Decoding Adipose Tissue Phenotypic Switching: From Mechanisms to Computational Drug Discovery
Yuqing Ye, Ruxin Yin, Junjie Sun, Yuwei Dai, Di Zhao, Xiaoling Zou

TL;DR
This paper reviews how brown fat can turn into white fat and uses computational methods to find potential drugs to prevent this process.
Contribution
The paper introduces a computational screening approach to identify candidate molecules like 4-hydroxybenzoic acid for combating BAT whitening.
Findings
BAT whitening is linked to mitochondrial dysfunction, which may connect various triggers to metabolic decline.
4-hydroxybenzoic acid is proposed as a promising candidate for further experimental validation.
Bioinformatic tools are highlighted as valuable for preliminary screening of intervention molecules.
Abstract
This review aims to explore the therapeutic potential of brown adipose tissue (BAT) to combat obesity and associated metabolic disorders by synthesizing the multifactorial influences and underlying mechanisms of BAT whitening and employing computational screening to identify promising candidate molecules for further investigation. BAT whitening is characterized by the loss of thermogenic capacity, representing a critical aspect of adipose plasticity. Although diverse physiological and environmental triggers have been identified, the mechanistic interconnections underlying this process remain poorly understood. Emerging evidence supports an integrated view of these factors, and bioinformatic approaches now provide a valuable tool for the preliminary screening of potential intervention candidates. This review synthesizes current understanding on BAT whitening, from influencing factors…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
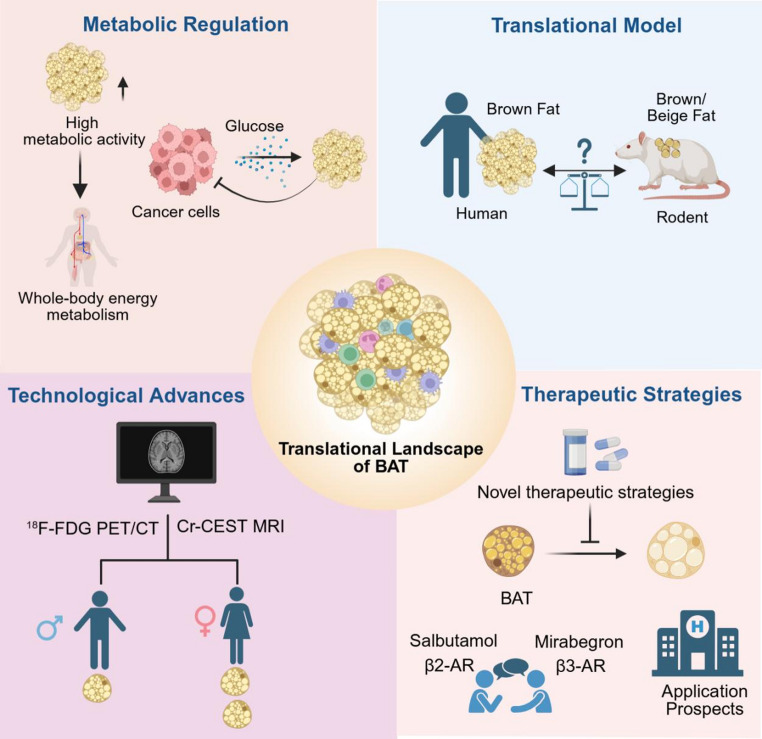 Figure 1
Figure 1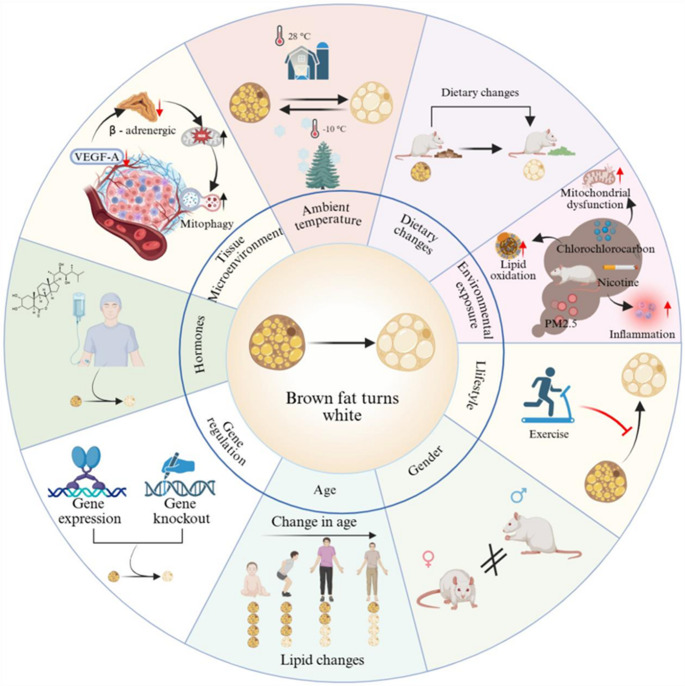 Figure 2
Figure 2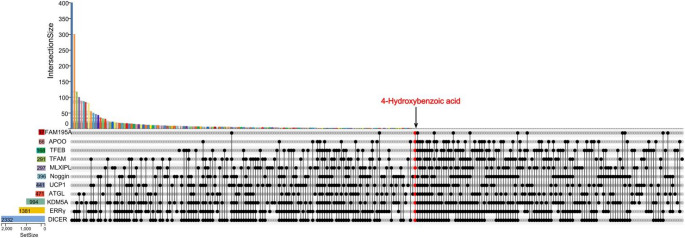 Figure 3
Figure 3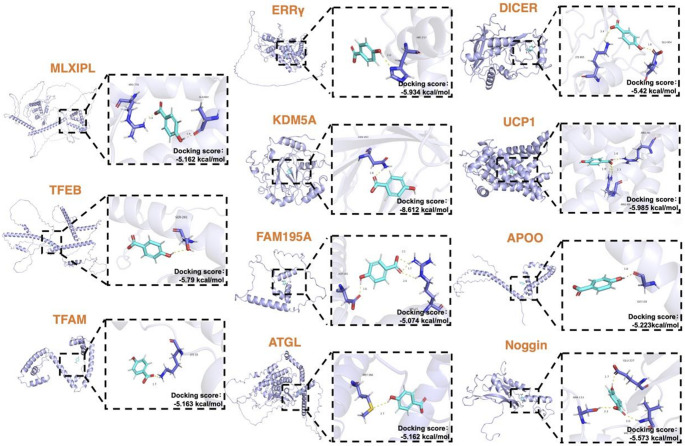 Figure 4
Figure 4- —the Scientific Research Fund Project of Hunan University of Chinese Medicine
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSingle-cell and spatial transcriptomics · Pluripotent Stem Cells Research · Adipose Tissue and Metabolism
Introduction
Obesity, a prevalent chronic disease driven by metabolic dysfunction, represents a major global public health challenge [1, 2]. Adipose tissue, a central regulator of metabolic homeostasis, plays a key role in the process of obesity [3]. Notably, brown adipose tissue (BAT) has emerged as a promising therapeutic target for obesity and associated disorders not only for its canonical role in non-shivering thermogenesis but also for its regulatory effects on systemic glucose and lipid metabolism [4, 5]. Translating this therapeutic potential into clinical applications requires a comprehensive understanding of BAT physiology, appropriate research models, advanced detection technologies, and therapeutic strategies focused on either activating BAT or preventing its functional decline.
Specifically, regarding strategies to prevent functional decline of BAT, the central challenge is the process of “whitening”—a process in which thermogenic BAT loses its specialized phenotype and function due to cellular plasticity [6–8]. Accordingly, this review delineates the multiple influencing factors of BAT whitening and synthesizes their converging pathophysiological mechanisms. Mitochondrial dysfunction is widely observed and may be proposed as a potential initiating event in this process, which could trigger a cascade of downstream alterations. These include disrupted thermogenesis and lipid metabolism, activated stress and inflammatory responses [9–12]. These processes, along with dysregulated autophagy (including mitophagy) and degradation of the vascular and neural microenvironment, may contribute to a self-perpetuating cycle that drives the functional decline of BAT [13–16].
Moreover, to explore the therapeutic potential of targeting BAT whitening, we performed virtual screening and molecular docking, identifying the gut microbiota-derived metabolite 4-hydroxybenzoic acid (4-HBA) as a candidate for further study in BAT whitening. It has shown anti-inflammatory and antioxidant properties in experimental models, with limited evidence indicating a potential role in promoting adipose browning and improving metabolic homeostasis, possibly via the AMP-activated protein kinase (AMPK) -Dynamin-related protein 1 (DRP1) pathway [17, 18]. However, these findings are primarily based on computational and experimental models, which may not fully reflect human physiology. Their translational relevance is further limited by the absence of systematic dose-response and long-term safety studies in mammals [18]. While 4-HBA may be of interest for metabolic research, its therapeutic potential and precise mechanisms in obesity-related conditions require substantial further investigation.
Translational Landscape of Brown Adipose Tissue
Metabolic Regulation
Evolutionarily, BAT helped ancestors survive cold and hunger and now acts as a buffer for blood glucose and lipids [19, 20]. BAT contributes to overall health by boosting energy expenditure, enhancing glucose clearance, and exerting anti-inflammatory effects [3, 21]. Furthermore, it exhibits potential anticancer properties by competing with tumor cells for glucose, and influences female reproductive functions through the secretion of factors such as irisin, fibroblast growth factor 21 (FGF21), and adiponectin [22, 23]. Retrospective analyses link BAT presence to favorable metabolic profiles, such as improved glucose and lipid homeostasis, as well as lower hepatic fat content [24]. This is further associated with reduced cardiometabolic risk, particularly in obesity [25]. These findings underscore the potential relevance of BAT in metabolic health.
Translational Model
The evolutionary relationship between human BAT and rodent models has been debated since the discovery of metabolically active BAT in adults. Early studies using cold-exposed young mice indicated a transcriptional similarity between human BAT and murine beige adipocytes [26, 27]. However, recent research demonstrates that the classical interscapular BAT in middle-aged mice closely models the key features of human supraclavicular BAT, supporting its translational relevance [28, 29].
Anatomical mapping studies have uncovered regional heterogeneity in human BAT [30]. Deep cervical BAT exhibits gene expression signatures closely resemble that of murine interscapular classical BAT, whereas BAT in intermediate regions displays moderate UCP1 expression, resembling murine inguinal beige fat [30, 31]. This distinction is further supported by infant adipose studies, where interscapular and perirenal BAT align with murine classical and beige signatures, respectively [32]. These findings underscore the necessity of using physiologically relevant models and considering depot-specific differences for translational BAT research.
Technological Advances
In humans, BAT activity is primarily assessed through 18F-fluorodeoxyglucose positron emission tomography-computed tomography (^18^F-FDG PET/CT). Evidence from this modality demonstrates that women generally possess greater BAT volume or activity than men [33, 34], and that cold exposure robustly activates BAT to increase energy expenditure [35]. Recently, Creatine Chemical Exchange Saturation Transfer Magnetic Resonance Imaging (Cr-CEST MRI) has emerged as an alternative approach. By exploiting the chemical exchange properties of creatine, Cr-CEST MRI sensitively detects cold-induced BAT activation with high concordance to ^18^F-FDG PET/CT. Given its advantages of no ionizing radiation, high reproducibility and multimodal capability, Cr-CEST MRI represents a promising tool for investigating BAT pathophysiology in metabolic disorders [36].
Therapeutic Strategies
Current therapeutic approaches for targeting BAT primarily focus on activating its thermogenic function or preventing its whitening. A central pharmacological debate concerns whether β2- or β3-adrenergic receptors (β2-ARs vs. β3-ARs) predominate in human BAT activation. Evidence favoring β2-ARs includes its higher expression in human BAT and the ability of agonists salbutamol to promote glucose uptake and thermogenesis; however, its activation is associated with additional cardiovascular effects [37]. In contrast, the β3-AR agonist mirabegron activates BAT via the β3-AR/cAMP/UCP1 signaling cascade and improves glucose and lipid metabolism, yet its clinically effective doses non-selectively activate other β-ARs, thereby increasing cardiovascular risk, while lower doses show limited efficacy [38, 39]. Therefore, clarifying receptor-specific roles and developing tissue-selective delivery systems are crucial for safer BAT-targeted therapies.
Another strategy aims to prevent the whitening of BAT. Supporting evidence from various clinical and experimental studies, summarized in Table 1, highlights the potential of the adrenergic agents mentioned above and specific anti-whitening compounds. Promising agents include the dual GIP/GLP-1 receptor agonist tirzepatide, which restores BAT morphology via improving mitochondrial dynamics, mitophagy, and biogenesis while suppressing endoplasmic reticulum (ER) stress and inflammation [40]; the senolytic combination of dasatinib and quercetin, which clears senescent cells, downregulates lipogenic genes, and enhances thermogenic gene expression to inhibit whitening [41]; the antidiabetic agent imeglimin counters whitening by inhibiting de novo lipogenesis and shifting mitochondrial substrate utilization toward lipids, independently of the gut microbiota [42]; the urate-lowering drug dotinurad reduces uric acid uptake and reactive oxygen species (ROS) production, thereby activating UCP1 and other thermogenic genes to restore BAT morphology [43], and the probiotic Roseburia hominis exerts protective effects partly through bacterial production of nicotinamide riboside, which elevates NAD^+^ to activate the Sirtuin1/mTOR pathway and subsequently leads to a reduction in lipid droplets along with the upregulation of thermogenic genes [44]. Together, these strategies highlight the therapeutic potential of preserving BAT identity and function for metabolic benefit.Table 1. Summary of therapeutic studies targeting BAT thermogenesis and whitening preventionRefAgentStudy TypeStudy designPopulation/ModelSample sizeIntervention/DoseComparatorDurationMain outcomes[37]SalbutamolHuman clinical dataSingle-center, randomized, double-blinded, crossover designHealthy white Caucasian men; Age: 19–35 years; BMI: 19.2–26.5 kg/m²Total: 10; EE analysis: 9Salbutamol 250 µg IV + placebo POSalbutamol 250 µg IV + propranolol 80 mg PO; crossover randomizationSingle dose; ~4 h per study visitSalbutamol stimulates BAT glucose uptake, which correlates with increased energy expenditure[39]MirabegronHuman clinical dataRandomized, double-blinded, placebo-controlled crossover studyHealthy lean men; Age: 18–30 years; BMI: 18–25 kg/m²Total: 20; MRI analysis: 19Single oral dose of mirabegron 200 mgPlacebo (oral);2-week washout༛crossover randomizationSingle dose; ~3.5 h per study visitMirabegron reduces BAT fat fraction and increases skin temperature in the supraclavicular region[40]Tirzepatideanimal modelControlled experimental design (induction + treatment phases); 8 groups (4 baseline + 4 treatment)3-month-old female C57BL/6 mice; Induced phenotypes: obesity + type 2 diabetes + estrogen deficiencyTotal: 80; 10 mice per groupTirzepatide 10 nmol/kg/day, subcutaneous injectionVehicle (Tris-HCl buffer, pH 8.0, 40 mM), subcutaneous injection16 weeks (12-week induction + 4-week daily intervention)Tirzepatide reverses BAT whitening; restores multilocular morphology; upregulates thermogenic markers/GLP-1R; improves mitochondrial function; reduces inflammation/ER stress[41]Senolytic cocktail (Dasatinib + Quercetin)animal modelControlled experimental design (induction + treatment phases); 4 parallel groups (2 baseline + 2 treatment)8-week-old female C57BL/6NTac mice; Induced phenotype: obesityTotal: ~24–28;6–7 per groupDasatinib 5 mg/kg + Quercetin 50 mg/kg, oral gavageVehicle (10% PEG400), oral gavage24 weeks (24-week diet induction; intervention in weeks 16–24)Senolytic cocktail prevents BAT whitening; increases thermogenic markers; reduces lipogenic genes; eliminates senescent cells[42]Imegliminanimal modelControlled experimental design (6-week treatment + microbiota ablation subgroup); 4 core groups (NCD, HFD+Vehicle, HFD+Imeglimin, HFD+Imeglimin+Antibiotics)18-week-old male C57BL/6 mice; Induced phenotype: obesityTotal: ~38 (max); 8–10 (max) per group;Imeglimin 300 mg/kg/day, oral administrationVehicle (HFD feeding without imeglimin); NCD group; HFD+Imeglimin+Antibiotics (subgroup comparator)6-week imeglimin treatment; Antibiotic co-treatment concurrent with imeglimin for 6 weeksImeglimin attenuates BAT whitening; decreases de novo fatty acid synthesis genes; inhibits mitochondrial basal respiration (pyruvate-stimulated) to enhance energy expenditure[43]Dotinuradanimal modelControlled experimental design (induction + treatment phases); 4 parallel groups (2 baseline + 2 treatment)8-week-old male C57BL/6 mice; Induced phenotypes: obesityTotal: ~62 (max); 15–16 (max) per group;Dotinurad 50 mg/kg/day, oral administrationVehicle (feeding without dotinurad)20–22 weeks total (16–18-week diet induction + 4-week dotinurad treatment)Dotinurad reverses BAT whitening; activates UCP1 (mRNA/protein upregulation) and thermogenic genes (Pgc-1α, Dio2); reduces BAT ROS production[44]Roseburia hominisanimal modelControlled experimental design; 3 parallel groups (ND-PBS, HFD-PBS, HFD-R. hominis)5-week-old male C57BL/6 mice; Induced phenotypes: obesityTotal: 15; 5 per groupR. hominis 10⁹ CFU/mouse, oral gavage, dailyVehicle (PBS), oral gavage15 weeks total (4-week co-housing + 11-week intervention)Roseburia hominis inhibits BAT whitening; upregulates BAT thermogenic genes (Cidea, Ucp1, Pgc-1α, Dio2)Abbreviations: BAT brown adipose tissue, BMI body mass index, CFU colony-forming units, Cidea cell death-inducing DFFA-like effector a, Dio2 type II iodothyronine deiodinase, EE energy expenditure, ER endoplasmic reticulum, GLP-1R glucagon-like peptide-1 receptor, HFD high-fat diet, IV intravenous, MRI magnetic resonance imaging, NCD normal chow diet, PBS phosphate-buffered saline, PEG400 polyethylene glycol 400, Pgc-1α peroxisome proliferator-activated receptor gamma coactivator 1-alpha, PO per oral, ROS reactive oxygen species, Tris-HCl tris(hydroxymethyl)aminomethane hydrochloride, UCP1 uncoupling protein 1.
These key aspects of BAT—metabolic regulation, translational model, technological advances, and therapeutic strategies—are summarized in Fig. 1. Concurrently, a critical translational perspective necessitates acknowledging the predominant reliance on murine models, which limits direct extrapolation due to interspecies differences like circadian biology—thus highlighting the need for validation in diurnal models and human-centric studies to bridge the preclinical-clinical gap.Fig. 1. Translational landscape of brown adipose tissue (BAT). Schematic illustration of the translational landscape of BAT, with an emphasis on four domains: metabolic regulation (its multiple functions) [19–25]; Translational model (similarities between mouse and human BAT) [26–32]; technological advances (^18^F-FDG PET/CT and Cr-CEST MRI) [33–36]; and therapeutic strategies (activate existing brown fat or prevent its conversion into white fat) [37–44]
Influencing Factors of Brown Adipose Tissue Whitening
As mentioned above, therapeutic strategies are directed toward either activating existing BAT or preventing its whitening. Understanding the process of BAT whitening is key to developing effective treatments. The following section systematically examines the key influencing factors of this process (Fig. 2), categorized as: (1) external inputs (ambient temperature, diet, environmental exposure, lifestyle); (2) intrinsic physiological modulators (age, gender, hormones); and (3) local executors (tissue microenvironment, genetic regulation). This progression from systemic triggers to local effectors sets the stage for identifying the convergent downstream pathways that drive the phenotypic switch.Fig. 2. Overview of the various factors leading to the whitening of BAT. The schematic illustrates nine major factors influencing BAT whitening: warm temperature induces BAT whitening while cold exposure maintains its thermogenic capacity [9, 14, 45, 46, 48]; high-fat diets accelerate the process [47–49]; environmental exposures (e.g., DP, PM2.5, nicotine) induce whitening via inflammation, mitochondrial dysfunction, and altered lipid oxidation [50–52]; exercise suppresses whitening progression [53–55]; aging exacerbates whitening [25, 56]; gender differences impact metabolic responses [57, 58]; hormone signaling modulates the process [59–64]; tissue microenvironment changes (e.g., reduced VEGF-A) drive whitening through increased ROS and mitophagy [65, 66]; and gene regulation by multiple key genes critically influences the process [11, 67–70, 91–97]
Ambient Temperature
Ambient temperature critically regulates metabolism by modulating the morphology and function of BAT. Reduced cold exposure dually suppresses energy expenditure, diminishing both thermogenic demand and intrinsic capacity. Since the 1960 s, increased indoor temperatures due to widespread climate control have lowered cold exposure, contributing to a decline in human thermogenic capacity and BAT activity [71]. Additionally, energy expenditure and cold-induced thermogenesis are higher in winter than in summer, with BAT metabolic activity peaking during the colder months [72].
In rodents, exposure to warm conditions (27–30 °C) promotes BAT whitening, characterized by reduced expression of thermogenic genes and proteins, increased macrophage infiltration and crown-like structure formation [9, 45]. This shift is associated with suppressed sympathetic activity, decreased norepinephrine turnover, downregulated tyrosine hydroxylase expression, and enhanced mitochondrial degradation alongside reduced mitochondrial mass [14, 67]. Conversely, cold exposure counteracts whitening, reducing triglyceride content and downregulating lipogenic factors in BAT [46].
Diet
Altered dietary composition is a key driver of BAT whitening in rodent [10]. Chronic high-fat diet (HFD) feeding promotes lipid deposition and whitening in BAT, characterized by the upregulation of adipogenic and pro-inflammatory genes, increased ER stress, and downregulation of thermogenic genes [47, 48]. Short-term HFD rapidly induces lipid accumulation via fatty acid influx-driven endocannabinoid synthesis, which blunts norepinephrine signaling and promotes mitochondrial fusion, thereby impairing fatty acid oxidation [49]. Similarly, a high-fructose diet (HFrD) induces this phenotype, which can be counteracted by PPAR-α agonists through enhanced thermogenesis, β-oxidation and angiogenesis [15]. In rats, both HFD and high-fat/high-sucrose diets (HFSD) impair UCP1-mediated oxidation and shift metabolism toward fatty acid esterification and triglyceride synthesis in BAT, thereby exacerbating obesity [73, 74].
Dietary interventions further influence BAT plasticity: positive modulators, including coffee, tart cherry supplementation, and omega-3 fatty acids, promote mitochondrial biogenesis, upregulate thermogenic gene expression, and reduce lipid infiltration [75–77]. Conversely, negative modulators such as palm and interesterified palm oil suppress thermogenic markers and promote inflammation, thereby accelerating BAT whitening [78].
Environmental Exposure
Environmental exposure significantly impacts BAT, inducing metabolic changes with obesity and related diseases. Chronic exposure to fine particulate matter (PM2.5) induces BAT whitening in rodents via mitochondrial dysfunction, ER stress and inflammation [50]. Similarly, chemical pollutants such as the flame retardant dechlorane plus (DP) and the fungicide prothioconazole (PTC) promote lipid accumulation and whitening in mice [51, 79]. Endocrine disruptors like bisphenol S (BPS) can trigger whitening through glucocorticoid receptor-mediated epigenetic alterations [68], while prenatal exposure to nicotine or polystyrene nanoplastics (PSNPs) results in impaired BAT function in male offspring [52, 80]. Notably, combined ozone and heat exposure exerts a synergistic effect on whitening, linked to activation of the stress axis and increased stress hormone secretion [81].
Lifestyle
Early-life exercise contributes to long-term metabolic health by suppressing BAT whitening, which helps prevent obesity and diabetes through preserved muscle mass and improved lipid metabolism [53]. Since BAT whitening and mitochondrial protein downregulation rapidly progress when exercise ceases, consistent physical activity is essential to sustain these protective effects [54]. Exercise-derived or dietary lactate counteracts BAT whitening in obesity by activating the G protein-coupled receptor 81 (GPR81)-Ca2+/calmodulin-dependent protein kinase (CaMK) pathway, which synergizes with adrenergic signaling to upregulate UCP1 expression and mitochondrial DNA (mtDNA) content [55]. Additionally, the exercise-induced myokine irisin exerts protective effects on BAT, contributing to maintaining its thermogenic capacity and resisting whitening [82, 83]. Furthermore, exercise can also mitigate whitening through sympathetic activation, enhanced mitochondrial function, and promoted BAT endocrine activity through the release of 12,13-dihydroxy-9Z-octadecenoic acid (12,13-diHOME) [84].
Age
Aging serves as a key driver of BAT functional decline and whitening. Large-scale human studies reveal a progressive, age-dependent decline in both BAT metabolic activity (measured via ¹⁸F-FDG PET/CT) and overall abundance [25, 56]. In rabbits, age-related BAT whitening involves progressive loss of thermogenic and mitochondrial function, enhanced autophagy, vascularization, innervation and immune cell infiltration [16]. Long non-coding RNAs (LncRNAs) participate in this process, with differential LncRNA mainly participating in purine metabolism, Wingless and Int-1(Wnt), Peroxisome Proliferator-Activated Receptor (PPAR), Cyclic Guanosine Monophosphate/cGMP-Dependent Protein Kinase (cGMP/PKG), and lipid metabolism pathways [85]. Besides, circular RNA (circRNA)-and lncRNA-mediated competing endogenous RNA (ceRNA) networks regulate age-related whitening through the Mitogen-Activated Protein Kinase (MAPK) and Rat sarcoma virus oncogene homolog (Ras) signaling pathways [86]. Temporal transcriptomic profiling further delineates two distinct phases of BAT whitening: an early stage marked by alterations in angiogenesis, mitochondrial function, and thermogenesis, followed by a late stage characterized by diminished ATP synthesis, reduced fatty acid oxidation, and a decline in progenitor differentiation potential [87].
In lambs, postnatal BAT whitening is associated with the downregulation of Vascular Endothelial Growth Factor A (VEGFA) and thermogenic genes, as well as the upregulation of Cytochrome P450 Family 1 Subfamily A Member 1 (CYP1A1) [88]. In mice, aging promotes BAT whitening through T cell-derived Interferon-gama (IFN-γ) [89]. Furthermore, α-lipoic acid ameliorates age-related metabolic decline and enhances mitochondrial fatty acylation to restore thermogenesis and glucose homeostasis [90].
Gender
Gender significantly impacts brown fat metabolism, showing different adaptations in various physiological states. In humans, BAT is markedly more prevalent in women than in men, and its age-related decline in both metabolic activity and abundance is more pronounced in men, whereas women exhibit a relatively attenuated reduction over time [25, 91]. Similarly, male mice exhibit morphological abnormalities and thermogenic dysfunction in brown fat during aging, while female rodents better maintain thermogenic capacity [57]. Maternal high-fat diet affects offspring with sex differences, causing brown fat whitening, inflammation, and abnormal oxidative phosphorylation in male offspring, while females show different metabolic adaptations [58].
Hormones
Emerging evidence indicates that glucocorticoid signaling (GCS) induces BAT whitening through multiple pathways, including miR-21-5p-mediated suppression of thermogenic genes, leptin resistance induction, BTG1-dependent autophagy, and disruption of mitochondrial homeostasis [59–61]. Estrogen inhibits whitening by modulating hypothalamic ceramide and ER stress to preserve BAT function [62], while androgen promotes lipid accumulation and the downregulation of UCP1 [63]. Hyperprolactinemia reduces thermogenic genes expression and may cause mitochondrial dysfunction and inflammation in BAT [64].
Tissue Microenvironment
BAT exhibits high vascularity and is regulated by the sympathetic nervous system. VEGF-A absence reduces capillaries, lowers β-adrenergic signaling, increases mitochondrial reactive oxygen species (mtROS) and promotes mitophagy [65]. Intravitreal injection of anti-VEGF antibodies in mice reduces brown fat VEGF levels, increases lipid accumulation, lowers vascular density and downregulates mitochondrial genes, causing whitening [66].
Genetic Regulation
The maintenance of brown fat function and identity is critically dependent on a multi-layered gene regulatory network. Alterations such as gene knockout or overexpression disrupt this network, leading to BAT whitening and functional decline. Negative regulators, including Carbohydrate-responsive element-binding protein β (ChREBP-β) and the histone demethylase KDM5A, suppress BAT identity when overexpressed by repressing mitochondrial dynamics and thermogenic gene programs, respectively [11, 68].
Conversely, the loss of positive regulators—including Transcription Factor EB (TFEB) [67], Mitochondrial Transcription Factor A (TFAM) [69], Estrogen-Related Receptor γ (ERRγ) [92], the RNA-binding protein Family with sequence similarity 195 member A (FAM195A) [93], Adipose Triglyceride Lipase (ATGL) [94], the miRNA-processing enzyme Dicer [95], UCP1 [96, 97], Apolipoprotein O (APOO) [98], and the secreted protein Noggin [70]—disrupts key cellular processes. Deficiency in any of these factors disrupts essential cellular processes—such as autophagic flux, lipid catabolism, mitochondrial function, oxidative stress response, and thermogenic gene expression—converging on the common pathological outcome of BAT whitening. Therefore, the stability of the brown adipocyte phenotype is not governed by a single gene but emerges from the integrity of this integrated, multi-tiered regulatory system, wherein an imbalance at any level can precipitate systemic functional collapse.
Integrated Molecular Mechanism of BAT Whitening
Moving beyond cataloguing individual factors, we propose that BAT whitening is orchestrated through a self-reinforcing, mitochondria-centric vicious cycle. Within this framework, mitochondrial dysfunction acts not merely as one of many defects, but as a critical integrative hub and a principal cellular executor of the phenotypic switch. It serves as both the common downstream consequence where diverse upstream insults (e.g., metabolic stress) converge, and a key proximate cause that initiates and amplifies the pathological cascade.
Within this framework, the contributors to BAT whitening largely converge to directly initiate and exacerbate mitochondrial dysfunction. This dysfunction manifests as impaired electron transport chain activity, disrupted dynamics, and structural damage, driving increased mtROS production and cellular stress [99–101]. Crucially, this damage establishes a self-reinforcing loop, where mitochondrial-derived signals further exacerbate the initial dysfunction, locking the cell in a state of progressive energetic deficit [102–105].
Downstream, this pathophysiological cascade is linked to the suppression of the thermogenic gene program and a loss of brown adipocyte identity [106, 107], alongside disruptions in lipid metabolism, such as reduced fatty acid oxidation and lipolysis [104, 108, 109]. Furthermore, attenuated thermogenic signaling is associated with the activation of cellular autophagy, particularly mitophagy. This selective degradation of mitochondria can lead to a reduction in mitochondrial mass [13]. Thus, functional impairment and quantitative loss synergistically deepen the metabolic crisis.
These cellular disturbances are accompanied by secondary tissue-level alterations, including impaired angiogenesis (promoting a hypoxic microenvironment), local inflammatory activation, and oxidative stress-associated sympathetic denervation, which collectively disrupt thermogenic activation [109–111]. This integrated perspective highlights whitening as a progressive, self-reinforcing cycle of energetic deficit and functional decline.
In summary, this integrated framework recasts BAT whitening as a hierarchical, vicious cycle initiated by diverse factors, executed and amplified through mitochondrial dysfunction, and often accompanied by downstream cellular and tissue-level sequelae. Clarifying this mechanism underscores that preserving mitochondrial function is not merely supportive but central to intercepting the entire pathological cascade.
Integrative Bioinformatic Exploration Highlights 4-Hydroxybenzoic Acid as a Candidate for Therapeutic Investigation
To identify potential therapeutic targets for mitigating the whitening of BAT, we focused on the key regulatory factors involved in this process as outlined previously. Eleven proteins critically associated with adipocyte plasticity and metabolic reprogramming were selected for analysis: MLXIPL (encoded by ChREBP-β), TFEB, TFAM, ERRγ, KDM5A, FAM195A, ATGL, DICER, UCP1, APOO and Noggin (see Methods for detailed selection criteria and rationale). Functional categorization revealed that these 11 proteins cluster into three distinct classes—transcriptional/epigenetic regulators (MLXIPL, TFEB, TFAM, ERRγ, KDM5A, FAM195A), metabolic enzymes (ATGL, DICER), and transporters and secreted proteins (UCP1, APOO, Noggin)—collectively representing the major regulatory layers of BAT whitening, ranging from nuclear transcriptional control and metabolic substrate switching to extracellular signal integration. To ensure clinical relevance, the three-dimensional crystal structures of these human target proteins were obtained from the UniProt database. Structural preparations were performed using AutoDockTools, including hydrogen addition, charge assignment, and atom type definition [112]. Molecular docking-based virtual screening was subsequently conducted against the U.S. Food and Drug Administration (FDA)-approved compound library using AutoDock Vina. Resulting docking poses were visualized and analyzed using PyMOL.
Using a binding energy threshold of ≤ − 5 kcal/mol—a criterion widely accepted in molecular docking studies as indicative of strong and stable binding [113–116], we initially identified candidate compounds exhibiting high binding affinity for each individual protein. Intersection analysis identified 4-Hydroxybenzoic acid (4-HBA) as the sole compound that exhibited significant binding affinity for all eleven target proteins (Fig. 3). Visualization of the molecular docking models (Fig. 4) indicated that 4-HBA forms stable molecular interactions with key residues within the binding pockets of each protein.Fig. 3. Identification of 4-Hydroxybenzoic acid by intersection analysis. An UpSet plot displays the intersections of hit compounds (predicted binding energy ≤ -5.0 kcal/mol) from an FDA-approved library across 11 target proteins. Each vertical bar represents the intersection for the combination of proteins connected by the dots below. The red column highlights the sole intersection of all 11 proteins, which exclusively contains 4-Hydroxybenzoic acidFig. 4Predicted binding modes of 4-Hydroxybenzoic acid with 11 target proteins. Molecular docking results showing the binding poses of 4-Hydroxybenzoic acid (displayed as cyan sticks) within the binding pockets of 11 target proteins (MLXIPL, TFEB, TFAM, ERRγ, KDM5A, FAM195A, ATGL, DICER, UCP1, APOO and Noggin), each represented in blue-purple cartoon representation. Putative hydrogen bonds are depicted as yellow dashed lines. Key interacting residues are shown as sticks, with oxygen atoms colored red and nitrogen atoms colored dark blue. The structural visualization was generated using PyMOLchematic illustration of the Translational L
4-HBA, an aromatic organic acid derived from benzoic acid, is one of the most abundant phenolic compounds produced by the human gut microbiota. It is generated endogenously via tyrosine metabolism and exogenously through microbial fermentation of dietary polyphenols present in foods such as green tea, berries, olives, and coconut [117, 118]. Preliminary evidence suggests 4-HBA may exhibit multiple bioactive properties, including antioxidant, anti-inflammatory, and antimicrobial effects, and been implicated in the regulation of gut microbial composition by promoting beneficial bacteria and suppressing harmful species [17]. Furthermore, 4-HBA serves as a precursor for coenzyme Q10 and has been shown to exhibit neuroprotective and cardioprotective functions in previous studies [119, 120]. Its metabolic fate involves conversion into several biologically active derivatives, including salicylic acid, vanillic acid, gallic acid and ellagic acid. This biotransformation is mediated through pathways involving both gut microbiota and host enzymatic activities [18]. Emerging evidence suggests that certain metabolites of 4-HBA, including vanillic acid and ellagic acid, can promote adipose tissue thermogenesis and mitigate metabolic disturbances in models of obesity [121, 122]. Besides, a study has reported that 4-HBA promotes adipose tissue browning in diet-induced obese mice through AMPK-DRP1 activation [18].
However, several important limitations of the current research on 4-HBA must be acknowledged. The existing evidence remains limited to short-term rodent studies, which lack clinical correlation. Furthermore, the pharmacokinetics and tissue-specific bioavailability of 4-HBA in mammals are still unclear, while its potential off-target effects and long-term safety profiles remain largely unexplored. Finally, the ecological relevance and generalizability of its gut microbiota-modulating effects across diverse human populations require further validation.
While these preliminary findings cautiously propose 4-HBA as a compound of interest for metabolic research, substantial additional studies are required to comprehensively evaluate its therapeutic potential and mechanistic underpinnings in obesity-related metabolic conditions.
Conclusions and Discussion
Given the crucial role of BAT in regulating systemic energy metabolism, preventing its whitening represents a promising therapeutic target against obesity and related metabolic diseases [6, 24]. This review systematically summarizes the translational landscape of BAT and provides a comprehensive overview of multiple factors influencing BAT whitening and underlying mechanisms (Table 2). Furthermore, we integrate these findings to propose that whitening progresses through a self-reinforcing cycle centered on mitochondrial dysfunction as an integrative hub, leading to progressive adipose tissue dysfunction.Table 2. Summary of the factors and potential mechanism linked to BAT whiteningRefMouse model/exp. conditionPhenotypes and mechanismAmbient Temperature[9]12-week-old female C57BL/6J mice were kept at 28 °C or at 6 °C for 10 daysWarm conditions: macrophage infiltration↑ crown-like structure formation↑[14]A/J mice were raised under either 22–30 °CWarm conditions: thermogenic program↓ tyrosine hydroxylase expression↓ norepinephrine turnover↓ sympathetic activity↓[45]10-week-old male C57BL/6J mice were kept at 27 °C or at 22 °CWarm conditions: Ucp1/Pgc-1α/Elovl3↓[67]The mice were housed under either 22–30 °C for 7 daysWarm conditions: mitochondrial biogenesis markers↓ mitochondrial degradation markers↑Diet[15]3-month-old male C57BL/6 mice were fed with CD、HFD or HFrDHFrD-induced BAT whitening was reversed by PPAR-α agonists: thermogenesis↑β-oxidation↑ VEGFA-driven angiogenesis↑[47]3-month-old male C57BL/6J mice were fed with HFD for 12、16 or 20 weeksHFD: Cidea/Plin1↑Pparα/Ucp1↓ pro-inflammatory (Tlr4/Nlrp3) ↑ER stress markers (Atf4/Chop/Gadd45)↑[49]15-week-old male C57BL/6J mice were fed with HFD for 1、3 or 7 daysHFD: substrate uptake↓ mitochondrial fusion↑ fatty acid oxidation↓[73]Male Wistar rats were fed with CD or HFSD for 8 weeksHFSD: Ucp1-driven oxidation↓ lipolysis↓ tyrosine hydroxylase↓[74]8-week-old male Wistar rats were fed with HCD、HFD or HFSD for 12 weeksHFD and HFSD diet: Ucp1-mediated oxidation↓ insulin receptor signaling↓ adipokine balance↓Environmental Exposure[50]Mice were exposed to chronic PM2.5 inhalation or PM2.5 intratracheal instillationPM2.5: lipid oxidation↑ mitochondrial/ER stress↑ inflammation↑ insulin resistance↑[68]Mice were fed diets supplemented with low, medium, and high levels of BPSBPS: KDM5A-dependent epigenetic reprogramming↑ estrogenic GR activation↑[52]Pregnant rats were randomly divided into a nicotine group and a control groupPrenatal nicotine exposure: brown fat-specific gene expression↓ mitochondrial structure/function↓[80]Pregnant C57BL/6J mice were randomly divided into a PSNPs group and a control groupPrenatal PSNPs exposure: lipogenic proteins (FASN, SREBP-1c/CD36/DGAT2)↑lipophagy pathway↓[81]8-week-old male C57BL/6 mice were divided into control, heat, ozone, and combined exposure groupscombined exposure: stress hormonesglucocorticoid↑ epinephrine↑Lifestyle[53]4-week-old male OLETF (obese) rats underwent exercise followed by detraining; LETO (control) rats remained sedentarySustained exercise: skeletal muscle mass↑ post-training lipid metabolism↑[54]4-week-old male OLETF (obese) rats were randomized into non-exercise sedentary group or exercise groups, along with LETO (non-obese) ratsExercise: fatty acid oxidation↑PGC-1α/UCP1↓[55]8-week-old male C57BL/6 mice were grouped by lactate supplementation and exerciseExercise-derived or dietary lactate: GPR81-Ca2+/CaMK pathway↑UCP1↑ mtDNA↑Age[16]Male New England rabbits of different ages, including 1 day, 14 days, 1 month, 2 months, 3 months, and 4 monthsAging: Ucp1/mitochondrial gene↓ mtDNA↓ autophagy/vascularization/innervation/immune cell infiltration↑[85]Tianfu Black rabbits of different ages, including 0 day, 15 days, 85 days and 2 yearsAging: purine/Wnt/PPAR/cGMP/PKG/lipid metabolism pathways↑[86]Tianfu Black rabbits of different ages, including 0 day, 15 days, 85 days and 2 yearsAging: MAPK/Ras signaling pathways↑[87]New Zealand white rabbits of different ages, including 1 day, 3 weeks, 6 weeks and 12 weeksAging: Ucp1/Dio2/Pgc-1α↓leptin↑ progenitor cell differentiation↓[88]Newborn Romney lambs at 12–24 h and 2 days of ageAging: VEGFA↓ thermogenic genes↓CYP1A1↑[89]3-month-old (young) and 18-month-old (old) male C57BL/6J miceAging: T cells-derived IFN-γ↑[90]10-week-old young and 76-week-old old miceAging: mitochondrial lipoylation levels↓Gender[58]Offspring of C57BL6/J female mice fed with a HFD which were separated by sexMaternal HFD affected male offspring: inflammation↑ abnormal oxidative phosphorylation↑Hormones[59]Male mice were divided into control and glucocorticoid-treated groupsGlucocorticoids: BTG1-dependent autophagy↑[60]Male rabbits were divided into control and glucocorticoid-treated groupsGlucocorticoids: leptin↑ leptin receptor↓ mitochondrial markers↓ ATP content↓[61]Male Wistar rats were divided into control and glucocorticoid-treated groupsGlucocorticoids: miR-21-5p↑ mitochondrial fission/fusion/mitophagy genes↑[62]Female Sprague-Dawley rats were divided into sham operation group, OVX or Estrogen treatment after OVX groupEstrogen: hypothalamic ceramide↓ ER stress↓[64]Drd2 floxed mice (control) and pituitary prolactin secreting cells-deficient Drd2 mice which were created by crossing with Prl-Cre miceProlactin: thermogenic genes↓ mitochondrial dysfunction↑ inflammation↑Tissue Microenvironment[65]VEGFA floxed mice (control) and adipose-deficient VEGFA mice which were created by crossing with aP2-Cre miceVEGFA deficiency: capillaries↓ β-adrenergic signaling↓ mtROS↑ mitophagy↑[66]Neonatal C57BL/6 mice with oxygen-induced retinopathy were intravitreally injected with PBS or anti-VEGF164 antibodyAnti-VEGF antibodies: VEGF↓ lipid accumulation↑ vascular density↓ mitochondrial genes↓Genetic Regulation[11]ChREBP-β floxed mice (control) and brown adipose-deficient ChREBP-β mice which were created by crossing with Ucp1-Cre miceChREBP-β overexpression: mitochondrial fission/fusion↓ thermogenic capacity↓[67]TFEB floxed mice (control) and brown adipose-deficient TFEB mice which were created by crossing with Ucp1-Cre miceTFEB deficiency: LC3B-II accumulation↑ autophagic flux↑ mitochondrial degradation↑[68]KDM5A floxed mice (control) and brown adipose-deficient Kdm5a mice which were created by crossing with Ucp1-Cre miceKDM5A overexpression: H3K4me3 at brown adipocyte-specific gene promoters↓[69]TFAM floxed mice (control) and adipose-deficient TFAM mice which were created by crossing with Adipo-Cre miceTFAM deficiency: mitochondrial electron transport↓ fatty acid oxidation↓ circulating fatty acids↑[92]ERRγ floxed mice (control) and adipose-deficient ERRγ mice which were created by crossing with Adipo-Cre miceERRγ deficiency: Ucp1 and Fabp3 promoter expression↓[93]FAM195A whole-body knockout C57BL/6J mice and wild-type littermate controlsFAM195A deficiency: branched-chain amino↓ fatty acid metabolism↓[94]ATGL floxed mice (control) and adipose-deficient ATGL mice which were created by crossing with aP2-CreATGL deficiency: lipolysis↓ fatty acid release↓ Pparα activity↓Ucp1↓[95]Dicer floxed mice (control) and adipose-deficient Dicer mice which were created by crossing with Adipo-Cre mice or tamoxifen-induced aP2-CreDicer deficiency: brown to white adipocyte differentiation↑Ucp1/Elovl3↓leptin↑[97]Ucp1 whole-body knockout C57BL/6J mice and wild-type littermate controlsUcp1 deficiency: mitochondrial subunits↓ inflammation↑ ER stress↑ oxidative stress genes↑[98]APOO floxed mice (control) and adipose-deficient APOO mice which were created by crossing with Adipo-Cre miceAPOO deficiency: mitochondrial fatty acid oxidation↓ peroxisome function↓[70]Noggin floxed mice (control) and adipose-deficient Noggin mice which were created by crossing with Adipo-Cre miceNoggin deficiency: differentiation↓ thermogenesis↓ lipid metabolism genes↓Abbreviations: Adipo adiponectin, APOO apolipoprotein O, ATP adenosine triphosphate, ATF4 activating transcription factor 4, ATGL adipose triglyceride lipase, aP2 adipocyte protein 2 (also known as Fabp4), BAT brown adipose tissue, BPS bisphenol S, BTG1 B-cell translocation gene 1, CaMK calmodulin-dependent protein kinase, CD36 cluster of differentiation 36, cGMP cyclic guanosine monophosphate, CHOP C/EBP homologous protein, ChREBP-β carbohydrate-responsive element-binding protein beta, Cidea cell death-inducing DFFA-like effector a, Cre Cre recombinase, CYP1A1 cytochrome P450 family 1 subfamily A member 1, DGAT2 diacylglycerol O-acyltransferase 2, Dicer Dicer ribonuclease III, Dio2 type 2 iodothyronine deiodinase, Drd2 dopamine receptor D2, Elovl3 ELOVL fatty acid elongase 3, ER endoplasmic reticulum, ERRγ estrogen-related receptor gamma, Fabp3 fatty acid-binding protein 3, FAM195A family with sequence similarity 195 member A, FASN fatty acid synthase, Gadd45 growth arrest and DNA damage-inducible 45, GR glucocorticoid receptor, GPR81 G protein-coupled receptor 81, H3K4me3 histone H3 lysine 4 trimethylation, HCD high-carbohydrate diet, HFD high-fat diet, HFrD high-fructose diet, HFSD high-fat/sucrose diet, IFN-γ interferon-gamma, IPO interesterified palm oil, KDM5A lysine demethylase 5A, LC3B-II microtubule-associated protein phosphatidylethanolamine 1A/1B-light conjugate, LETO chain 3 Long-Evans Tokushima Otsuka (rat strain), MAPK mitogen-activated protein kinase, miR-21-5p microRNA-21-5p, mtDNA mitochondrial DNA, mtROS mitochondrial reactive oxygen species, NLRP3 NLR family pyrin domain containing 3, OLETF Otsuka Long-Evans Tokushima Fatty (rat strain), OVX ovariectomy, PBS phosphate-buffered saline, PGC-1α peroxisome proliferator-activated receptor gamma coactivator 1-alpha, PKG protein kinase G, PLIN1 perilipin 1, PM2.5 fine particulate matter, PO palm oil, PPAR peroxisome proliferator-activated receptor, PPARα peroxisome proliferator-activated receptor alpha, Prl prolactin, PSNPs polystyrene nanoplastics, Ras rat sarcoma virus, SREBP-1c sterol regulatory element-binding protein 1c, TFAM mitochondrial transcription factor A, TFEB transcription factor EB, TLR4 toll-like receptor 4, UCP1 uncoupling protein 1, VEGFA vascular endothelial growth factor A, Wnt Wingless/Integrated, n-3 FAs omega-3 fatty acids.
Moreover, through an integrated approach combining literature review and computational bioinformatics, 4-HBA was identified as a candidate compound for BAT modulation. Our computational predictions showed partial alignment with a recent study reporting AMPK-DRP1 pathway activation by 4-HBA [18], suggesting possible mechanistic relevance that warrants experimental confirmation. We recognize that the observed interaction profile of 4-HBA may partly reflect target selection bias, as our protein panel was deliberately constructed around functionally validated regulators, enzymes, and effectors of BAT whitening. All targets were treated with equal weight to assess pathway-centric engagement rather than target prioritization, and redundancy or pathway overlap was retained to capture the multi-layered regulation of BAT whitening. This design enables assessment of broad engagement within this functionally defined network but does not permit conclusions regarding global binding selectivity. Accordingly, the finding that 4-HBA interacts with all 11 selected targets should be interpreted as evidence of pathway-wide engagement within this defined biological context. More quantitative, network-based approaches could further refine target prioritization in future studies.
Several critical limitations constrain the interpretative scope of these findings:
Methodological constraints:
Despite its utility in initial screening, molecular docking has inherent limitations: its static modeling and predefined binding thresholds may not fully capture biological complexity. Consequently, these findings should be supplemented with dynamic simulations and experimental mutagenesis for robust validation.
Polypharmacological risks:
The multi-target activity of the compound raises concerns about potential off-target interactions and tissue-specific toxicity, particularly through its engagement with mitochondrial and nuclear receptors. This complexity is further heightened by its metabolic conversion into active derivatives, which may exhibit distinct biological profiles.
Clinical relevance gaps:
Although studies in rodent models provide foundational insights, the translational relevance of targeting BAT whitening, and specifically of the candidate metabolite 4-HBA, must be contextualized within key limitations. First, direct extrapolation to humans is constrained by interspecies variations in adipose tissue biology, depot distribution, and gut microbiota composition. Second, and more critically, no human pharmacokinetic or chronic safety data exist to inform the therapeutic dosing and risk profile of 4-HBA.
Therefore, while BAT modulation represents a theoretically promising strategy against metabolic disorders, the proposed role of 4-HBA remains exploratory and mechanistically suggestive. The integrated framework presented herein demonstrates a systematic strategy for prioritizing metabolites from bench to bedside. To bridge the current translational gap, future work must prioritize: (1) biophysical validation of metabolite-target interactions through structural biology approaches; (2) comparisons of 4-HBA efficacy across human and murine systems to elucidate translational potential; (3) multi-omics characterization of metabolite crosstalk across metabolic tissues to address interspecies physiological differences.
Methods
Literature Reviews
This review employed a structured narrative approach to synthesize evidence on BAT whitening. To ensure comprehensive and transparent literature coverage, a literature search was conducted across PubMed and Web of Science (2000–2025) using tailored Boolean operators (e.g., (brown adipose tissue OR BAT OR brown fat) AND (whiten* OR plastic* OR phenotypic switch)). All identified records were collated, deduplicated, and screened by title/abstract for relevance, followed by full-text assessment against predefined inclusion criteria: (i) original research or review articles; (ii) studies reporting key drivers, regulators, or determinants of BAT plasticity at the molecular, cellular, or physiological level; (iii) human and other mammalian models; (iv) English language publications.
Figures 1 and 2 were generated using BioRender.com, and no generative AI was involved in the figure design or creation process.
Protein Panel Selection
A functional classification framework relevant to brown fat whitening was constructed by integrating function and compartment-based annotations from the Human Protein Atlas (v18.0) [123, 124] with the genome-scale functional annotation strategy [125]. Three categories were predefined—transcriptional/epigenetic regulators, metabolic enzymes, and transporters and secreted proteins—and within each category, candidate proteins were retained based on two sequential criteria: (i) documented functional association with brown fat whitening, especially concerning mitochondrial regulation, and (ii) availability of a high-resolution human crystal structure in UniProt to support downstream structural modeling. This selection process yielded 11 proteins: six transcriptional/epigenetic regulators—MLXIPL, TFEB, TFAM (transcription factors), ERRγ (nuclear receptor), KDM5A (histone demethylase), and FAM195A (RNA-binding protein); two metabolic enzymes—ATGL (lipase) and DICER (ribonuclease); and three transporters and secreted proteins—UCP1 (mitochondrial transporter), APOO (mitochondrial structural and secreted protein), and Noggin (secreted protein). All functional annotations for the selected proteins were retrieved from the UniProt Knowledgebase [126].
Bioinformatics Analyses
Based on the protein panel selection, eleven proteins critically associated with adipose tissue plasticity and metabolic reprogramming (MLXIPL, TFEB, TFAM, ERRγ, KDM5A, FAM195A, ATGL, DICER, UCP1, APOO and Noggin) were selected as targets. Their three-dimensional structures were obtained from the UniProt database and prepared using AutoDockTools. Molecular docking-based virtual screening against the U.S. FDA-approved compound library was performed using AutoDock Vina. Docking poses were visualized and analyzed with PyMOL. Applying a binding energy threshold of ≤ − 5 kcal/mol, 4-HBA was identified as the sole compound demonstrating significant binding affinity across all eleven targets.
Key References
- Peng Y, Zhao L, Li M, Liu Y, Shi Y, Zhang J : Plasticity of Adipose Tissues : Interconversion among White, Brown, and Beige Fat and Its Role in Energy Homeostasis. Biomolecules 2024, 14(4).
- ○ This study provides a comprehensive overview of adipose tissue metabolic plasticity, encompassing phenomena such as BAT whitening, and highlights the therapeutic potential of targeting these processes for obesity treatment.
- Kim SH, Park WY, Song G, Park JY, Jung SJ, Ahn KS, et al : 4-hydroxybenzoic acid induces browning of white adipose tissue through the AMPK-DRP1 pathway in HFD-induced obese mice. Phytomedicine 2025, 137 :156353.
- ○ This study provides direct in vivo evidence that 4-Hydroxybenzoic acid combats high-fat-diet-induced obesity by activating adipose thermogenesis.
- Mori MP, Lozoya OA, Santos JH: BAT mitochondria: whitening meets softening. Nat Metab 2025, 7(8):1503-1504.
- ○ This study provides key evidence that mitochondrial stress and dysfunction drive BAT whitening through D-2HG-mediated epigenetic and nuclear mechanical remodeling, which offers crucial support for the main mechanism in this review.
- UniProt C : UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res 2025, 53(D1): D609-D617.
- ○ This study provides the essential reference for the UniProt database, which supplied both the crystal structure and functional annotations of protein.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Karanfil AS, Louis F, Matsusaki M. Brown adipose tissue engineering: advances, challenges, and future directions. Trends Biotechnol; 2025.10.1016/j.tibtech.2025.08.01041006179 · doi ↗ · pubmed ↗
- 2Dabrowska AM, Dudka J. Mirabegron, a selective beta 3-adrenergic receptor agonist, as a potential anti-obesity drug. J Clin Med. 2023;12(21):6897.10.3390/jcm 12216897 PMC 1064961537959362 · doi ↗ · pubmed ↗
- 3Santiago PJD, Shimada BK, Alfulaij N, Hallam KA, Soares AG, Young V, Swanson SM, Remedios GL, Toh P, Seale LA. Targeted disruption of selenocysteine lyase in brown adipocytes controls glutathione peroxidase 1 and 4 expression in males. Biol Trace Elem Res. 2025.10.1007/s 12011-025-04904-7PMC 1314958141366172 · doi ↗ · pubmed ↗
- 4Lee SY, Fontana F, Sugatani T, Portales Castillo I, Leanza G, Coler-Reilly A, Civitelli R. Connexin 43 in mesenchymal lineage cells regulates body adiposity and energy metabolism in mice. JCI Insight. 2024;9(6):e 170016.10.1172/jci.insight.170016 PMC 1106394538349739 · doi ↗ · pubmed ↗
- 5Hurtado-Barroso S, Quifer-Rada P, Marhuenda-Munoz M, Rinaldi de Alvarenga JF, Tresserra-Rimbau A, Lamuela-Raventos RM. Increase of 4-hydroxybenzoic, a bioactive phenolic compound, after an organic intervention diet. Antioxid (Basel). 2019;8(9):340.10.3390/antiox 8090340 PMC 676975831450569 · doi ↗ · pubmed ↗
- 6Human Protein Atlas. Protein Atlas v 18.0: Protein classes [Internet]. Available from: https://v 18.proteinatlas.org/humanproteome/proteinclasses