fe, [26, 27, 28, 29, 30] Co, [31] Ni, [32, 33, 34, 35] Zn [36, 37, 38, 39, 40, 41, 42], and Cu [43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54]. The latter is the most widely studied first row transition metal for photonic applications due to its d^10^ electron configuration and resulting absence of MC states. In addition, the convenient oxidation potential of the Cu^I/II^ pair leads to the formation of low energy MLCT states, increasing the density of states and providing SOC so that formally spin‐forbidden ISC processes S_n_→T_m_ become fast up to a few ps.
In the last decade, linearly heteroleptically coordinated Cu^I^ carbene complexes with a donor‐acceptor (D–Cu–A) structure have been extensively studied [52, 53, 54, 55, 56]. This class of emitters provides access to low energy ligand‐to‐ligand charge transfer states (LLCT) in the visible region of the electromagnetic spectrum (EMS), which often results in thermally activated delayed fluorescence (TADF) as the dominating emissive path. The LLCT nature of the excited states lower the singlet‐triplet energy gap ΔE ST by increasing the charge separation so that thermal population of higher vibronic states of the T_1_ leads to reverse (r)ISC into the S_1_ state and formation of an excited state equilibrium at ambient temperature [57]. The S_1_ excitons can efficiently decay by spin‐allowed fluorescence circumventing the slow, spin‐forbidden phosphorescence. Sophisticated ligand design allows for dedicated fine‐tuning of the TADF‐relevant parameters [45, 52, 54, 56, 57, 58, 59, 60, 61] and highly efficient emitters with emission kinetics and efficiency competitive to commercially employed Ir^III^ compounds have been obtained [44].
In such D–Cu–A compounds, the carbene ligands typically serve as acceptor moieties, and modification of their electrophilic character could be exploited to achieve deep red to NIR emission of interest for future IT applications or NIR OLEDs [45, 62, 63, 64]. Such low energy emission can also be realized by modification of the π‐basicity of the donor ligand, for which aromatic amides have been used as the most common motif because of their delocalization of the anionic charge enhancing the CT nature of the excited states, which is beneficial for the TADF process. Further increasing the π‐basicity of the amide moiety by employing phenazine derivatives indeed leads to a significant decrease of the emission energy. Interestingly, and to the best of our knowledge, heavier congeners of the pnictogens have not been considered yet for deep red luminescent coinage metal complexes although one would expect them to be more potent donor ligands due to their higher polarizability.
Photophysical investigation of phosphide complexes of coinage metals, especially of copper, was impeded by their tendency to form a wide variety of oligomers or clusters depending on the steric and electronic influences both of the phosphide as well as of the neutral ligands. For example, Tuck reported that the electrolysis of an acetonitrile solution containing both diphenylphosphine and bis(diphenylphosphino)methane (dppm) as a neutral tertiary phosphine ligand yields crystalline tetranuclear [Cu_4_(dppm)4(μ‐PPh_2_)4] [65, 66]. The presence of non‐chelating phosphines in the reaction of CuCl with Me_3_SiPPh_2_ can lead to clusters of the general formula [Cu_m_(PR_3_)n(μ‐PPh_2_)q] with great structural diversity as demonstrated by Fenske et al. [67] Waterman and coworkers investigated Cu‐catalyzed hydrophosphination reactions and in the course of their work they isolated [Cu_4_(P(t‐Bu)3)2(μ‐PPh_2_)4] as efficient pre‐catalyst [68]. Other neutral chelating ligands such as phenanthroline (phen) were used to stabilize defined complexes, such as [Cu_3_(phen)3(μ‐PPh_2_)3], which showed to be synthetically useful in the preparation of highly functionalized alkyl phosphines and arsines [69] The bridging properties of the phosphido ligand could be attenuated by classical protection in the form of a borane adduct yielding the tetrahedral monocopper(I) complex [Cu(phen)(PHPh_2_)(PPh_2 ⋅ BH_3)] [70]. The first synthesis of linear phosphide Cu^I^ complexes that are structurally related to the above mentioned TADF‐active systems was reported by Whittlesey, Nolan and Cui with classical imidazole‐based N‐heterocyclic carbene (NHC) ligands via internal base method, σ‐bond metathesis and one pot reaction [71, 72, 73, 74]. However, it is important to note that the coordination geometries are dependent on the steric demand of the carbene and can lead to monomeric, dimeric or trimeric structures [73, 74]. Interestingly, linear [Cu(IDipp)(PPh_2_)] (IDipp = Bis(2,6‐diisopropyl)imidazolylidene) has been shown to photocatalyse the hydrophosphination reaction of styrene derivatives with HPPh_2_ when irradiated with 360 nm UV light [75]. Linear, monomeric [Au(cAAC)(PPh_2_)] was reported to be a nucleophilic superbase undergoing selective complexation of various metal fragments to form compounds of the type [(cAAC)Au(μ‐PPh_2_)Rh(acac)(CO)] [76]. Based on the demonstrated accessibility of defined linear complexes and bearing in mind the higher polarizability (and thus higher π‐basicity) of the heavier pnictogens in comparison to commonly used nitrogen‐based donors, we were interested in the influence of phosphide donors on the CT excited states and the resulting photophysical properties.
Results and Discussion
2
Synthesis and Characterization
2.1
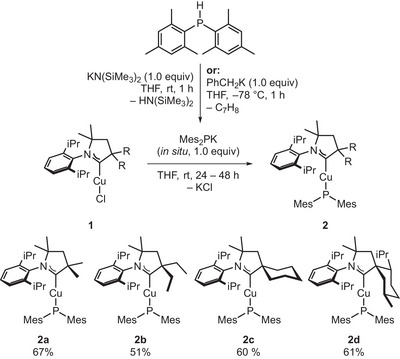
The linearly coordinated copper(I) phosphido complexes [Cu(^R^cAAC)(PMes_2_)] were prepared by salt metathesis starting from the respective chlorido complexes and in situ formed potassium dimesitylphosphide (Scheme 1). Single Crystals of 2a–2c were obtained by slow vapor diffusion of n‐pentane into a solution of the complexes in diethyl ether/THF (3:1) at –35°C, while 2d was crystallized by cooling a saturated solution in n‐pentane to –35°C. The chiral complex 2d was obtained enantiopure from chiral and enantiopure 1d. All complexes were fully characterized by multinuclear NMR spectroscopy, single crystal X‐ray diffraction (SCXRD) studies, high resolution mass spectroscopy (ESI‐HRMS), elemental analysis, and mid‐infrared (MIR) absorption spectroscopy (see Supporting Information). Noteworthy, the high steric demand of the mesityl substituents is crucial for the stability of the desired compounds as analogous attempts to synthesize linear [Cu(cAAC)(PPh_2_)] complexes led to decomposition in solution after several days and during crystallization experiments. However, the isolated PMes_2_ complexes 2a‐d are highly reactive towards oxygen (see below), water, acetonitrile, and dichloromethane.
General synthesis of the complexes of type [Cu(RCAAC)(PMes2)] (2a‐d) by salt metathesis using [Cu(cAAC)Cl] (1a‐d) as precursor. Mes = 2,4,6‐Me3C6H3.
The successful coordination of the PMes_2_ is confirmed by a ^31^P NMR resonance between –75.5 ppm and –76.1 ppm with a large halfwidth of 7.0 Hz due to coupling with the quadrupole moment of the ^63^Cu nucleus. The ^13^C resonance of the carbene atom is slightly shifted towards lower field in 2a‐d compared to the chlorido complex 1a (e.g. for 2a, 252.1 vs. 248 ppm) [77], and appears as a doublet (^2^ J P,C = 34.9 – 38.3) due to coupling to the ^31^P nucleus, similar to related NHC complexes [74]. We note that the coordination of PMes_2_ does not lead to an increase of π‐back‐bonding Cu^I^ → cAAC in the ground state as the ^15^N NMR data are very similar to those reported for [cAAC⋅LiOTf] (–159.9 ppm) or protonated cAAC⋅H^+^ (–148.1 ppm) [78]. Complexes 2a‐c crystallize in a monoclinic lattice with centrosymmetric space group P2_1_/n with 4 molecules per unit, while 2d crystallizes in the monoclinic space group P2_1_ with 6 molecules per unit and equimolar amounts of n‐pentane [79]. The presence of n‐pentane was also confirmed by ^1^H and ^13^C{^1^H} NMR spectroscopy. In all complexes, the bond length between the carbene carbon atom and the copper atom is slightly elongated by ca. 0.03–0.05 Å in comparison to the respective chloride complexes 1 (e. g., 1.9136(25) Å for 2a vs. 1.878(2) Å for 1a, see Figure 1) due to the electron donating effects of the PMes_2_ ligand. The Cu–P bond distances in 2a and 2c of 2.1897(7) and 2.1843(5) Å, respectively are shorter than found in structurally related [Cu(IDipp)(PPh_2_)] (2.2076(6) Å) [74], whereas it is comparable in 2b (2.2077(4) Å) and elongated in 2d (2.2222(5) Å), which can be explained by the varying repulsion of the ligands. Depending on the steric influence and flexibility of the substituents at the quaternary carbon of the cAAC ligand, the coordination geometry of the Cu^I^ center as well as the conformation of the phosphide change dramatically. While for 2a, 2c, and 2d, a nearly linear ligand arrangement is observed in the solid state (C–Cu–P angle 168–170°), compound 2b shows pronounced bending (156.98(4)°) presumably due to packing effects as the buried volume of the carbene ligand in 2b of 45% is identical to 2a/c, while the menthyl substituent in 2d provides larger coverage of the copper center with 55% (Figure 1). The bending of the PMes_2_, indicated by the angle between the C14–P–C14 and the N–C15–Cu planes, strongly depends on the steric influence of the cAAC ligand, leading to only 38° for 2a and 2c. In contrast, the freely rotating ethyl groups in 2b as well as the sterically rigid spiromenthyl substituent in 2d increase the angles between the planes to 51°, which correlates with the emission properties and kinetics (see below).
Molecular structure of 2a‐d in the solid state determined by SCXRD (ellipsoids given for 50% probability; hydrogen atoms omitted for clarity) and steric maps showing the buried volume V bur in % for the carbene (top) and phosphide (bottom) ligands, respectively. The numbering scheme applies to all structures [79].
Optoelectronic Properties
2.2
The compounds appear as bright yellow (2b, d) to orange red (2a, c) solids depending on the cAAC ligand, and their solutions in THF, benzene or toluene are deep orange. The UV/vis absorption spectra of 2a‐d in toluene solution are dominated by two major broad bands (Figure 2). The weakly allowed low energy absorption at λ_max_ = 478‐484 nm with extinction coefficients of ε = 1,400 – 2,000 M^−1^ cm^−1^ can be assigned to a LLCT p_P_+π_Mes_ → πCN(cAAC) transition with some MLCT (σ_PCu_ → πCN(cAAC)) admixture according to our TD‐DFT calculations performed for 2a. We note that the experimental values for ε represent an average over all available conformational geometries due to rotation around the Cu–L σ bonds. In comparison to previously reported [Cu(^Ment^cAAC)(NPh_2_)] (A) absorbing at λ_max_ = 440 nm [44], the lowest energy absorption is bathochromically shifted in 2a–d. The more intense high energy bands at λ_max_ = 326‐339 nm (ε = 9,900–15,300 M^−1^ cm^−1^ for 2b–d) are the result of allowed LLCT and ILCT(phosphide) (S_0_ → S_5_ and S_0_ → S_6_) transitions overlapping with less allowed MLCT and LLCT transitions (S_0_ → S_3_ and S_0_ → S_4_). Interestingly, the less sterically encumbered 2a exhibits a much higher extinction coefficient (ε = 15,300 M–^1^ cm–^1^) for this band in comparison to the other derivatives.
Absorption spectra of compounds 2a–d in toluene solution (solid lines), TD‐DFT (D3BJ‐PBE0/ZORA/def2‐TZVP) calculated oscillator strengths of the first 11 vertical S0→Sn transitions (red bars) and depiction of the electron density difference between the S0 and S1/S5 states for 2a in geometry optimized ground state structure.
The phosphide complexes exhibit weak near‐IR photoluminescence with λ max = 784 nm–818 nm upon irradiation at 480 nm in toluene solution, which is drastically bathochromically shifted by > 200 nm in direct comparison with the lighter N‐congener A (Figure S104 and Table S2) [44]. However, bright orange emission at λ max = 625 and 600 nm for 2a and 2b‐d, respectively, with photoluminescence quantum yields of ϕ = 0.30–0.52 is observed in the solid state at room temperature (Figure 3 and Table 1). The large Stokes shift of Δν̃≈4200 cm^−1^ in combination with the broad spectral appearance indicate pronounced structural reorganization in the excited state of dominantly LLCT character. Interestingly, the emission of 2a is slightly red shifted in comparison to the other derivatives.
Normalized emission intensity (NEI) of compounds 2a‐d in the solid state at room temperature (left), emission and excitation spectra of 2d in different media (middle) and exemplary excited state lifetime analysis (in counts per second) of compound 2c (λex = 450 nm, λem = 600 nm) in the solid state at 297 K showing a multiexponential decay (right).
The transient luminescence decays are of bi‐ or tri‐exponential nature and their deconvolution gives averaged lifetimes in the µs regime, which suggests that spin‐forbidden processes are involved (Figure 4 and Table S3). The mean radiative rate constants <k r> are similar for complexes 2a (5.9⋅10^4^s^−1^), 2c (4.3⋅10^4^s^−1^) and 2d (3.1⋅10^4^s^−1^). However, 2b exhibits a much lower radiative efficiency (ϕ = 0.30, k r = 1.9⋅10^4^s^−1^), which may be due to the bend structure in the single crystalline solid state (Figure 1), reducing orbital overlap between the donor and acceptor ligands and, consequently, reducing the oscillator strength f. The emission in different environments was investigated for compound 2d, showing a bathochromic shift in comparison to the solid state when doped in polar PMMA (λ max = 636 nm), and even more in non‐polar poly(styrene) (PS) (λ max = 660 nm) films (Figure 3 and Table 1). A similar relative trend is observed for the excitation spectra, which suggests that the ground state of 2d is more polar than the emitting excited state. Again, we notice that the luminescence of 2d in PS is bathochromically shifted by 130 nm (3645 cm^−1^) compared to A, highlighting the influence of the heavier pnictogen [36, 37, 38, 39, 40, 41, 42]. Surprisingly, nonradiative decay in the polymer films is very facile and k r is significantly reduced in comparison to the solid‐state measurements for all compounds due to a less efficient TADF process (Table 1). In the solid state, the phosphide complexes are very densely packed and the conformation fixed, providing optimal prerequisites for TADF. However, the higher conformational flexibility of 2a‐d in viscous polymers and intermolecular interactions between the emitter complexes and the environment apparently influence the excited state coupling and, according to the energy gap law at lower emission energies, enhance vibrational coupling of the S_1_ state with the ground state.
Normalized emission of compounds 2a‐d in the solid state at 77 K (left), tr‐VT data of 2c showing the temperature dependence of the average k r and relative contributions of the observed lifetime components to the multiexponential transient decays (middle, values not obtained for 87 K, see also Supporting Information), and mono‐exponential decay (in counts per second) observed for 2c (λex = 450 nm, λem = 600 nm) at 77 K (right).
As d^10^ coinage metal complexes can emit from both their S_1_ and T_1_ states, we were curious whether TADF or phosphorescence is the major radiative path and thus we carried out steady‐state and time‐resolved variable temperature luminescence measurements (Figure 4). Upon cooling 2a‐d to 77 K in the solid state, the emission onset of all compounds is bathochromically shifted to 550 nm indicating a different lower energy state to be responsible for the luminescence than at 297 K. However, the spectral appearance reveals for 2a and 2c a vibrational progression, but remains broad and featureless for 2b and 2d. We attribute this to a more rigid solid‐state structure favoring certain vibrational modes for compounds 2a, c. For all complexes, a significant increase of the luminescence lifetime up to the millisecond range with concomitant decrease of ϕ is observed, resulting in a drastic decrease of <k r> between 297 K and 77 K (Table 1). Such a behavior hints at TADF as the dominant emission mechanism at room temperature. Fitting k r to the Boltzmann distribution function for a three‐state‐model (Equation (1)) [44] indeed reveals a sigmoidal temperature dependence:
with the beginning of a plateau at room temperature, while at T < 150 K, no significant further decrease of k r is observed, indicating pure phosphorescence from the T_1_ state. The relatively large ΔE ST = 139 ± 5 meV (1119 cm^−1^) nicely coincides with the values obtained from the energetic shift of the emission onset at 297 and 77 K (Figure S107) and can be explained by the spatial proximity and more localized nature of the frontier orbitals involved in the vertical transitions (Figure 2), in contrast to structurally related carbazolate and diarylamide complexes, where the HOMO is delocalized over the entire π system [44].
Chemiluminescence
2.3
During the preparation and photophysical investigation of 2a‐d, we inadvertently discovered orange chemiluminescence in the solid state as a result of a chemical reaction upon contact with air (Figure 5 and Video SI). Further studies showed that 2a‐d do not react with N_2_O as an oxidizing agent even after several days, but exposure to dry molecular O_2_ gave immediate intense luminescence and yielded a white solid, which is insoluble in most standard solvents. ^31^P NMR spectroscopic studies in dry 1,2‐difluorobenzene showed one narrow singlet at –5.73 ppm, and HR‐MS analysis of the reaction product indicates the formation of, Cu(I)‐diarylphosphinites and Cu(I)‐diarylphosphinates supported by a cAAC ligand, reprotonated carbene and tetramesityldiphosphine (Figure S95). Unfortunately, attempts to isolate or crystallize the reaction products for further characterization were not successful. However, it is important to note that the chemiluminescence also occurs when PMMA or PS films doped with 2a‐d are exposed to air and is not related to differences in the steric protection of the copper(I) center as 2d with the highest V Bur is equally reactive as the other derivatives (Figure 1). These properties are highly unusual, as we are not aware of any previously reported copper(I) amide analogs or copper(I) phosphide clusters displaying chemiluminescence upon oxidation, which appears to be due to the higher reactivity of the phosphide ligands coordinated to the electron‐rich copper(I) cAAC fragment in comparison to other typically employed donor ligands.
Normalized chemiluminescence spectra of 2a‐d in the solid state upon reaction with atmospheric oxygen.
Chiroptical Properties
2.4
Given the current interest in chiroptical properties for photonic applications, we further investigated in this regard the chiral and enantiopure complex [Cu(^Menth^cAAC)(PMes_2_)] (2d) in different media. The CD spectrum in toluene (Figure 6) shows a relatively small absorption dissymmetry for the lowest energy transition (g abs≈10^−3^) and enhanced dissymmetry values of g abs = 10 ^−2^ for higher energy transitions with small extinction coefficients ε. According to Equation (2) [51], the dissymmetry for the transition from state i to state f maximizes when the electronic (** μ ) and magnetic ( m **) transition dipole moment vectors are in antiparallel orientation. Our TD‐DFT calculations suggest an angle of 79° between ** μ ** and ** m ** for the S_0_→S_1_ transition, similar to related [Cu(cAAC)(Cz)] derivatives, [50] while the prominent CD bands at higher energy are due to or for symmetry‐forbidden electronic transitions with small ** μ ** and high ** m **.
Left: CD (red) and absorption spectrum (blue) of compound 2d in toluene at room temperature with absorption dissymmetry (grey). Middle: CPL‐spectrum of compound 2d in solid state (yellow), in PMMA matrix (brown) and in PS matrix (pink). Right: Emission dissymmetry of the compound 2d (λ ex = 450 nm).
The luminescence dissymmetry of 2d in the solid state is weak with g lum≈1.2·10^−3^, comparable to other structurally related CPL‐TADF complexes [50, 51]. As 2d exhibits intermolecular non‐polar C(sp^3^)–H** ^…^ **π London dispersion interactions in the crystals similar to previously reported [Cu(BINAP)(Cz)] (Figure 7), it is not surprising that the chiroptical properties in polar and rigid PMMA capable of hydrogen bonding are very similar. However, more flexible and nonpolar PS not only leads to a bathochromic emission shift, but we also observe a drastic increase of the dissymmetry by a factor of 10 (g lum≈10–^2^) as well as a sign change. We attribute this finding to stabilization of a different excited state geometry and interactions between 2d and the matrix. For 2d in PS we calculated a CPL‐brightness [80] of B CPL = 1.31 M^−1^ cm^−1^ according to Equation (3):
and using k CPL = (k r·g lum)/2 as a new way to quantify the efficiency of CPL emission [51], we obtained k CPL = 41.5 s^−1^ and 15.6 s^−1^ for PS and the solid state, respectively. The comparatively high CPL‐brightness is probably due to a reduced k nr compared to similar complexes (e.g. [Cu(BINAP)(Cz)]: B CPL = 2 M^−1^ cm^−1^ [51], [Cu(^Ment^cAAC)(PZN)]: B CPL = 1.9 M^−1^ cm^−1^ [50], Pt^II^: B CPL = 0.01‐4.7 or Ir^III^: B CPL = 0.63‐1.1 [80]) but considerably less than in compounds with Laporte‐forbidden transitions, like some Cr^III^ complexes (B CPL = 6.3·10^−5^ – 174 M^−1^ cm^−1^) [20, 80, 81], allowing high Φ with reduced k r.
Csp3–H…π interactions between H5 from the cAAC backbone and the π system of the mesityl substituent; bond distances (in Å) d(C13B, H5) = 2.9059(22), d(C2B,H5) = 3.1030(24).
Photocatalysis
2.5
The significant absorbance in the visible blue spectral region renders complexes 2 as suitable candidates for photocatalytic hydrophosphination reactions, which have been reported to occur under thermal conditions with complexes of several metals [82, 83, 84]. A few studies employing other earth‐abundant metal complexes based on Ti^IV^ [85], Zr^IV^ [86, 87], Fe (II [88, 89]) or Ni (II [90]) as catalysts suggest that UV or visible light irradiation in the presence of a HPR_2_ or HP(O)R_2_ species and unsaturated organic substrates can initiate radical reactions, which finally provide access to the desired hydrophosphination products. Waterman et al. have shown that Cu^I^ complexes are generally also suitable for this purpose [68, 75, 91] but do not require radical reactions. Instead, high energy UV excitation with 360 nm light, for example of [Cu(NHC)(PPh_2_)], populates an LMCT state weakening the Cu–P bond, which allows for 2,1‐insertion of the alkene or nucleophilic attack [75]. Considering the low energy absorption of 2, the previous reports motivated us to investigate the potential of the cAAC based Cu^I^ complexes.
Irradiation of a C_6_D_6_ or THF‐d_8_ solution containing phenyl acetylene and diphenylphosphine with 480 nm light for 6 h in the presence of 2–10 mol % of 2c gave nearly quantitative conversion, forming both (E) and (Z) hydrophosphination products 4a and 4b as detected by ^1^H and ^31^P{^1^H} NMR spectroscopy together with traces of styrene, while the gem‐product is not observed (Scheme 2, left, and Table 2). Further addition of substrates continues the photocatalytic hydrophosphination reaction (see Supporting Information). Under the same reaction conditions in the dark, only minor product formation < 5% is observed even after 12 h.
Photocatalyzed hydrophosphination reaction of phenylacetylene with HPPh2 employing 2c or 2d as precatalysts (left), or using 1c and 1d for in situ formation of catalytically active phosphide complexes with subsequent isolation of 7a/b after oxidative work‐up (right), and molecular structure of 7a determined by SC‐XRD (right, ellipsoids given for 50% probability; hydrogen atoms are omitted and one phenyl ring is depicted as wireframe for clarity). Selected bond lengths (Å) and angles (°): P1‐O1: 1.491(3), C1A‐P1: 1.807(5), C1B‐P1 1.810(4), C1‐P1: 1.774(4), C1‐C2: 1.342(6), C2‐C3: 1.472(6), C1‐P1‐O1: 114.9(2), P1‐C1‐C2: 119.9(4), C1‐C2‐C3: 125.2(4). For the Full ORTEP/PLATON plot and unit cell, see Figure S32.
Several observations suggest that the mechanism of this photocatalytic transformation is significantly different than observed for prominent photoredox catalysis or bond homolysis mechanisms. At high catalyst loading of 10 mol % 2c, the formation of 4a is generally more favored than 4b, but the yield of the E‐isomer is significantly higher in nonpolar C_6_D_6_ with 95:2 than observed for THF‐d_8_ (80:10) (Table 2). While with 5 mol % of 2c the E/Z‐ratio is reduced to 66:34 in THF, further lowering of the catalyst loading to 2 mol % surprisingly favors the Z‐isomer independent of the solvent. The catalyst concentration dependent product distribution indicates that formation of the E‐isomer 4a requires aggregation of the copper(I) species, while 4b appears to be the result of a mono‐copper(I) complex acting as photocatalyst. In line with this interpretation is the fact that 2d bearing a sterically more demanding carbene with a higher V bur (Figure 1) gives a higher yield of 4b at 2 mol % than 2c. This interpretation is supported by the finding that 2d bearing the bulky ^Menth^cAAC with a higher V bur of 55% than ^Cy^cAAC with 45% (Figure 1) further shifts the E/Z‐ratio towards the latter with 11:82 in comparison to 2c even at low catalyst loading of 2 mol %. We note that the photocatalytic hydrophosphination does not occur with sterically more demanding HPMes_2_ as a substrate under the same conditions, which rules out an outer‐sphere product forming step in the catalytic cycle or radical reactions (Figure S20).
Deeper insight into the mechanism is provided by ^31^P NMR spectroscopic measurements of 2c in the presence of free HCCPh in the dark, suggesting fast equilibrium between the copper(I) phosphide and the related Cu–acetylide complex of 0.75:1.00 (Figure S24). However, structurally related and [Cu(^Ad^cAAC)(CCPh)] has been reported to not absorb light at the irradiation wavelength and such a species is thus not relevant for the photoinitiation, but could be a resting state [77]. Furthermore, TD‐DFT calculations of [Cu(cAAC)(η^1^‐CCPh)(PHMes_2_)] reveal only absorption in the UV, but [Cu(cAAC)(η^2^‐HCCPh)(PPh_2_)] exhibits S_0_ → S_1_/S_2_ excitation of (ML)LCT (p_P_+d_Cu_ → π*(HC≡CPh)) character for S_1_ and LLCT (p_P_+π_Mes_ → π*CN(cAAC)) character for S_2_ at 474 and 461 nm, respectively (Tables S24, S25, and S31). As fast exchange of ^−^PMes_2_ for ^−^PPh_2_ will occur under the reaction conditions, we therefore infer either [Cu(cAAC)(PPh_2_)] as the derivative of original 2c/d to be the decisive photocatalytically active species via Cu–P bond weakening due to LLCT excitation and concomitant distortion, or a trigonal Cu^I^ complex with both phosphide and side‐on π‐bond acetylene to be involved as the critical light absorbing and product forming species. As we have described above, the copper(I) carbene diphenylphosphide complex could not be isolated due to its high reactivity and decomposition upon purification, which is the reason why we investigated the PMes_2_ analogues. However, the equilibrium reaction with free phenylacetylene provides a resting state for stabilization to continue the photocatalytic reaction cycle upon addition of further substrate to the reaction mixture (see above).
Investigation of the time‐dependence of the product yield using 2 mol % 2c reveals very fast formation of the Z‐isomer 4b, which is nearly finished after 60 min, while E‐isomer 4a requires an induction period within the first 30–45 min (Figure 8). Apparently, the assumed aggregation of the photocatalytically active metal complexes is time‐dependent and after 1 h continues the formation of 4a as the dominant process. We note that styrene (6) is observed as a by‐product. Interestingly, its formation is increased after 60 min when the Z‐isomer 4b is barely produced anymore, indicating that 6 is the result of the aggregation process, which is complete after 120 min. These findings further support our hypothesis of inner‐sphere mechanisms of the hydrophosphination, of which the product distribution is under kinetic control of an equilibrium between a photocatalytically active mono‐copper(I) species and its catalytically active aggregates.
Time vs. product yield plot for the reaction of diphenylacetylene (3) with diphenylphosphine catalyzed by 2 mol‐% of 2c under irradiation with 475 nm light. For additional details see Figure S8.
Furthermore, 1c/d can be used as precatalysts by in situ formation of catalytically active phosphide complexes with HPPh_2_ in the presence of a mild mesityl Grignard reagent and subsequent addition of phenylacetylene. After 16 h and oxidative work‐up using H_2_O_2_ / NaOH, the desired vinylphosphine oxide could be isolated in 91% yield based on phenylacetylene (Scheme 2, right). For the (E)‐vinylphosphine oxide 7a, the molecular structure was analyzed by SC‐XRD further confirming the double bond configuration [79]. Again, the steric requirements of the carbene ligands and the photocatalyst concentration appear to control the degree of copper(I) complex aggregation and thus the E/Z‐product distribution. Employing 2d in high concentration of 10 mol % inverts the E/Z‐ratio, highlighting the potential reaction control for future application. Further studies of other substrate classes are beyond the scope of this work and are currently being studied in our ongoing projects.
Conclusion
3
The new copper(I) complexes demonstrate that stable monomeric diarylphosphide coordination compounds can selectively be obtained in pure form, thus expanding the chemical space of donor ligands for new luminescent materials. However, the unexpected chemoluminescence of 2c in the presence of molecular oxygen shows that they are sensitive to O_2_. We anticipated that introduction of the diarylphosphide ligand in cAAC‐copper(I) complexes should lead to smaller energy gaps between the ground state and the excited states based on the higher polarizability of phosphorous in comparison to well‐known diarylamide complexes, and indeed pronounced bathochromic shifts of the emission (Δλ≈130 nm) have been observed. Although an unexpected large ΔE ST reduces the efficiency of TADF, the generally promising photophysical results suggest that further optimization by chemical modification of either the phosphide or the carbene moiety can lead to efficient deep red to NIR emitters. While there are reported cases of CPL active compounds with the C 1 symmetrical carbene [^Menth^Caac] [92], most of its carbene‐metal‐amide (CMA) derivatives do not show this technologically relevant property [50]. The fact that structurally related 2d does exhibit for small molecules high emission dissymmetry up to g lum = 10^−2^ demonstrates that not only the chiral ligand, but the entirety of the ligand system has direct influence on the chiroptical properties, although also environmental effects are relevant as shown by our matrix dependent luminescence studies. Finally, the phosphide complexes 2c and 2d proved to be efficient visible‐light photocatalysts for the hydrophosphination of terminal alkynes. In contrast to previous copper based hydrophosphination photocatalysts, the cAAC ligand allows lower energy excitation in the blue spectral region. The observed direct influence of the carbene sterics on the diastereoselectivity of the reaction suggests the prevalence of an inner‐sphere product forming step, of which formation of the E‐isomer requires metal complex aggregation and the Z‐isomer appears to be formed by a mono‐copper(I) species. Further investigation of this reaction regarding functional group tolerance, selectivity and mechanism may render this reaction a synthetically useful tool.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting File 1: The authors have cited additional references within the Supporting Information [1–22, 44, 51].
Supporting File 2: anie71068‐sup‐0002‐SuppMat2.pdf.
Supporting File 3: anie71068‐sup‐0003‐VideoS1.mp4.
Supporting File 4: anie71068‐sup‐0004‐Data.zip.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1O. S. Wenger , “Photoactive Complexes With Earth‐Abundant Metals,” Journal of the American Chemical Society 140 (2018): 13522–13533, 10.1021/jacs.8b 08822.30351136 · doi ↗ · pubmed ↗
- 2B. M. Hockin , C. Li , N. Robertson , and E. Zysman‐Colman , “Photoredox Catalysts Based on Earth‐Abundant Metal Complexes,” Catalysis Science & Technology 9 (2019): 889–915, 10.1039/C 8CY 02336 K. · doi ↗
- 3C. Wegeberg and O. S. Wenger , “Luminescent First‐Row Transition Metal Complexes,” Journal of the American Chemical Society Au 1 (2021): 1860–1876.34841405 10.1021/jacsau.1c 00353 PMC 8611671 · doi ↗ · pubmed ↗
- 4G. Giobbio , R. D. Costa , and S. Gaillard , “Earth‐Abundant Transition Metal Complexes in Light‐Emitting Electrochemical Cells: Successes, Challenges and Perspectives,” Dalton Transactions 54 (2025): 3573–3580, 10.1039/D 4DT 03210 A.39835838 · doi ↗ · pubmed ↗
- 5N. Kumar , T. Sharma , N. Thakur , R. Jain , and N. Sinha , “Abundant Transition Metal Based Photocatalysts for Red Light‐Driven Photocatalysis,” Chemistry—A European Journal 31 (2025): e 202500365, 10.1002/chem.202500365.40135511 · doi ↗ · pubmed ↗
- 6R. V. Eldik , P. C. Ford , V. W.‐W. Yam , et al., Advances in Inorganic Chemistry (Elsevier, 2024).
- 7V. W.‐W. Yam and W.‐K. Kwok , Advances in Inorganic Chemistry, ed. Rudi van Eldik , Peter C. Ford (Elsevier, 2024), S. 1–3.
- 8V. Ferraro , C. Bizzarri , and S. Bräse , “Thermally Activated Delayed Fluorescence (TADF) Materials Based on Earth‐Abundant Transition Metal Complexes: Synthesis, Design and Applications,” Advancement of Science 11 (2024): e 2404866, 10.1002/advs.202404866.PMC 1142600938984475 · doi ↗ · pubmed ↗