Periodic Hirshfeld Atom Refinement
Kanghyun Chu, Dylan Jayatilaka, Lorraine A. Malaspina, Alessandro Genoni, Georgia Cametti, Stefan Mebs, Dieter Lentz, Hans-Beat Bürgi, Sergey V. Churakov, Simon Grabowsky

TL;DR
This paper introduces a new method called periodic Hirshfeld Atom Refinement (pHAR) that improves the accuracy of structural data for periodic materials containing B–H and N–H bonds.
Contribution
The novel contribution is pHAR, a periodic version of HAR that extends its applicability to periodic-network structures.
Findings
pHAR results show close agreement with neutron-diffraction data for X–H bond lengths.
pHAR nearly doubled the available reliable experimental data on B–H bonds.
The method is compatible with conventional HAR using Gaussian orbitals and Bloch wave formalism.
Abstract
Hirshfeld Atom Refinement (HAR) is a quantum crystallographic method for analyzing single-crystal X-ray diffraction data, providing accurate and precise structural parameters. Despite its success in predicting hydrogen-atom parameters, the application of HAR is fundamentally limited to molecular crystals. Inspired by two recently developed HAR versions that employ periodic-boundary conditions, here we introduce a new variant of periodic HAR (pHAR) that is applicable to any periodic-network structure while remaining compatible with conventional HAR by using atom-centered Gaussian orbitals with a Bloch wave formalism. pHAR was tested against high-quality single-crystal diffraction data for boranes and borates comprising N–H and B–H bonds in different chemical environments. The results demonstrate a close agreement of X–H bond lengths with reference data from neutron-diffraction…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
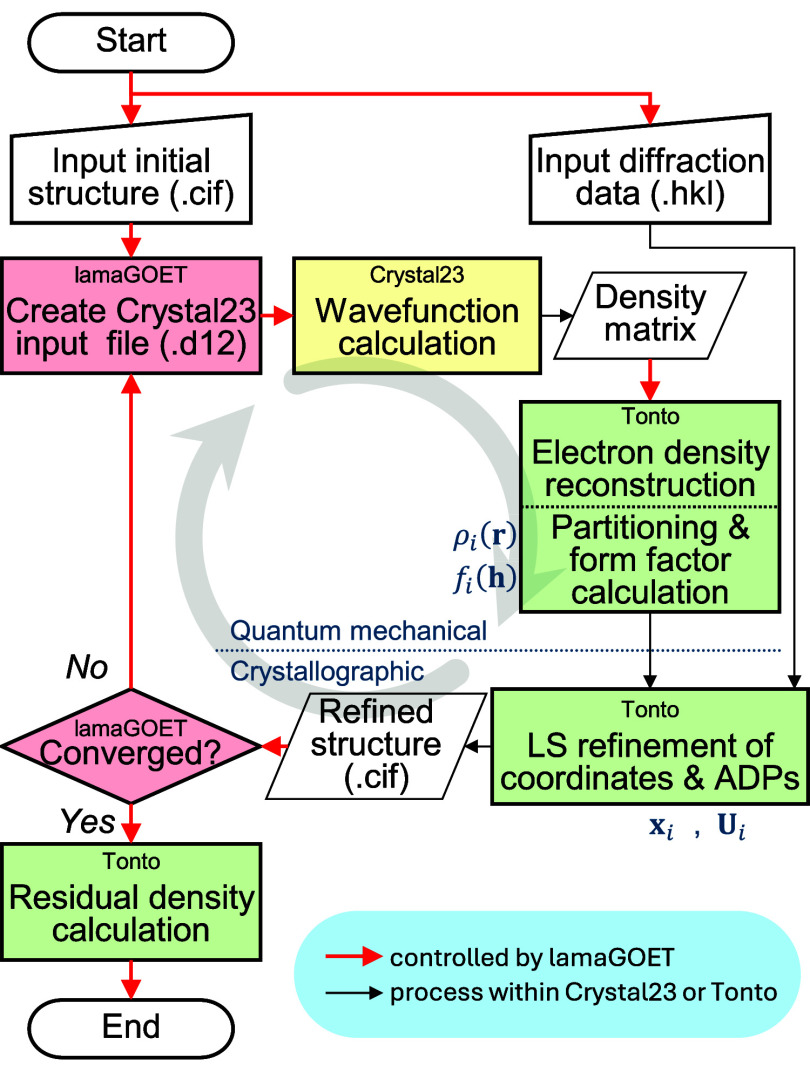 Figure 1
Figure 1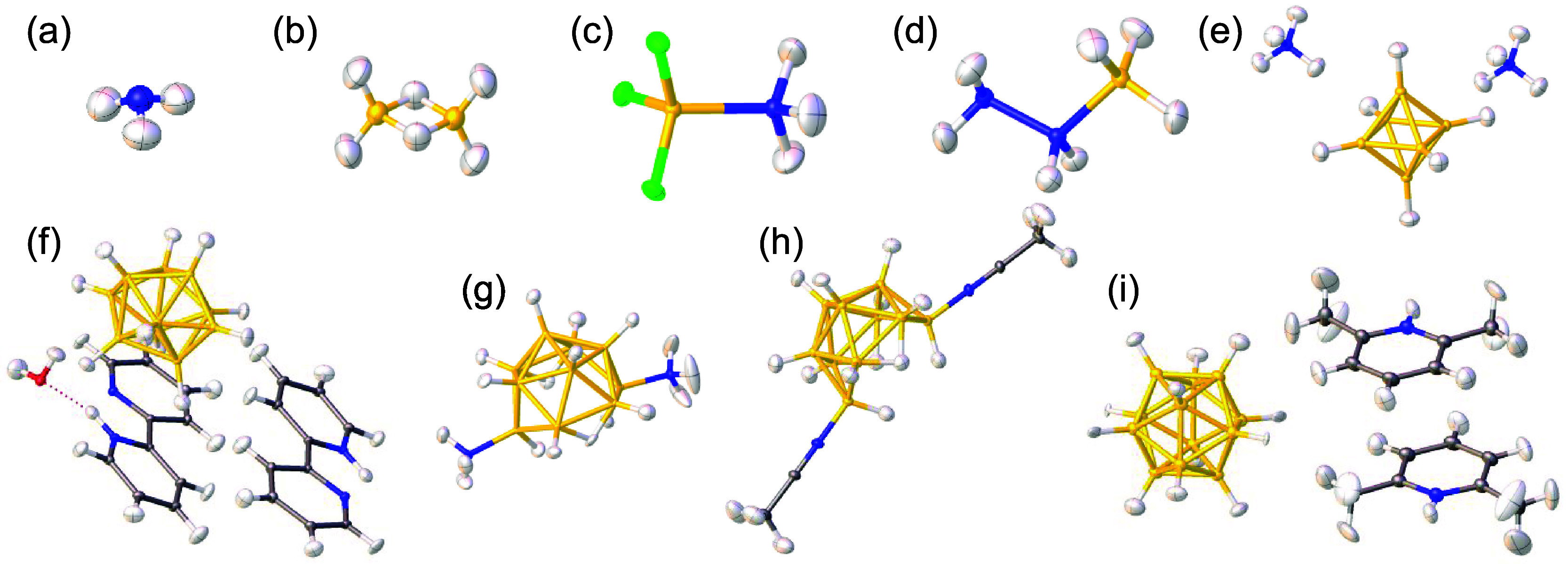 Figure 2
Figure 2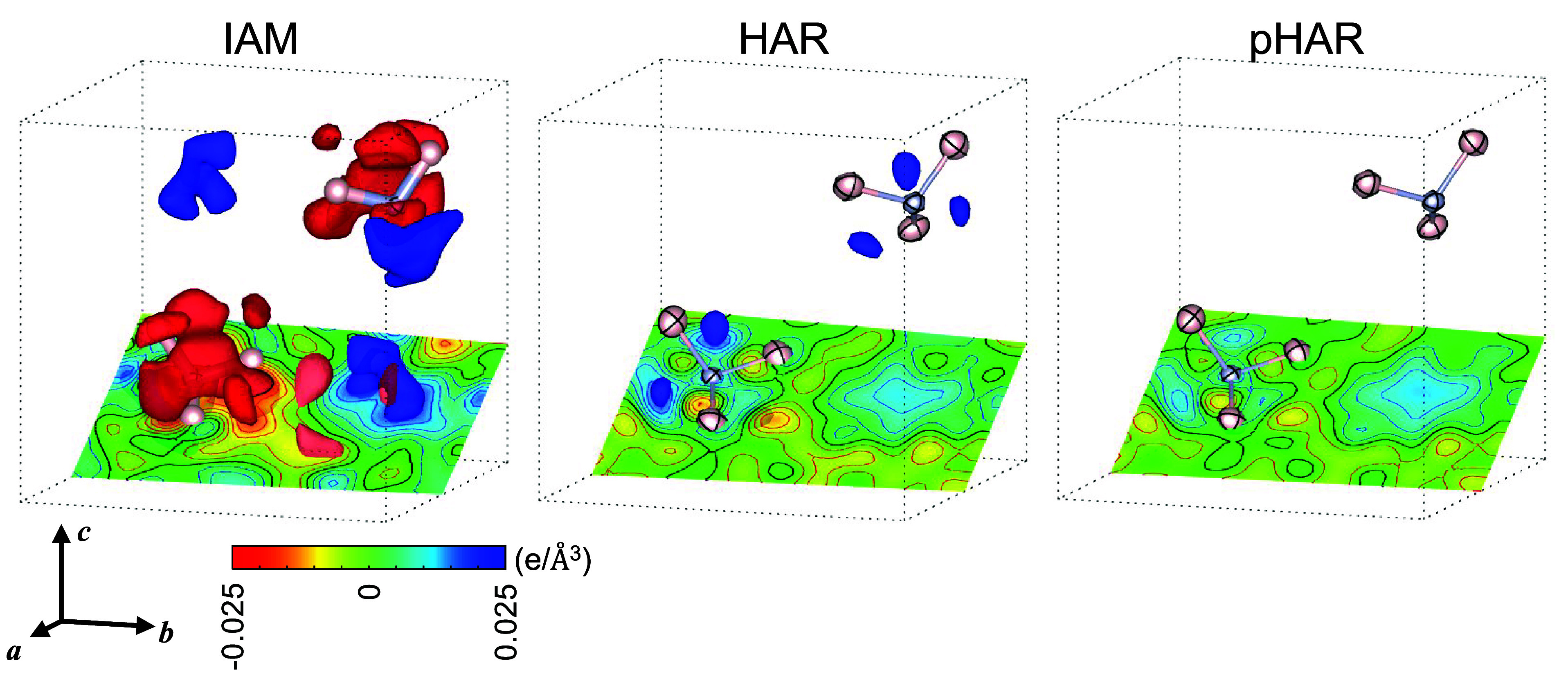 Figure 3
Figure 3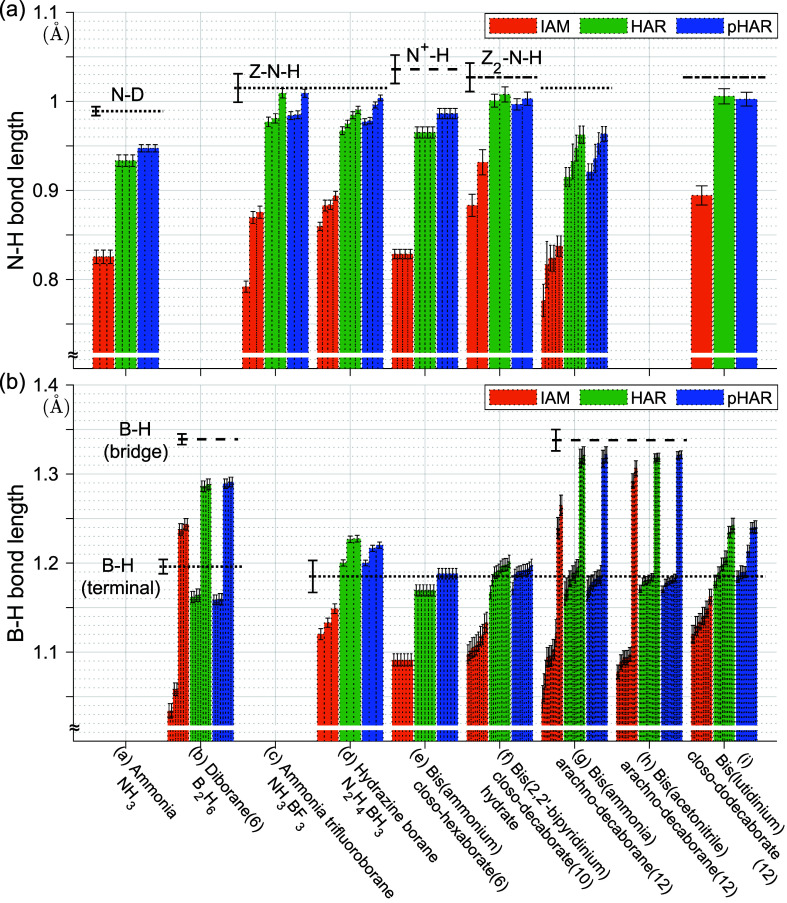 Figure 4
Figure 4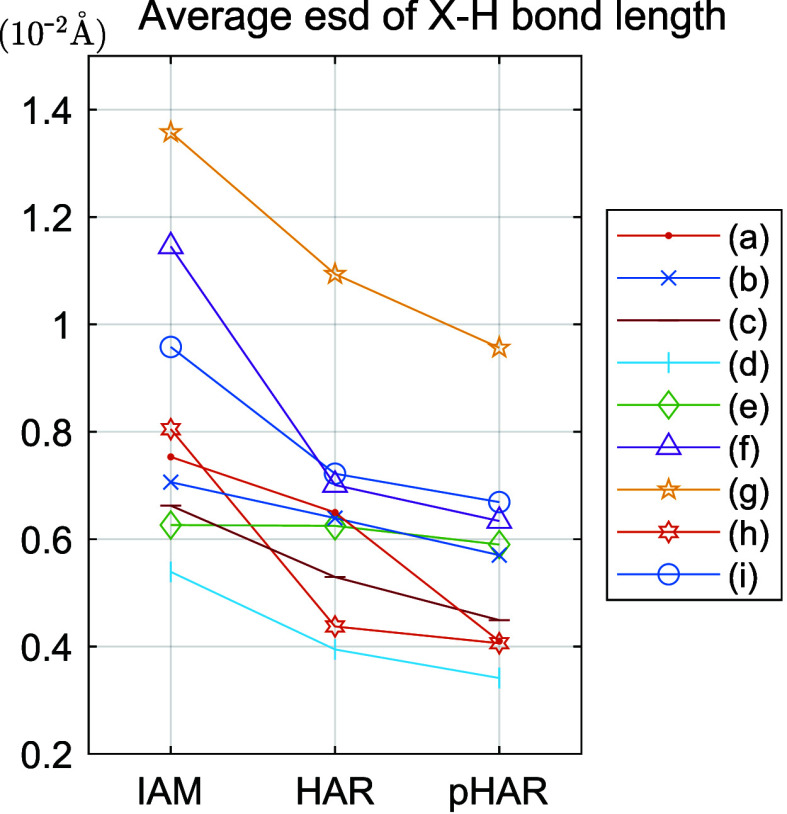 Figure 5
Figure 5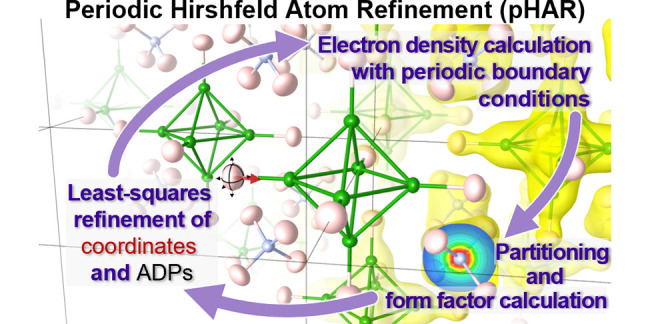 Figure 6
Figure 6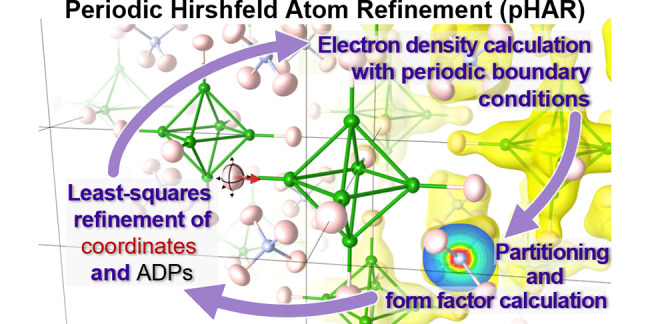 Figure 7
Figure 7- —University of Bern10.13039/100009068
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCrystallography and molecular interactions · Enzyme Structure and Function · Machine Learning in Materials Science
Single-crystal X-ray diffraction is the leading technique for atomically resolved structure elucidation. The technique places models of atomic electron densities in the crystal unit cell and optimizes their positions and mean-square displacements (atomic displacement parameters, ADPs) with a least-squares procedure to best fit the experimental diffraction pattern. The simplest and most widely adopted model is the Independent Atom Model (IAM).? It represents the crystal electron density with spherically symmetric, noninteracting atomic densities calculated quantum mechanically; their Fourier transforms are the so-called spherical atomic form factors.? This approach neglects bonding-induced anisotropy, leading to a bias in atomic positions and ADPs. This is especially important for hydrogen atoms with their highly polarized single valence electrons. ?,? X-H bond lengths in light-atom structures obtained with an IAM are typically 0.1 Å shorter than corresponding values obtained by neutron diffraction.?
Two modern techniques of quantum crystallography overcome these limitations: the multipole model (MM) and Hirshfeld Atom Refinement (HAR). ?−? ? The MM complements spherical atomic densities with weighted spherical harmonic functions representing electric dipole, quadrupole, octupole, and hexadecapole moments (deformation functions). ?−? ? The weight factors (multipole populations) are either refined against experimental X-ray diffraction data (together with atomic positions and ADPs) ?−? ? or taken from databases derived from experiments? or from quantum chemical calculations. ?,? The additional parameters can account for chemical bonding and crystal-field effects; they improve the hydrogen atom parameters significantly, all without much loss in computational efficiency. ?,?
HAR models are based on tailor-made, quantum-chemically calculated electron densities of unit-cell building blocks, i.e. molecules or clusters of molecules. ?−? ? ? The electron densities of the blocks are partitioned into general nonspherical Hirshfeld atomic densities of all symmetry-unique atoms. Their Fourier transforms are used in conventional least-squares refinement of the atomic positions and ADPs. The electron densities are recalculated with the refined atomic positions and the process is iteratively cycled until the atomic parameters and the quantum chemical energy converge. HAR is far more computationally expensive than MM but can yield more accurate structure models because a high-quality and rigorous quantum-chemical model of the underlying electron density is used. ?−? ? For light atom molecular structures, hydrogen-atom positions can attain precision and accuracy comparable to those from neutron diffraction. ?,?
The various models in the HAR family differ in several aspects (see the references for details).
- IChoice of the atomic assembly for the quantum-chemical calculation: isolated molecule, or a molecule embedded in a dielectric medium; ?,?,? molecule surrounded by a cluster of molecules represented by self-consistent point charges and dipoles simulating an approximate crystal field; ?,? explicit cluster of molecules around a central unit; ?,? content of complete unit cell with crystal periodicity. ?,?
- IIRepresentation of the electron density: plane-wave basis;? local atomic basis functions; ?,? pseudopotential descriptions of core electrons; ?,? projections onto multipole functions. ?,?
- IIIHamiltonian: Hartree–Fock; ?,? Post Hartree–Fock;? Density Functional Theory; ?,?,?,? Semiempirical.?
- IVPartitioning scheme: fragmentation Hirshfeld atom approaches; ?−? ? atomic partitioning alternative to Hirshfeld atoms. ?,? Here, we focus on the most general choice of atomic assembly, namely a periodic electron-density distribution. This allows treating infinite network compounds without arbitrarily cutting interatomic (covalent or long-range Coulomb) interactions at the boundaries of the unit cell. Such approaches have been tested by Wall,? Ruth et al. (XHARPy)? and Patzer and Lehmann (ReCrystal).?
For our version of periodic HAR (pHAR), we avoid the use of a plane-wave basis (Wall?). While it is well suited to describe valence electron densities, a plane-wave basis becomes impractical for modeling highly localized core-level electrons which represent an important part of the atomic scattering density. A comparison between plane-wave- and Gaussian-basis-set-derived electron densities can be found in ref ?. Pseudopotential descriptions were excluded because they cannot account for core polarization (Ruth?). ?−? ? For heavy elements, the use of effective core potentials can be useful in HAR, but it requires correction functions.? We also decided not to consider an approach in which the quantum chemical electron density is first projected onto multipole functions followed by a multipole refinement (Patzer and Lehmann?). In our pHAR method, we treat all electronsboth core and valenceon an equal footing. Electronic wave functions are expressed in terms of Bloch functions of atom-centered Gaussian orbitals. This choice allows meaningful comparisons with results from traditional quantum chemistry and conventional HAR on finite atomic assemblies provided Hamiltonians and basis sets are the same. It also provides density matrices for complementary chemical bonding analysis.?
The pHAR procedure is visualized in more detail in Figure. Starting from a tentative crystal structure (e.g., IAM), Crystal23? calculates its electronic wavefunction under periodic-boundary conditions imposing the correct crystallographic symmetry. The software Tonto? reads the Crystal23 density matrix, calculates the electron density ρ(r) and partitions it into nonspherical atomic densities ρ_ i _(r) which are Fourier transformed into nonspherical atomic form factors f _ i _(h). If the initial and refined parameters do not agree within the predefined tolerances, the refined structure becomes the new tentative structure and the procedure is repeated until convergence. Crystal23 and Tonto are interfaced via lamaGOET.? More details of the implementation are given in the section Experimental and Computational Methods and in the Supporting Information.
The performance of the newly developed pHAR was tested with high-quality single-crystal data for molecules with X-H bonds (XB, C, N, O). Here, we concentrate primarily on B–H and N–H bonds which have been less studied than C–H or O–H bonds. There are only three published HAR studies of boranes/borates: diborane, B_2_H_6_, in ref ?; bis(ammonium) closo-hexaborate(6), (NH_4_)2_B_6_H_6, in ref ?; and m-terphenylhydridoborates in ref ?. There are no studies of bigger cage structures and no systematic comparisons of the refined structures obtained with HAR or with neutron-diffraction data. We have included six neutral borane and ammonium borate compounds for which high-quality data sets are available and which were previously studied with MMs. ?−? ? We also included bis(ammonium) closo-hexaborate(6), (NH_4_)2_B_6_H_6, from a previous HAR study, which consists of highly symmetric, small molecular ions.? Ammonia and diborane are included as small neutral parent compounds for the N–H and B–H bonds, respectively. The single crystal X-ray diffraction data of ammonia (160 K) and of diborane (94 K) are taken from the literature. ?,?
Figure shows all the B–H and N–H compounds included in this study after pHAR treatment. It covers a wide variety of chemical environments. The compounds contain B–H bonds in both terminal (t) and bridging (b) positions, in anionic and neutral species, as well as N–H bonds, with or without hydrogen bonding, in cationic and neutral species.
The refined bond distances are compared with values from a compilation of corresponding distances obtained by neutron diffraction. For B–H, distances are compared with the mean (and standard uncertainty) of the distribution of terminal and bridging B–H bond distances determined with neutron diffraction at various temperatures: d(B–H_t_) = 1.185(18)Å and d(B–H_b_) = 1.338(12)Å.? The N–H distances chosen as references are bond-length populations coming from neutron-diffraction experiments: d(N^+^–H) = 1.036(16) Å, d(Z–N–H) = 1.015(16) Å, and d(Z_2_–N–H) = 1.027(16) Å.? Note that these uncertainties do not reflect the precision of individual refinements, but are sample standard deviations representing both measurement uncertainty and the variance of chemical environments across neutron diffraction measurements. It is also worth noting that the number of observations in ref ? is much smaller for B–H bonds than for N–H bonds: 27 and 10 for B–H_t_ and B–H_b_, respectively, compared with 187, 233, and 74 for N^+^–H, Z–N–H, and Z_2_–N–H.
In a few cases, structural data are available from both X-ray and neutron diffraction, measured on the same molecule or its isotopologues at approximately the same temperature, thus allowing the most conclusive test on the performance of pHAR. Ammonia (NH_3_) at 160 K is compared with ND_3_ studied by powder neutron diffraction at 180 K (d(N–D) = 0.989(5) Å).? Note that, due to the isotope effect, d(N–H) > d(N–D) by ∼0.004 Å (as estimated from a gas-phase electron-diffraction experiment).? The comparison for diborane is with values from a gas phase electron diffraction experiment interpreted with spherical atomic electrostatic potentials: d(B–H_t_) = 1.196(8) Å, d(B–H_b_) = 1.339(6) Å.? Note, though, that X–H distances from single-crystal electron-diffraction experiments interpreted with spherical form factors tend to be too long. ?,?
First, the performance of various models (including IAM) was investigated for ammonia? varying the basis sets and the ways of simulating the crystal field (Table). The R values decrease as the level of basis set sophistication increases and the simulated crystal fields become more realistic. The maximum, minimum, and root-mean-square (rms) residual densities follow the same trend, with three minor exceptions. The corresponding three-dimensional maps for IAM, HAR, and pHAR confirm this finding (Figure): the residual density becomes progressively smaller and smoother, as apparent from the shrinking isosurfaces and the reduced undulation of the contour lines. The density at the bond sites is also reduced.
The refined bond lengths increase for every basis set as the crystal field model becomes more realistic, approaching the neutron ND_3_ reference value of 0.989(5)Å. Likewise, agreement with the neutron bond angle improves progressively from HAR to HAR/CC to pHAR. However, there is no obvious correlation between R value and bond length. The best match in N–H distance (pHAR, 6-311G(d,p), d = 0.968 Å, R = 0.0074) is not associated with the lowest R value (pHAR, pob-TZVP-rev2, d = 0.947 Å, R = 0.0069). For STO-3G, the electron-density prediction is extremely pooreven worse than IAMyet the structure prediction is not nearly as bad. Earlier refinements with a basis set containing diffuse functions or the simpler Hartree–Fock Hamiltonian showed better geometrical agreement, although with slightly larger R values. ?,? Superior results with the simpler HF method compared to DFT methods were also found recently in a study that benchmarked methods and basis sets for HAR.? The authors ascribed this finding to a cancellation of errors. The different behavior of R and d(N–H) for ammonia in our study indicates that, even with the present sophistication of modeling electron density using pHAR, the hydrogen-atom position still depends on how accurately the quantum-mechanical model describes the electron density in the vicinity of H and of N. This suggests that the choice of the basis set and Hamiltonian influences the geometric refinement results, but not necessarily in a foreseeable way. For ammonia, their effect appears to be larger than the effect due to the different models of the crystal field.
This conclusion is further substantiated by a comparison of pHAR with the best XHARPy results using two test examples reported in the literature.? It is found that pHAR showed lower performance for most but not all investigated parameters (see the Supporting Information, section 5). E.g., for the five O–H bonds in the molecule xylitol the average absolute difference between neutron and X-ray derived O–H distances from pHAR with the B3LYP/pob-TZVP-rev2 level of theory is 0.043(9)Å, whereas it is 0.011(7)Å from XHARPy with the revSCAN functional. The highly polar O–H bonds are most susceptible to the choice of method and basis set as we will discuss below. Considering that valence electrons in XHARPy are described by plane-wave basis functionswhich are by definition completely delocalizedand in view of the data in Table, the choice of basis set should be carefully examined to achieve the best structural agreement with neutron data. Currently, the use of basis sets with diffuse functions is limited in Crystal23 and thus in pHAR, because, as it is well-known, they generally cause numerical instabilities in periodic calculations with atom-centered Gaussian orbitals.
The borane/borate compounds were refined with IAM, conventional HAR, and pHAR, using the B3LYP/pob-TZVP-rev2 level of theory for both HAR and pHAR (more details are given in the section Experimental and Computational Methods). Table reports the R factors and residual electron densities for each compound. Consistent improvements are observed from IAM to HAR/pHAR across all the eight compounds. However, the differences between HAR and pHAR in terms of R values and residual densities are not significant. It suggests that the polarization due to the crystal field for these molecular crystals has no measurable impact on these metrics. There are some cases that exhibit significant residual electron density (Δρ_max_ > 0.5 e Å^–3^) even after HAR or pHAR refinements. Therefore, we have compared the residual densities between HAR, pHAR and the original multipole models ?,? in the Supporting Information, section 6. For some cases, the high residual densities are a chemical and not a model effect, for some cases HAR/pHAR show lower values, for some cases higher values than MM. Overall, a fair comparison between MM and HAR/pHAR is difficult because only merged data are available, the numbers of reflections used in MM and HAR/pHAR are not the same, and the multipole populations in MM are adjusted to the experimental data during the refinement, whereas the charge distributions in HAR/pHAR are fixed during the refinement.
As seen in the ammonia case, improvements in R-statistics do not necessarily ensure closer agreement of bond lengths with the reference data, and vice versa. Therefore, we examine trends in the N–H and B–H bond distances in more detail. Figure shows the distribution of refined d(N–H) and d(B–H) for each kind of molecule in the unit cells. Only (a) ammonia and (e) bis(ammonium) closo-hexaborate(6) exhibit differences in individual bond lengths between HAR and pHAR beyond a single standard uncertainty. On the other hand, aggregate analysis shows that the averages of the estimated standard deviations (esds) of d(X–H) gradually decrease from IAM to HAR to pHAR for all compounds (Figure). Although the reduction from HAR to pHAR may be as small as 0.001 Å, the trend is unambiguous, with a reduction ratio of roughly 10%, thereby improving statistical inferences on X–H bonds.
Comparison with the neutron reference values (horizontal dotted/dashed lines in Figure) reveals that the refined N–H bond lengths are systematically underestimated, whereas most terminal B–H bond lengths agree with the reference values within a single standard uncertainty. This motivated us to extend the analysis to C–H and O–H bonds, thereby completing the B, C, N, and O series. Among the nine compounds in this study, 28 independent C–H bonds but only two O–H bonds are found. To enable aggregate analysis of the X–H bond series, we therefore added xylitol, which contains five O–H as well as seven C–H bonds, and has both X-ray and neutron diffraction data available (see the Supporting Information, section 8). ?,? Analysis of these bond lengths refined with HAR shows the same trend as N–H and B–H: O–H bond lengths are clearly underestimated, whereas C–H bond lengths match the reference values.?
Table shows the average absolute deviations, Δ_X‑H_, relative to neutron referenceswhich can be regarded as an estimate of accuracy?for X = B, C, N, and O. For both HAR and pHAR, the underestimation of d(X–H) becomes more pronounced from B to O as the electronegativity increases. We speculate that the observed differences among bond types are related to bond polarity, which is low for B–H/C–H bonds and high for N–H/O–H bonds. Since Hirshfeld atoms are based on neutral spherical atoms, this may be a source of this trend. At the same time, there is a consistent reduction in Δ_X‑H_ from HAR to pHAR, which is more apparent for more electronegative atoms that are more involved in hydrogen bonding and, thus, benefit more from the accurate treatment of the periodic environment. Although the difference in individual refined bond lengths are insignificant, the aggregate analysis reveals closer agreement with the neutron reference for pHAR than for HAR for every bond type.
An interesting, clearly significant chemical feature can be observed for bis(2,6-lutidinium) closo-dodecaborate(12) in Figure. The 12 terminal B–H bonds in the dodecahedron are not identical. Six bonds match the literature neutron-diffraction data for terminal B–H bonds, whereas six are clearly overestimated by about 0.05 Å. Upon closer inspection of the crystal packing, four overestimated bonds are those that are in contact with N–H bonds in a B–H···H–N dihydrogen interaction, since the borane hydrogen atoms are hydridic but the ammonium N^+^-H hydrogen atoms are protic/acidic. This phenomenon has already been described for small Lewis acid–base adducts by several authors of this study elsewhere.? For the other two overestimated bonds, H atoms point toward the regions with high residual density, which could be attributed to a possible minor substitutional disorder with a halogen atom. Using the newly developed pHAR method, such subtle chemical features can now be detected in terms of accurate and precise B–H bond distances from X-ray diffraction.
Concluding Remarks
A new periodic Hirshfeld atom refinement variant, pHAR for short, has been developed. This technique can now be applied to periodic network compounds, which were previously beyond the reach of HAR (compare ref ?). Since pHAR is a natural extension of HAR, changes in chemical features induced by crystal fields can now be consistently explored at the same level of theory.
pHAR was tested on nine different borane, borate, ammonia, and ammonium systemsmolecular crystals or molecular saltsthereby expanding the range of available compounds containing accurate structural information on B–H bonds. pHAR yields significant improvements in refinement statistics and residual densities over IAM, but only marginal gains compared with conventional HAR. Since the impact of the basis setespecially the use of diffuse functionsappears to be more important than the method used to simulate the crystal field, further basis set optimization is needed to ensure the utility of pHAR.
Although the influence of periodic-boundary conditions on individual X–H bond lengths is insignificant for the tested systems, aggregate analysis shows a clear decrease in average absolute deviations from neutron-diffraction values and in estimated standard deviations. The underestimation of X–H bond lengthsmore pronounced with increasing bond polarityis also mitigated in pHAR. The more polar a bond type is, the more it benefits from the correct periodic description of the environment in pHAR.
Experimental and Computational Methods
pHAR combines Crystal23? and Tonto.? Crystal23 performs quantum-chemical calculations with periodic-boundary conditions using atom-centered Gaussian-type atomic orbitals. Tonto carries out electron-density partitioning and least-squares refinement of structural parameters. In this study, the self-consistent field (SCF) convergence threshold for the total energy was set to be 10^–7^ Hartree. The irreducible part of the Brillouin zone was sampled using the Pack-Monkhorst method with a shrinking factor of 6 for the construction of the Hamiltonian matrix. The corresponding numbers of sampling points in the Brillouin zone for triclinic, monoclinic, orthorhombic, simple cubic and face-centered cubic systems were 112, 80, 64, 24 and 16, respectively. The resulting supercell required for calculating the electron density of the reference cell typically had a radius of about 20 Å to absorb electron-density tails of the neighboring cells into the reference cell.
The data exchange between the two packages Crystal23 and Tonto was implemented at the level of density matrices. The keyword CRYAPI_OUT of Crystal23 is used to print density matrices in XML format. Tonto reads those density matrices and reproduces the electron density for each atom-centered grid system? of unique atoms in the unit cell. To improve computational efficiency in calculating electron density from density matrices within Tonto, the calculation of the product of two basis functions is skipped in two cases: when the corresponding density-matrix element is zero, or when the distance between the basis-function centers is sufficiently large so that their tails fall below the threshold value of e ^–20^ (≈2 × 10^–9^) at the center of their counterpart. The validity of the reproduced electron density was tested by comparing density maps produced by Tonto and Crystal23 on identical grids, and agreement to at least five decimal places at every grid point was confirmed.
After reproducing electron density within Tonto, stockholder partitioning? is performed to obtain Hirshfeld atoms, and the corresponding atomic form factors are calculated by Fourier transform. Structural least-squares refinement is then carried out. The CIF output from Tonto and the output file from Crystal23 are processed by lamaGOET? for convergence testing. The thresholds are set to be 10^–5^ hartree for the energy change and 0.01 for the ratio parameter shift/standard uncertainty. The pHAR cycle is repeated until the output passes the convergence test. Once the iteration has converged, the residual density is calculated from the final CIF. The overall procedure is illustrated in Figure. Detailed instructions and known issues are provided in the Supporting Information, sections 1 and 2.
All data used in this study stem from the literature; references are given in the caption of Figure. The temperatures of the determinations were between 9 and 100 K, and resolutions between 1.0 and 1.3 Å^–1^. For most compounds, only merged data sets were available, and the merging procedures are described in the original publications. For IAM in Tonto, hydrogen-atom ADPs were set to be isotropic. For HAR and pHAR, all structural parameters of hydrogen atoms were refined freely, including their anisotropic ADPs. Unless otherwise specified, HAR and pHAR were performed by using the pob-TZVP-rev2 basis set? and the B3LYP hybrid functional, ?,? which is motivated by their widespread use. For HAR, the smallest neutral formula unit was chosen, and no cluster charges were used for the simulation of the crystal environment. For the cocrystal compounds, whose molecular configurations of neutral formula units are not unique, the geometries for the electron-density calculation are provided in the Supporting Information, section 4.
HAR in Tonto requires atomic form factors that respect the site symmetries for atoms in special positions. For pHAR, this is now automatically given; and was part of the motivation for this method development. However, for conventional HAR, when molecules are selected for electron-density calculations without preserving each atom’s site symmetry, the corresponding atomic form factors become asymmetric, often causing problems in the refinement of high-symmetry systems. A practical procedure for the symmetrization of the Hirshfeld atom density was suggested in ref ?, which stated in section 2.4 that “details of the efficient implementation of this important procedure in Tonto will be reported elsewhere in due course”. In this study, we have now equipped Tonto with a subroutine for form-factor symmetrization. For all atoms in special crystallographic positions, atomic form factors are averaged over their site symmetries. It makes conventional HAR applicable to structures with atoms in special positions irrespective of their environment. Only with this implementation, all the comparisons between HAR and pHAR in this paper have become possible. Meanwhile, for pHAR, this procedure enhances the numerical stability of least-squares matrices (see the Supporting Information, section 3).
Similarly, ADP tensors are symmetrized by averaging over their site symmetry. This restores any broken symmetry that may have occurred during the rounding process of ADPs when recording in CIF, and reinforces the numerical stability.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Compton A. H.The Distribution of the Electrons in Atoms Nature 19159534334410.1038/095343 b 0 · doi ↗
- 2Brown, P. J. ; Fox, A. G. ; Maslen, E. N. ; O’Keefe, M. A. ; Willis, B. T. M. In International Tables for Crystallography; Prince, E. , Ed.; International Union of Crystallography: 2006; Vol. C, Chapter 6.1, pp 554–595.
- 3Cooper R. I.Thompson A. L.Watkin D. J. CRYSTALS enhancements: dealing with hydrogen atoms in refinement J. Appl. Crystallogr.2010431100110710.1107/S 0021889810025598 · doi ↗
- 4Malaspina L. A.Edwards A. J.Woińska M.Jayatilaka D.Turner M. J.Price J. R.Herbst-Irmer R.Sugimoto K.Nishibori E.Grabowsky S.Predicting the Position of the Hydrogen Atom in the Short Intramolecular Hydrogen Bond of the Hydrogen Maleate Anion from Geometric Correlations Cryst. Growth Des.2017173812382510.1021/acs.cgd.7b 00390 · doi ↗
- 5Allen F. H.Bruno I. J.Bond lengths in organic and metal-organic compounds revisited: XH bond lengths from neutron diffraction data Acta Crystallogr., Sect. B 20106638038610.1107/S 010876811001204820484809 · doi ↗ · pubmed ↗
- 6Grabowsky S.Genoni A.Bürgi H.-B.Quantum crystallography Chem. Sci.201784159417610.1039/C 6SC 05504 D 28878872 PMC 5576428 · doi ↗ · pubmed ↗
- 7Genoni A.BučinskỳL.Claiser N.Contreras-García J.Dittrich B.Dominiak P. M.Espinosa E.Gatti C.Giannozzi P.Gillet J.-M.Jayatilaka D.Macchi P.Madsen A. Ø.Massa L.Matta C. F.Merz K. M.Nakashima P. N. H.Ott H.Ryde U.Schwarz K.Sierka M.Grabowsky S.Quantum crystallography: Current developments and future perspectives Chem. Eur. J.201824108811090510.1002/chem.20170595229488652 · doi ↗ · pubmed ↗
- 8Krawczuk A.Genoni A.Current developments and trends in quantum crystallography Acta Crystallogr., Sect. B 20248024927410.1107/S 2052520624003421 PMC 1130189938888407 · doi ↗ · pubmed ↗