Vaccine elicitation of HIV-1 neutralizing antibodies against both V2 apex and fusion peptide in rhesus macaques
Hongying Duan, Joseph P Nkolola, Shuishu Wang, Jayeshbhai Chaudhari, I-Ting Teng, Christy Lavine, Danealle K Parchment, George S. Sellers, Krisha McKee, Sijy O’Dell, Misook Choe, Haijuan Du, Baoshan Zhang, Alejandro A. Espinosa Perez, Annika Rossler, Ninaad Lasrado, Andrea Biju

TL;DR
A new HIV vaccine design successfully induces antibodies that neutralize multiple virus strains in monkeys.
Contribution
A dual-epitope vaccine elicits cross-strain neutralizing antibodies against both the fusion peptide and V2 apex of HIV-1 in rhesus macaques.
Findings
Macaques immunized with the dual-epitope vaccine showed >1,000-fold higher autologous tier 2-neutralizing titers than wild-type Env trimers.
FP- and V2 apex-directed monoclonal antibodies isolated from immunized macaques exhibited heterologous neutralization.
The vaccine demonstrates proof of concept for eliciting antibodies against multiple HIV-1 Env vulnerabilities simultaneously.
Abstract
Broadly neutralizing antibodies (bNAbs) targeting multiple sites of HIV-1 Env vulnerability can be induced by infection, but simultaneous elicitation of bNAbs against multiple epitopes has not been achieved by vaccination. In this study, we designed a dual-epitope vaccine targeting both the fusion peptide (FP) and the V2 apex and evaluated its capacity to induce bNAbs against both epitopes in rhesus macaques. This vaccine combined an FP conjugate with a cocktail of engineered Env trimers with enhanced V2 apex recognition and increased antigen retention in lymph nodes. Macaque immunization with the dual-epitope vaccine elicited >1,000-fold higher autologous tier 2-neutralizing titers than wild-type Env trimers and enhanced heterologous neutralization. Both FP- and V2 apex-monoclonal antibodies were isolated from immunized macaques and showed heterologous neutralization with genetic and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
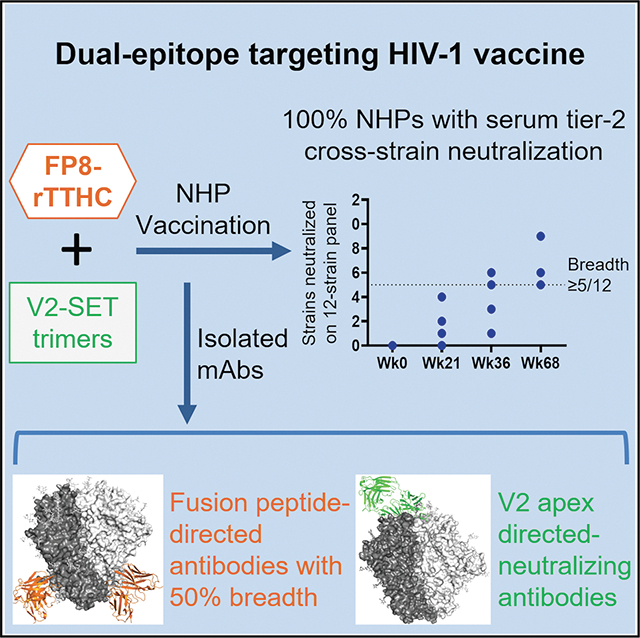 Figure 1
Figure 1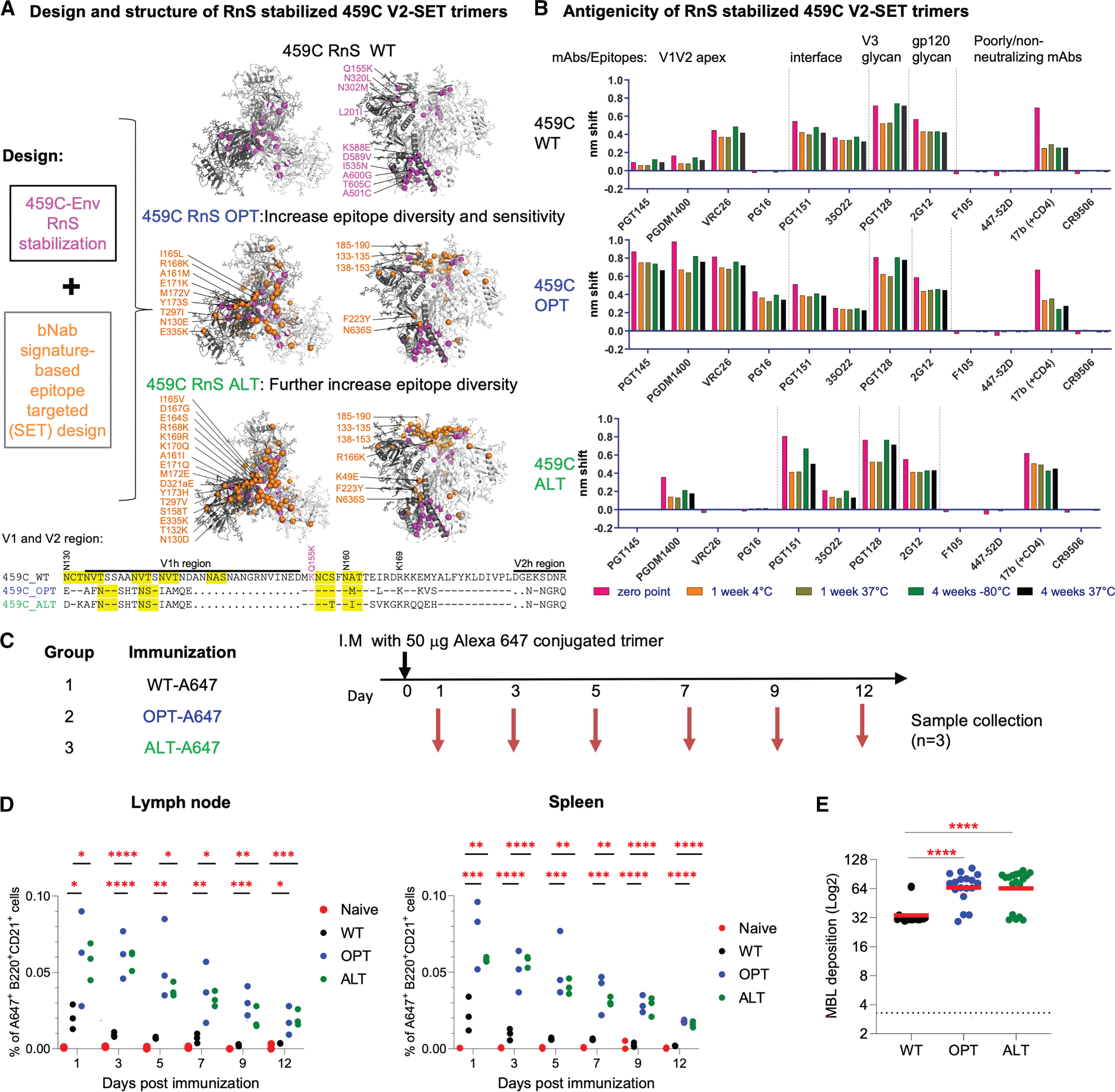 Figure 2
Figure 2 Figure 3
Figure 3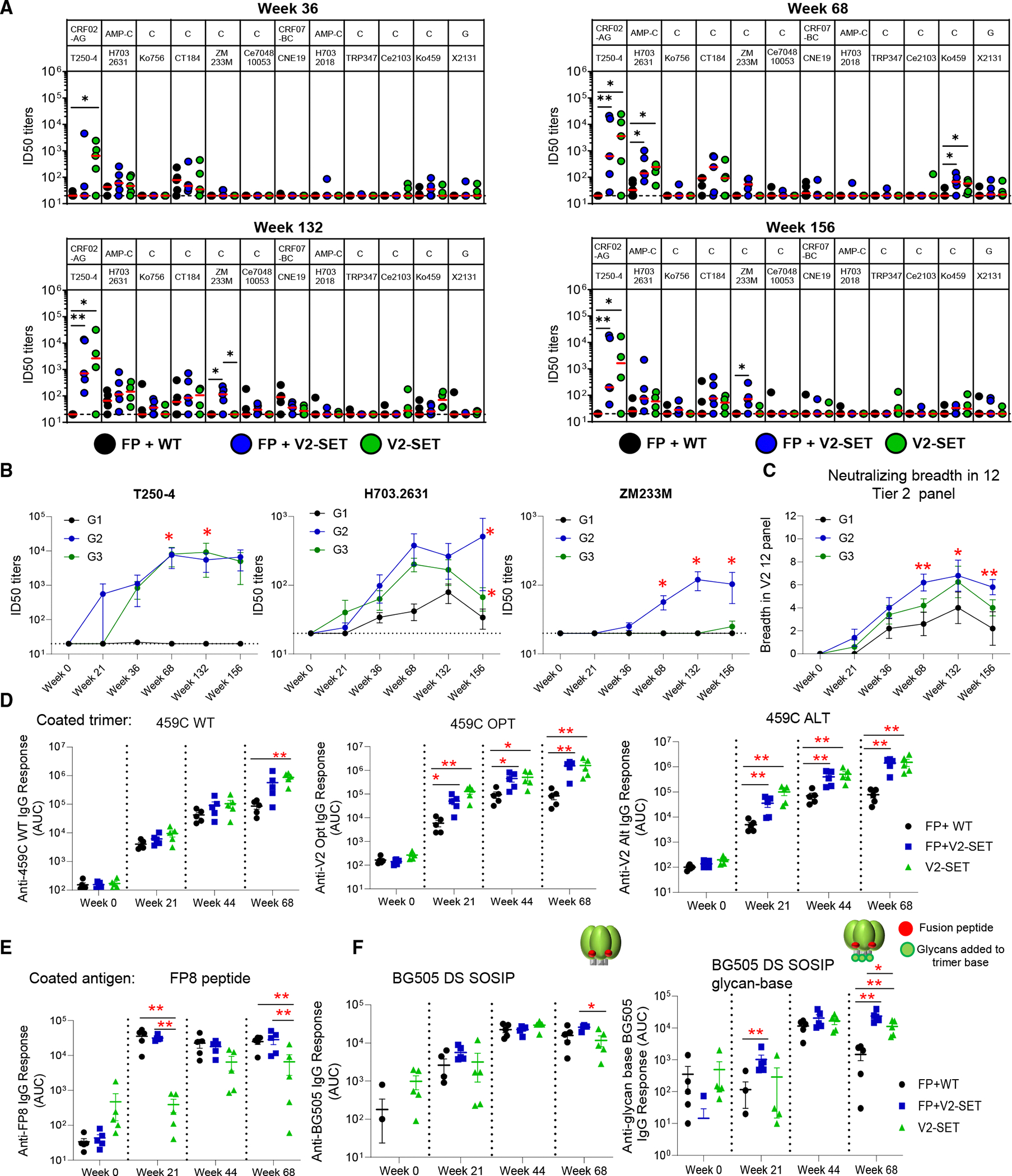 Figure 4
Figure 4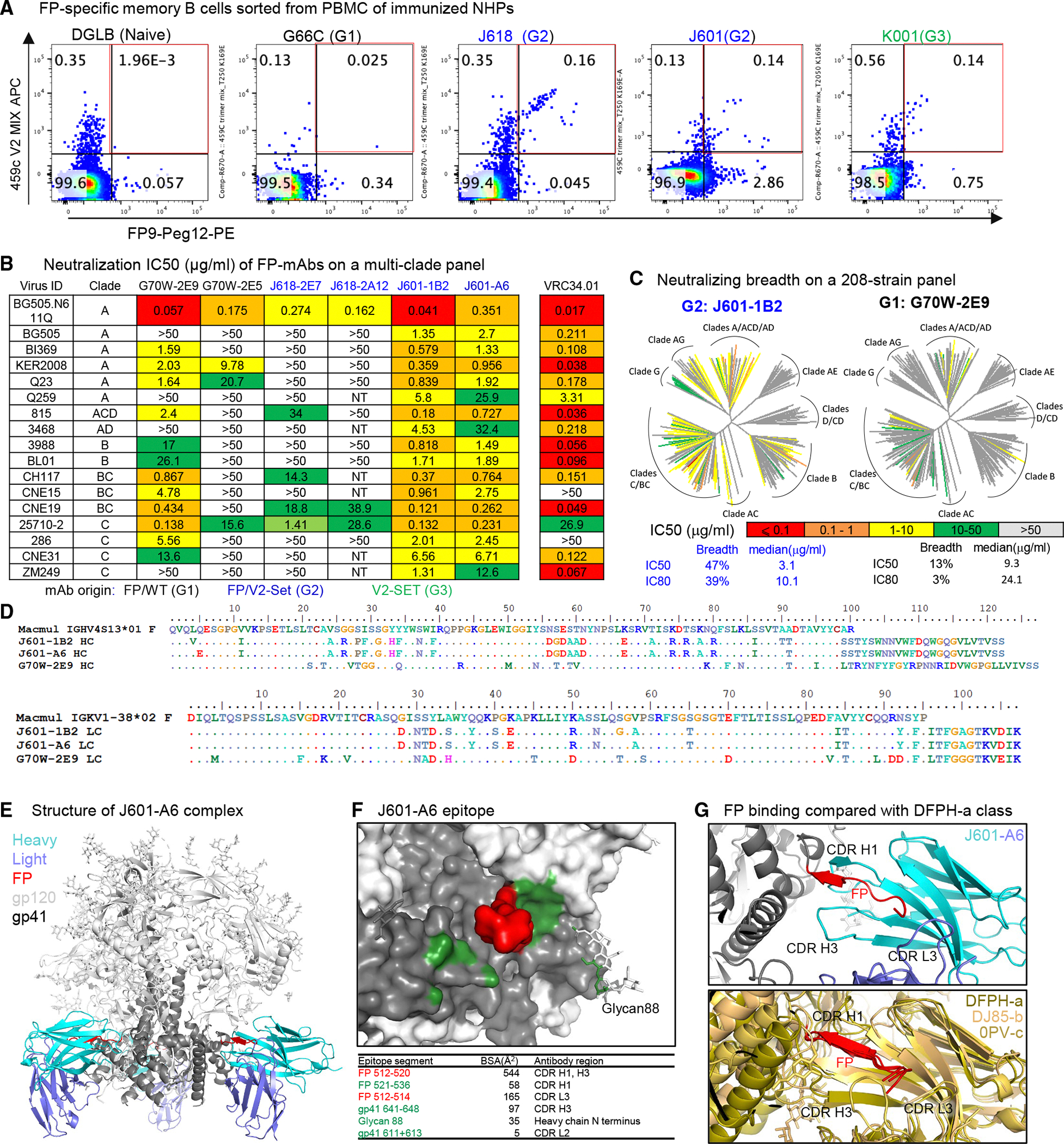 Figure 5
Figure 5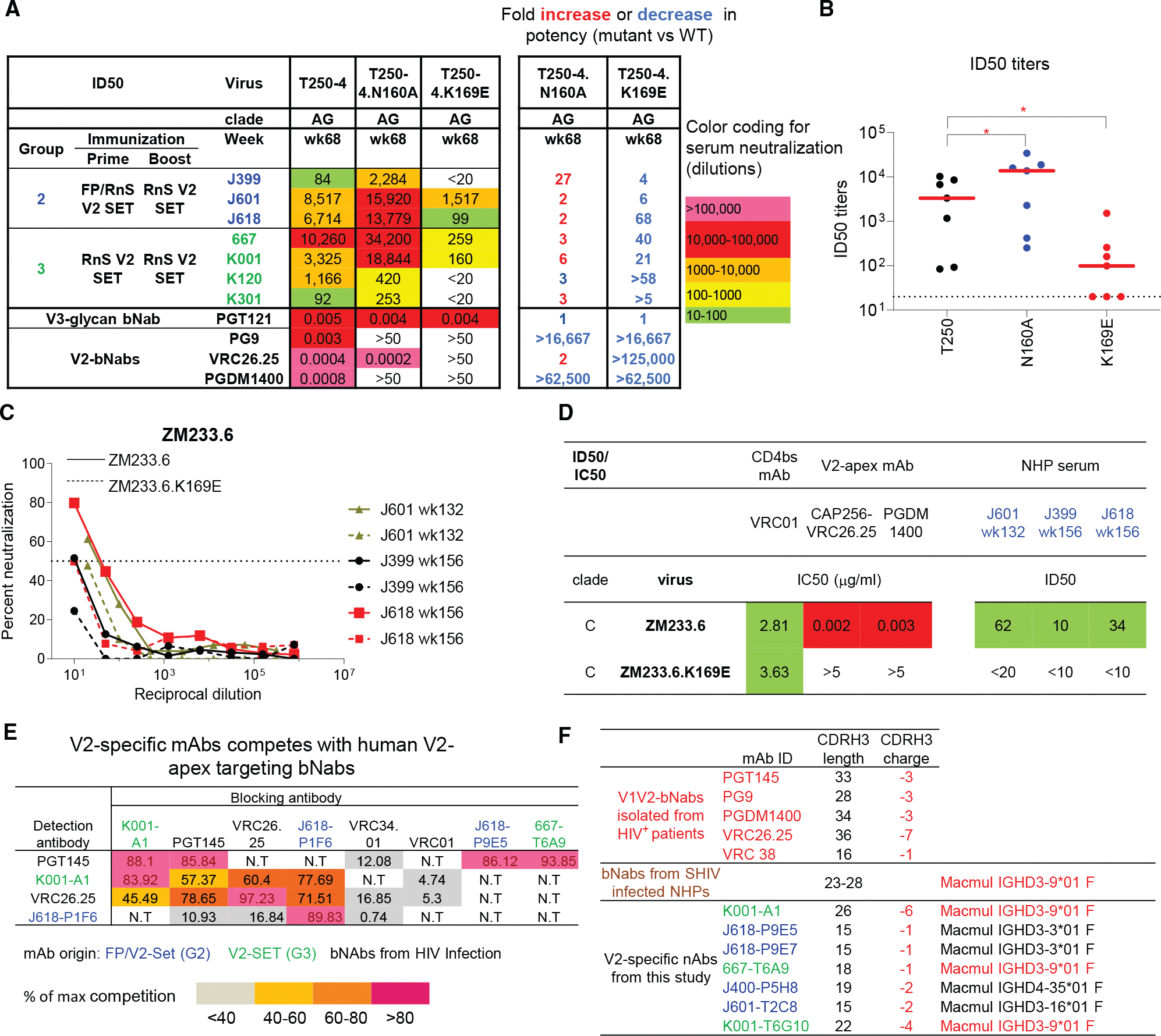 Figure 6
Figure 6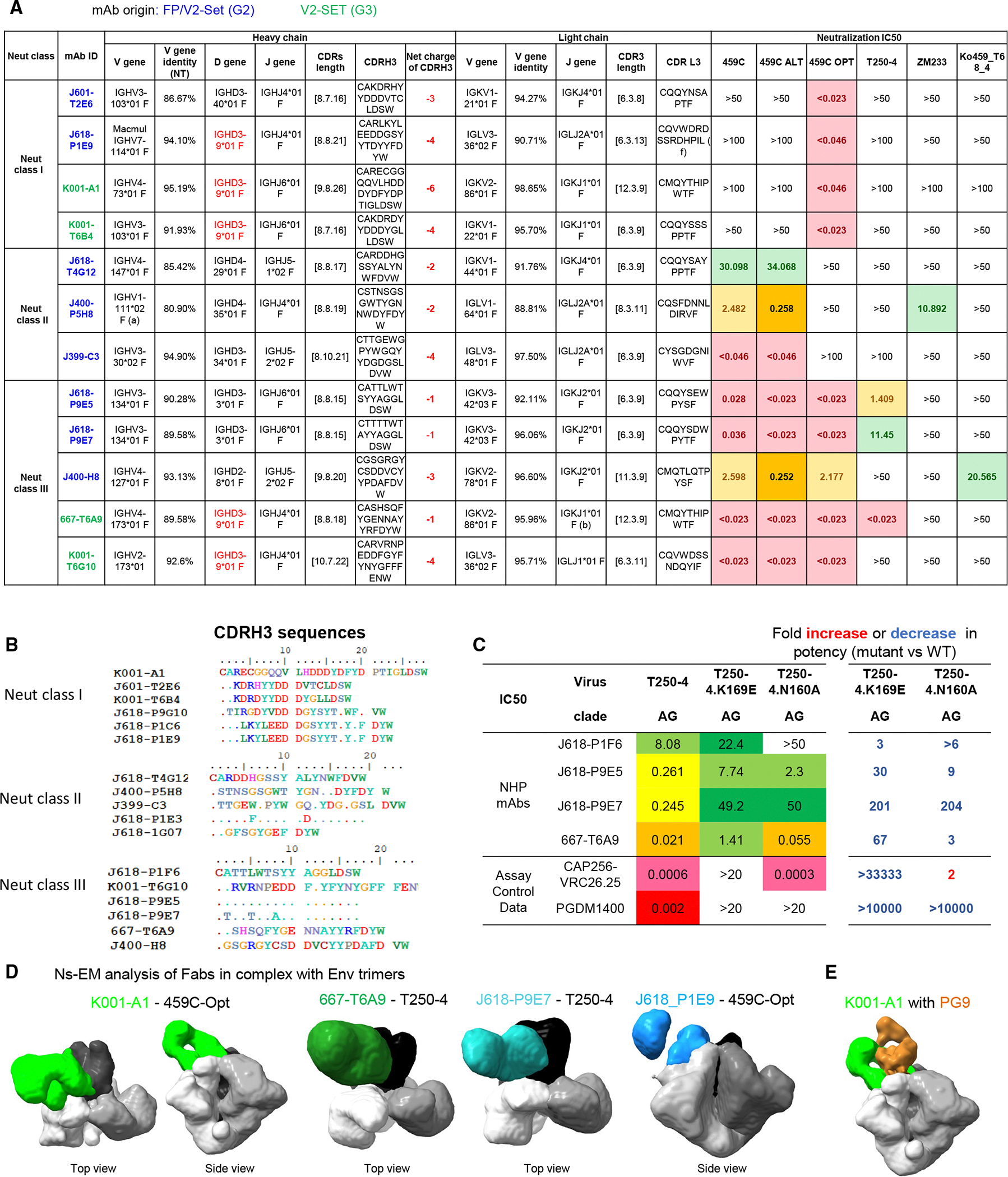 Figure 7
Figure 7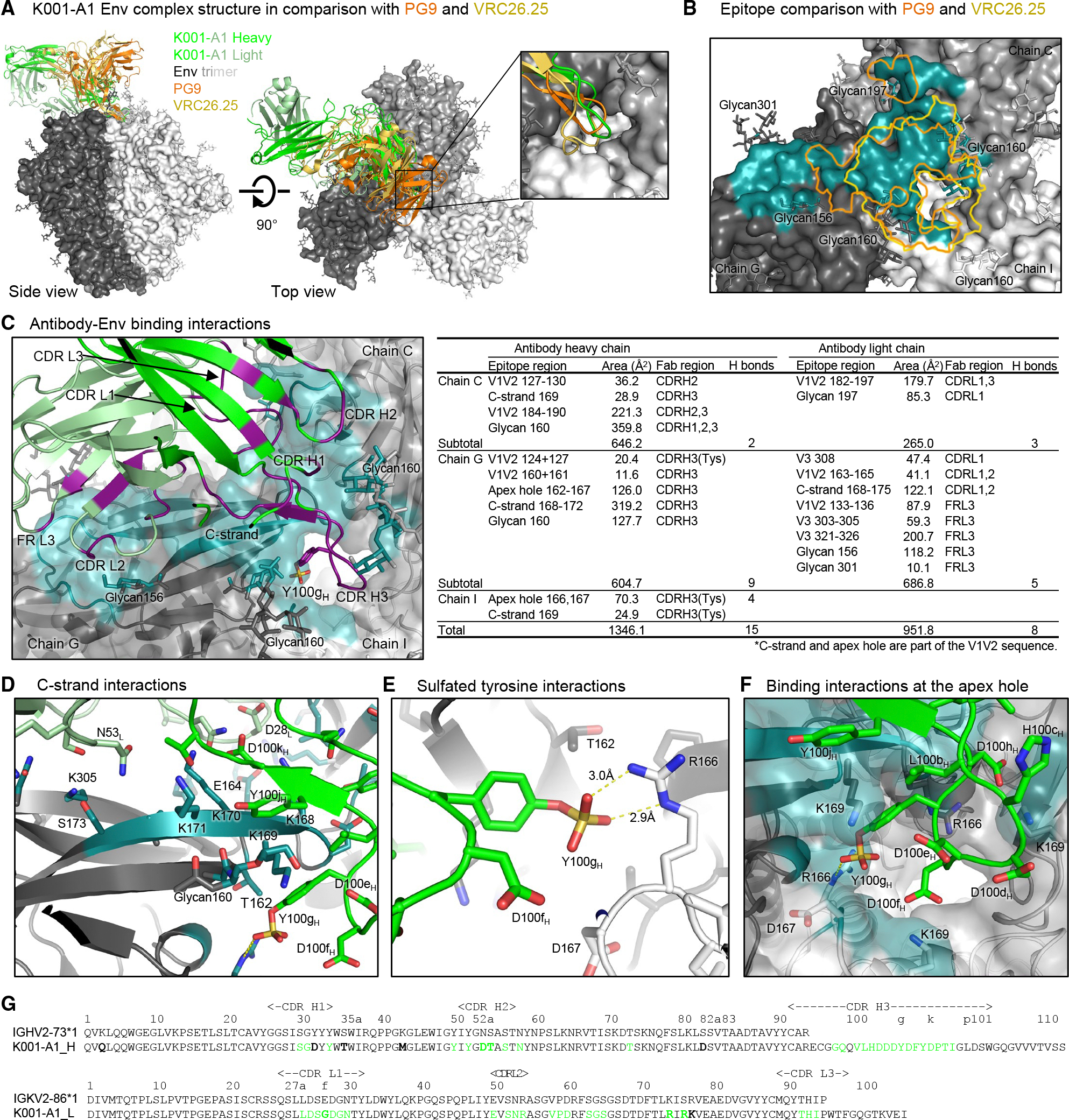 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHIV Research and Treatment · HIV/AIDS drug development and treatment · HIV/AIDS Research and Interventions
INTRODUCTION
Although HIV-1 was identified over four decades ago, a protective vaccine against HIV-1 remains elusive. The HIV-1 envelope (Env) glycoprotein trimer is the only target of virus-specific neutralizing antibodies (NAbs), making it an attractive focus for vaccines. Progress toward an effective HIV-1 vaccine has been hindered by the high rate of mutation and recombination during viral replication,^1,2^ a dynamic Env structural ensemble,^3–5^ and the extensive glycan shield of ~90 N-linked oligosaccharides.^6^ Soluble Env trimers from multiple HIV-1 strains have been stabilized in prefusion-closed conformations,^7^ but immunization with stabilized soluble trimers has generally failed to elicit robust heterologous neutralizing titers.^8–11^
Several NAb epitopes on HIV-1 Env have been targets for vaccine development, including the fusion peptide (FP), V2 apex, V3 glycan supersite, CD4 binding site, and membrane-proximal external region (MPER) region.^9,11–29^ Current efforts for the development of an HIV-1 vaccine have largely focused on the induction of NAbs to a single target epitope,^24,26,30–33^ but an effective HIV-1 vaccine will almost certainly need to target multiple sites of Env vulnerability given the challenge of HIV-1 diversity. Previously, broadly neutralizing antibodies (bNAbs) targeting multiple epitopes have been described in persons with HIV-1,^16,34–39^ but vaccines that induce NAbs against multiple epitopes simultaneously have not yet been reported.^40^
We previously reported that priming with FP-carrier conjugates together with or followed by soluble prefusion-closed stabilized Env trimers elicited FP-specific bNAbs in nonhuman primates with up to 59% neutralizing breadth as assessed on a cross-clade panel of 208 HIV-1 strains.^26,41,42^ In addition, recombinant tetanus toxoid heavy chain fragment (rTTHC) linked to FP8 (FP8-rTTHC) was evaluated in animal models as a suitable FP-conjugate vaccine immunogen^20^ and has recently been advanced into a phase I clinical trial. We also previously defined a V2 bNAb signature associated with bNAb sensitivity and resistance, and we selected and modified the V2 sequence of an acute clade C isolate 459C to create V2 signature-based epitope-targeted (V2-SET) 459C trimers, with the 459C Optimal (OPT) trimer modified with V2 bNAb sensitive mutations and the 459C alternative (ALT) trimer modified to capture additional natural V2 diversity that included the signatures of resistance. A trimeric cocktail of 459C wild-type (WT), OPT, and ALT trimers comprised the 459C V2-SET Env vaccine, and immunization with this trimeric cocktail conferred enhanced neutralizing breadth and potency as compared to the 459C WT Env vaccine in guinea pigs.^14^
In this study, we stabilized the V2-SET trimers with repair and stabilization (RnS) modifications.^43^ RnS-stabilized OPT and ALT trimers showed improved V2 binding and higher capture and retention in lymph nodes compared with the WT trimer. We explored the potential of combining the FP8-rTTHC immunogen with V2-SET 459C Env trimers in nonhuman primate (NHP) and found that the dual-epitope vaccine induced potent autologous and lower but detectable heterologous serum-tier 2 NAbs. Isolated FP- and V2 apex-specific neutralizing monoclonal antibodies (mAbs) showed genetic and structural signatures of well-characterized bNAbs.
RESULTS
Production of RnS-stabilized 459C V2-SET trimers
Previously, we reported viral signatures associated with V2 apex-specific bNAb sensitivity and diversity and created the 459C V2-SET vaccine,^14,43^ which is a set of three trimers designed to cover the most common amino acid variants within the V2 epitope region in the globally circulating viral population. The cocktail included a 459C WT trimer, a natural acute clinical strain that showed intrinsic capacity to elicit NAbs with some heterologous breadth in guinea pigs; a 459C OPT trimer, which included amino acid modifications that represented common variants not found in 459C and that were associated with V2 apex-specific bNAb sensitivity sequence signatures; and a 459C ALT trimer, which included V2 apex-specific bNAb sensitivity signatures outside of the contact region and captured natural diversity in V2 signature sites (Figures 1A and S1A). Stabilizing the prefusion conformation was important to reduce the exposure of non-neutralizing epitopes and to elicit a more focused immune response toward neutralizing epitopes.^18,44^ Since we were unable to create a stable disulfide and Ile559Pro (SOSIP) stabilized ALT trimer,^14^ we incorporated RnS mutations^43^ into the sequences of the 459C WT, OPT, and ALT trimers (Figures 1A and S1B) and produced RnS-stabilized WT, OPT, and ALT trimers (Figure 1A).
All three trimers were expressed and purified with high yields and retained prefusion conformations at 37°C for up to 4 weeks (Figures S1C and S1D). Furthermore, the stability of all three trimers revealed minimal antigenic disruption after storage at −80°C, 4°C, or 37°C for 4 weeks (Figure 1B). In binding assays, the 459C WT trimer showed little reactivity to V2 apex bNAbs PGT145,^45^ PGDM1400,^46^ and PG16,^47^ and moderate reactivity to CAP256/VRC26.25.^48^ As expected, the 459C OPT trimer, which included V2 antibody sensitivity signatures, showed higher binding to V2 apex-specific bNAbs PGT145, CAP256/VRC26.25, PGDM1400, and enabled binding to PG16 (Figures 1A, 1B and S1E). The key signature differences incorporated into 459C OPT that were associated with enhanced V2 bNAbs sensitivity included shorter and more positively charged V1 and V2 loops and the elimination of the N-linked glycosylation site at position N130. In contrast, the 459C ALT trimer was less efficiently bound by V2 apex-bNAbs as designed (Figures 1A, 1B, and S1E). All three trimers showed similar binding to gp120/gp41 interface antibodies, FP and V3-glycan antibodies, and gp120 glycan and CD4bs antibodies, and little binding to non-neutralizing F105 and 447–52D mAbs, suggesting that they were folded properly in the prefusion-closed conformation (Figures 1B and S1E).
To characterize the effects of the RnS and V2-SET modifications on the structural features of the trimers, we determined cryo-electron microscopy (cryo-EM) structures of the individual 459C WT, OPT, and ALT RnS trimers. All three trimers were flash frozen, and single particle cryo-EM data were obtained using a Titian Krios electron microscope. We obtained a structure of the 459C WT RnS trimer at 3.3 Å and a structure of the 459C ALT RnS trimer at 3.9 Å (Data S1.1–4). We failed, however, to obtain an interpretable map for the 459C OPT RnS trimer as the particles appeared more heterogeneous. Multiple classes of 3D reconstructions with partially disordered trimers suggested that the 459C OPT RnS trimer structure, while intact, was more flexible and dynamic (Data S1.1). Overall, the structures of the 459C ALT RnS and 459C WT RnS trimers were similar, with a root-mean-square deviation of 1.6 Å for 1,586 aligned Cα atoms. Additionally, the WT and ALT structures were most similar to each other in the gp41 core helices, but shifted substantially in the V1V2 region, so that the 459C ALT RnS structure expanded slightly outwards (Data S1.1D and E). These data suggest that, in the absence of antibody binding, the 459C OPT RnS trimer is flexible, whereas the 459C WT RnS trimer is more stable in a prefusion-closed conformation than the 459C ALT RnS and 459C OPT RnS trimers.
RnS modified V2-SET OPT and ALT trimers showed enhanced capture and retention in lymph nodes and spleens of mice
To investigate the impact of the V2-SET modifications in vivo, we conjugated the RnS-stabilized V2-SET trimers with an Alexa 647 fluorophore and immunized BALB/c mice with 50 μg of Alexa 647-labeled WT, OPT, or ALT trimer in three groups (Figure 1C). After a single immunization, we collected spleen and draining lymph nodes from three mice per group on days 1, 3, 5, 7, 9, and 12 and measured Alexa 647-positive populations of B220^+^CD21^+^ cells in the lymph nodes and spleens. We found that both OPT and ALT trimers demonstrated significantly higher associations with B220^+^CD21^+^ cells in lymph nodes and spleens than WT trimers. Furthermore, OPT and ALT trimers remained detectable on B220^+^CD21^+^ cells up to day 12 after immunization, whereas the WT trimer signal declined to baseline levels by day 7. These findings suggest that V2-SET-modified trimers exhibited higher capture and longer retention on follicular dendritic cells (FDCs) and B cells (Figure 1D). To elucidate the potential mechanism behind the enhanced antigen capture and retention of the OPT and ALT trimers in lymph nodes and spleen, we evaluated these three trimers in vitro for their ability to interact with mannose-binding lectin (MBL) using an MBL deposition assay. Binding to MBL has been shown to enhance antigen-specific germinal center (GC) responses and serum antibody titers, leading to improved vaccine efficacy.^49^ For the MBL assay, fresh serum from naive mice, which contains MBL protein, was collected and used to quantify MBL deposition on WT, OPT, or ALT trimers, and the MBL response was detected with an anti-mouse MBL mAb. The 459C OPT and ALT trimers showed significantly higher MBL binding than the WT trimer (Figure 1E), which is consistent with the antigen tracking results and suggests a potential mechanism by which OPT and ALT show higher capture and better retention over WT in lymph nodes and spleen.
Dual epitope vaccine induced potent serum autologous NAb titers with cross-strain breadth in NHPs
Three groups of rhesus macaques were immunized three times with FP8-rTTHC + 459C WT trimer (group 1; G1), FP8-rTTHC + 459C V2-SET trimers (group 2; G2), and 459C V2-SET trimers alone (group 3; G3), followed by six boosts with the trimers alone without FP8-rTTHC. At week 112, a similar boosting series was repeated (Figure 2A). We tested sera for neutralization and detected a significant difference between 459C WT only (G1) and V2-SET 459C immunized animals (G2 and G3) in terms of autologous tier 2 459C serum neutralization titers. At week 36, after 5 immunizations, all NHPs in V2-SET immunized animals in G2 and G3 showed >1:2,000 50% inhibitory dilution (ID50) titers against 459C virus, compared with borderline ID50 titers against 459C virus in FP + 459C WT immunized animals in G1. At week 68, V2-SET immunized animals in G2 and G3 had high median neutralizing ID50 titers of 1:29,384 and 1:201,496, respectively (Figures 2B–2D). In contrast, FP + 459C WT immunized animals in G1 showed a median ID50 titer of 1:193. Similar results were observed with sera obtained at week 132, with median ID50 titers of 1:96, 1:150,616, and 1:339,443 for G1, G2, and G3 NHPs, respectively (Figures 2B–2D). These data demonstrate that the 459C V2-SET Env vaccine elicited autologous tier 2 NAb titers > 1,000-fold greater than did 459C WT Env trimers.
In addition, earlier and higher neutralizing activity against pseudotype viruses incorporating 459C OPT and ALT trimers was observed (Figures 2B and 2C). Interestingly, although G2 and G3 groups were immunized with only one-third of the dose of 459C WT trimer as compared to G1, sera from these animals showed far greater neutralization activity against WT 459C pseudovirus than did sera from animals immunized with WT 459C trimers only. To evaluate the cross-strain neutralization, we selected 12 tier 2 viruses, which are sensitive to previously identified human V2 apex bNAbs.^14,50^ We tested the NHP sera on the 12 virus panel and found that 459C V2-SET immunization also elicited earlier, higher, and broader heterologous tier 2 neutralizing activity (Figures 3A, 3B, and S2A–S2C), with heterologous neutralization detected as early as week 21 (Figure S2D), including the capacity to neutralize multiple tier 2 strains, including T250–4, H703.2631, and CT184.D3.16 (Figures 3A, 3B, and S2E).
G2 differs from G3 by the addition of the FP8-rTTHC immunogen. To assess the impact of the FP8-rTTHC on the V2-SET vaccine, we compared G2 versus G3 sera for neutralizing and binding antibody responses. We observed that for autologous 459C neutralizing activity, FP8-rTTHC + V2-SET immunization in G2 exhibited slightly lower ID50 titers compared to V2-SET alone immunization in G3. However, for heterologous tier 2 virus neutralization, FP8-rTTHC + V2-SET in G2 elicited earlier and higher neutralizing activity against tier 2 viruses H703.2631, ZM233M (Figure 3B), and CT184.D3.15 (Figures 3A and S2B–S2E). Of note, only FP8-rTTHC + V2-SET immunized animals in G2 showed neutralizing activity against ZM233M (Figure 3B). Furthermore, FP8-rTTHC + V2-SET elicited broader neutralizing activity in the 12 tier 2-virus panel (Figures 3C, S2B, S2C, and S3).
We next looked at binding antibody responses and observed that V2-SET immunized animals in G2 and G3 elicited significantly higher serum binding antibodies to vaccine-matched 459C WT, OPT, and ALT trimers compared with 459C WT immunized animals in G1 (Figure 3D). As expected, FP8-rTTHC with WT trimer or with V2-SET trimers elicited significantly higher FP8-specific serum responses than did V2-SET alone, although V2-SET alone also elicited detectable FP-specific responses (Figure 3E). Interestingly, the addition of FP8-rTTHC resulted in higher cross-strain responses to soluble BG505 disulfide-linked 201–433 (DS) SOSIP trimer, and the differences were more pronounced at week 68 against a BG505 DS SOSIP glycan-based trimer,^19,51^ which has glycans added on the trimer base to block base-directed antibodies, suggesting that the addition of FP8-rTTHC increased non-base antibody responses (Figure 3F). As higher non-base antibody responses have been correlated with higher neutralizing activity,^10,51^ we analyzed the correlation between serum responses to BG505 DS SOSIP glycan-based trimer and neutralizing activity. We found positive correlations between the week 68 serum binding antibody responses to BG505 glycan-based trimer and neutralizing titers and breadth (Figures S2F and S2G). Moreover, the ratio of binding antibody responses in G2 and G3 to G1 was higher to the BG505 glycan-based trimer than to the BG505 trimer (Figure S2H). Electron microscopy-based polyclonal epitope mapping (EMPEM) analysis further indicated that FP8-rTTHC with V2-SET vaccine in G2 elicited binding antibodies mainly targeting the trimer apex, the FP site, and the base (Data S1.5).
FP-specific mAbs from FP-rTTHC + V2-SET Env-immunized animals demonstrate up to 47% neutralization breadth
We isolated FP-specific mAbs from all three vaccinated groups using FP8 and 459C V2-SET trimer probes as previously described.^26,41,42^ Utilizing peripheral blood mononuclear cells from week 68 after the first immunization series, FP-specific memory B cells were isolated using FP9-PEG12 and 459C V2-SET trimers as probes (Figure 4A). Antibody variable domains of the isolated B cells were sequenced, and selected clones were synthesized and expressed. Multiple mAbs isolated from FP-primed G1 and G2 NHPs exhibited neutralizing activity on BG505.N611Q virus, which is sensitive to FP-specific NAbs^26,41^ (Figure S4A). Furthermore, cross-clade neutralizing activity was detected with tier 2 FP-sensitive virus panels,^26,41,52^ with two mAbs J601-A6 and J601–1B2 isolated from the G2 animal J601, neutralizing all 17 FP-sensitive strains tested, including human FP-specific bNAb VRC34.01-resistant strains CNE15 and 286.36 (Figure 4B). One mAb G70W-2E9, isolated from the G1 animal G70W, also showed cross-clade neutralizing activity.
We tested J601–1B2 and G70W-2E9 on a 208-strain HIV-1 panel. J601–1B2 neutralized 47% of tested viruses with a half-maximal inhibitory concentration (IC50) < 50 μg/mL and 39% of tested viruses with an IC80 < 50 μg/mL (Figure 4C). G70W-2E9 neutralized 13% and 3% of viruses with an IC50 < 50 μg/mL and IC80 < 50 μg/mL, respectively (Figure 4C). In addition, J601–1B2 was more potent than G70W-2E9 (median IC50 3.1 μg/mL vs. 9.3 μg/mL, median IC80 10 μg/mL vs. 24 μg/mL), although both were less potent than the human FP-specific bNAb VRC34.01 (Figures S4A and S4B). Sequence analysis showed that J601-A6 and J601–1B2 belong to the same lineage, while G70W-2E9 was from a different lineage but utilized the same V-genes (Figures 4D and S4C). Of note, all neutralizing FP-specific mAbs were isolated from FP8-rTTHC immunized animals in G1 and G2. Some FP-specific mAbs were also isolated from G3 animals immunized with V2-SET trimer alone, but these mAbs were not neutralizing (Figure S4A). In a binding assay, isolated FP-specific mAbs bound to FP8 peptide similarly, regardless of the neutralizing activity (Figures S5A and S5B). As 459C V2-SET trimers were used in G2, we tested the binding of FP-neutralizing mAbs to 459C WT, OPT, and ALT trimers, and all these mAbs bound to 459C WT slightly less than to OPT and ALT trimers (Figures S5A–S5C).
FP-specific mAbs bind to Env trimer with binding modes similar to the previously identified DFPH-a class FP-specific bNAbs
To elucidate the structural interactions of the isolated FP-specific mAbs with Env, we determined cryo-EM structures of J601-A6 and J601–1B2 Fabs, each in complex with the BG505 DS-SOSIP Env trimer. J601-A6 and J601–1B2 were derived from the same lineage, with differences in only three residues in the heavy chain and one in the light chain (Figure 4D). The cryo-EM structure of J601-A6 was resolved at 3.5 Å and that of J601–1B2 at 3.8 Å (Figures 4E and 4F; Data S1.4, 6, 7). The two structures were essentially identical, with the distinguishing residues away from the antibody interface with the Env trimer (Data S1.7G, H). Therefore, the effect of these mutations on binding and neutralization was indirect and subtle. J601-A6 bound at the FP site, interacting primarily with the FP N-terminal residues 512–520 (Figures 4E–4G, S5D, and S5E). The dominance of binding to the FP N-terminal segment allowed flexibility in the antibody approach angles, resulting in multiple 3D classes of cryo-EM particles with slightly different binding orientations of the antibodies on the trimer (Data S1.6C, D; Data S1.7E, F). The FP N-terminal segment was held between the complementarity determining region 3 of the heavy chain (CDR H3) and CDR H1 and interacts with CDR L3 (Figure 4G). The binding mode of J601-A6 and J601–1B2 was similar to the broadest vaccine-elicited antibody DFPH-a.01; this similarity as well as similarities in antibody immunogenetics suggested these antibodies to be members of the reproducible DFPH-a antibody class (Figure 4G; Data S1.6G, H).^41,52^ The dominant binding to the relatively conserved FP N-terminal segment allowed for the broad recognition of HIV-1 strains, although the moderate binding surface area and low somatic hypermutation appeared to limit the potency of neutralization.
V2-SET Env immunized G2 and G3 animals show V2-specific serum neutralization
As we observed higher autologous and broader heterologous neutralization with the V2-SET trimers compared with 459C WT alone, we tested sera for neutralization against V2 epitope mutants N160A and K169E on a T250–4 Env backbone, with the V2 apex-specific bNAbs PG9 and CAP256/VRC26.25 included as positive controls and the V3-glycan bNAb PGT121 as the negative control. As expected, PGT121 was not impacted by N160A or K169E mutations, whereas V2 apex-specific bNAbs lost neutralization activity on N160A and K169E mutants, except CAP256/VRC26.25, which retained strong neutralization on N160A (Figure 5A). Week 68 sera from V2-SET immunized G2 and G3 animals were tested, and all showed substantial reductions of neutralization to the K169E mutant, indicating that serum neutralization against T250–4 was largely due to V2-specific NAbs (Figures 5A and 5B). However, all animals except K120 showed enhanced neutralization on the N160A mutant, indicating that this mutation is beneficial for serum neutralization (Figures 5A and 5B). We also assessed serum neutralization against ZM233.6 and similarly found that animals from V2-SET immunized G2 showed reduction of neutralization to the K169E mutant, indicating that serum neutralization against ZM233.6 was also due to V2-specific NAbs (Figures 5C and 5D). These data suggest that V2-specific NAbs contributed substantially to the observed tier 2 serum neutralization.
V2-specific mAbs isolated from V2-SET Env immunized animals showed neutralization with binding modes similar to the previously identified V2 bNAbs
To further investigate V2-specific neutralization activity in 459C V2-SET immunized G2 and G3 animals, we used 459C WT, OPT, ALT, T250–4, ZM233.6, H703.2631, and CT184 gp140 DS SOSIP trimer probes and K169E mutant probes for V2-specific mAb isolation (Figure S6A) and confirmed that these mAbs do not bind to FP8 peptide (Figure S6B). We identified V2 apex-targeted mAbs that effectively competed with V2 apex bNAbs on binding to Env trimers (Figures 5E and S6C). Antibody sequence analysis revealed well-characterized V2 bNAb signatures, including a long and negatively charged CDR H3 and the usage of the Mamu-IGHD3–9^***^01 gene (Figure 5F).
Next, we tested the V2-specific mAbs in neutralization assays and classified them based on the neutralization pattern of 459C WT, OPT, and ALT pseudoviruses. We defined neutralization class I mAbs as those that neutralized 459C OPT but not WT and ALT pseudoviruses, neutralization class II mAbs as those that neutralized 459C WT and ALT but not OPT pseudoviruses, and neutralization class III mAbs as those that neutralized all three autologous pseudoviruses and some heterologous pseudoviruses, including T250–4, ZM233, and Ko459 (Figure 6A). CDR H3 sequence alignment of three classes of mAbs showed similar motifs (Figure 6B). In binding assays, these antibodies generally showed stronger binding to 459C OPT and ALT than WT trimers (Figure S6D). The V2-specific mAbs showed much more potent autologous neutralization against 459C WT and T250–4 viruses compared with FP-specific mAbs, but lower heterologous neutralization against other tier 2 viruses (Figure S6E). All T250.4 neutralizing mAbs showed reduced activity against N160A and K169E mutants in neutralization assays, confirming that these mAbs were V2 apex-specific mAbs (Figure 6C). Indeed, negative-stain electron microscopy (nsEM) 3D reconstruction maps of several of these antibodies revealed their binding to the V2 apex of Env trimers (Figure 6D). Comparison of the nsEM 3D map of the K001-A1 Env trimer complex with the cryo-EM map of PG9 Env trimer complex (EMDB: EMD-25736)^53^ indicated a similar binding mode as PG9 but with a slightly different angle of approach (Figure 6E).
K001-A1 binds the V2 apex of the Env trimer, interacting with all three protomers at the V2 apex hole and tilting to contact primarily the C-strand of one protomer
To characterize the molecular interactions between K001-A1 mAb and Env trimer, we determined a cryo-EM structure of K001-A1 Fab in complex with 459C-OPT Env trimer at 3.0 Å (Figure 7, Data S1.4, 8). There was a single copy of K001-A1 Fab binding at the V2 apex of the Env trimer interacting with the three gp120 subunits around the apex hole (Figure 7A and Data S1.9A). The cryo-EM map had sufficient density for the K001-A1 constant domains, albeit weaker than the rest (Data S1.9E), which enabled modeling the Fab constant domains. The antibody binding mode was analogous to PG9^47^ and CAP256/VRC26.25,^48^ with the CDR H3 inserting into the apex hole and the rest of the binding epitope predominantly but not exclusively on one gp120 subunit.
K001-A1 binding differed from PG9 and CAP256/VRC26.25 in the relative position of the heavy and light chains and a larger tilt angle, and its light chain contacted the trimer. Consequently, the K001-A1 epitope area was larger with an additional patch distal to the apex hole, unlike PG9 and CAP256/VRC26.25 (Figure 7B). CAP256/VRC26.25 had its CDR H3 loop penetrating deeply into the apex hole and thus interacted strongly with all three gp120 subunits around the apex hole. K001-A1 was more like PG9, with a rather asymmetric epitope surface, interacting with two of the three glycans at 160 and reaching to glycans 197 and 156. The interactions with glycan 156, however, were different between PG9 and K001-A1 (Data S1.9C). PG9 interacted with glycan 156 favorably, maintaining its ligand-free conformation. K001-A1, however, would clash with glycan 156 if this glycan were to adopt the same conformation as in the PG9 complex or the glycan 156 of the other two protomers in the K001-A1 complex; instead, the K001-A1 light chain pushed away glycan 156, which was mostly disordered in the cryo-EM density map. Therefore, K001-A1 interaction with glycan 156 was likely energetically unfavorable.
K001-A1 binding to the Env trimer involved both heavy and light chains. The heavy chain interactions were similar to other V2 apex bNAbs, involving all three CDRs binding the V1V2 region, with the long CDR H3 dominating the binding (Figure 7C). The heavy chain contact regions were around the apex hole, involving mostly chains C and G (the three gp120 subunits were designated as chains C, G, and I, with chain G containing most of the binding epitope; Figures 7B and 7C). The K001-A1 light chain interactions involved all three CDRs and framework region 3, interacting with mostly chain G and covering an additional patch away from the apex hole. The total epitope surface area was nearly 2,300 Å^2^, which was larger than that of PG9 and CAP256-VRC26.25 (Data S1.9D and E).
Despite the substantial contribution to the epitope from light chain binding, heavy chain binding mimicked the binding of the well-characterized V2 apex bNAbs, with its long, highly negatively charged CDR H3 interacting with the positively charged residues at the apex hole and the lysine-rich V1V2 β-strand C (C-strand) (Figures 7C–7G). The C-strand plays a dominant role in binding interactions of V2 apex bNAbs.^48,53,54^ The 459C OPT Env had four lysine residues at positions 168–171. The chain G gp120 subunit contributed to the majority of the epitope, and all the C-strand lysine side chains were in close contact with K001-A1 (Figure 7D). K168 was 5.2 Å away from D28_L_; K169 was 4.6 Å from D100e_H_; K170 was 2.8 Å from D100k; and K171 was 3.2 Å from the carbonyl of P100l_H_ and had hydrophobic interactions with P100l_H_ and Y100j_H_. Most of these lysine residues within the epitope were variable among 459C WT, OPT, and ALT trimers, as well as among the circulating HIV-1 strains (Data S1.9B and 10), partially explaining the limited heterologous neutralization by K001-A1; the variability in Env positions 168–171 also generally constrained the breadth and potency of V2 bNAbs.^14^ Moreover, Y100 of the CDR H3 was sulfated and had a salt bridge with R166 of chain G (Figure 7E). This sulfated tyrosine added to the highly negative charges of the CDR H3, which interacted with the positively charged R166 and K169 residues at the apex hole from the three protomers (Figure 7F). Overall, the K001-A1 binding had substantial similarities to PG9, using an axe-like recognition mode, with parallel β-strand and charge pairing with the C-strand and the apex hole.
DISCUSSION
Elicitation of bNAbs by vaccination has been a long-sought goal of the HIV-1 vaccine field, and induction of bNAbs to multiple sites of vulnerability will almost certainly be required for an effective HIV-1 vaccine, given the challenge of HIV-1 variability. However, most HIV-1 vaccine studies have to date focused on the induction of bNAbs to a single epitope, such as the FP, V2 apex, V3 supersite, CD4 binding site, and MPER region.^24,26,30–32^ In this study, we designed a combined dual-epitope vaccine targeting both FP and V2 and demonstrated the elicitation of potent autologous and low heterologous serum neutralizing activity to both epitopes in rhesus macaques.
Our dual-epitope vaccine included the FP-rTTHC conjugate and three related 459C V2-SET Env trimers with V2 modifications.^14,26^ The FP-rTTHC immunogen was co-administered with trimers at the start of the vaccination regimen,^10,15^ and the trimers had compatible FP sequences. The 459C OPT trimer showed enhanced binding to V2-specific bNAbs and longer retention in draining lymph nodes compared with the WT trimer, and the trimeric V2-SET vaccine induced >1,000-fold higher autologous neutralizing titers than did the 459C WT trimer. Vaccine-elicited autologous tier 2 NAb titers reaching >1,000,000 and even >100,000 are uncommon for neutralization-resistant HIV-1 strains such as 459C. These NAb titers contrast with the relatively modest autologous neutralizing titers elicited by the 459C WT trimer and previous studies with the BG505 trimer. It is possible that the difference in the length of V1V2 loops, the removed glycans, and the structural differences may have contributed to the differences in immunogenicity between the V2-SET vaccine and the WT trimer. Structural differences were observed in the EM analysis (Data S1), with the wild-type 459C trimer producing well-ordered reconstruction at 3 Å resolution, while the OPT trimer was too flexible to enable residue-level tracing. It remains to be seen whether the enhanced lymph node retention and the increased Env immunogenicity with the V2-SET vaccine will be generalizable to other Env strains.
In addition to autologous and heterologous tier 2 serum NAb responses, both V2 apex-specific and FP-specific mAbs were isolated from the immunized animals and showed autologous and some heterologous neutralizing activity. V2 apex-specific mAbs showed competition against human V2-specific bNAbs for binding to Env trimers, sequence analyses revealed similar antibody signatures, including long and negatively charged CDR H3 loops, and structural analyses revealed V2 apex binding of these isolated mAbs. However, V2-specific mAbs showed only modest heterologous neutralizing breadth, likely because of limited maturation and increased specific light chain contacts. These data suggest a path forward for improving the induction of V2-specific NAb breadth by adding immunogens that facilitate antibody maturation. In contrast, FP-specific mAbs showed broad cross-strain neutralizing activity with 47% breadth on a 208-strain HIV-1 panel.
In summary, our data demonstrate that FP-rTTHC and 459C V2-SET Env elicited NAbs against both FP and V2 epitopes that exhibited potent autologous neutralizing activity and some cross-strain heterologous neutralizing breadth in rhesus macaques. Although most HIV-1 vaccine studies have to date focused on the induction of NAbs against a single epitope, our strategy to target multiple sites of HIV-1 Env vulnerability simultaneously will likely be critical for an effective HIV-1 vaccine for humans.
Limitations of the study
Our analyses of epitope-specific antibody responses involved macaques immunized with FP8-rTTHC and 459C Env trimers; so it is possible that immunization with different FP immunogens, Env trimers, and adjuvants may show different results. Moreover, the addition of heterologous immunogens from different strains may improve maturation of the V2 NAb responses and improve V2-specific neutralization breadth. Finally, clinical trials will be required to demonstrate the translatability of our findings to humans.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dan H. Barouch ([email protected]).
Materials availability
All new reagents are available by materials tranfer agreement for non-commercial research.
Data and code availability
Cryo-EM structures have been deposited to the Protein Data Bank with accession codes PDB: 9PQ2, 9PQ3, 9PNN, 9PNI, and 9PNU, and cryo-EM maps have been deposited to the EMDB with accession codes EMDB: EMD-71781, 71782, 71767, 71766, and 71772.This study does not generate new code.Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
CONSORTIA
The VRC Production Program includes Nadia Amharref, Nathan Barefoot, Christopher Barry, Elizabeth Carey, Ria Caringal, Kevin Carlton, Naga Chalamalsetty, Adam Charlton, Rajoshi Chaudhuri, Mingzhong Chen, Peifeng Chen, Nicole Cibelli, Jonathan W. Cooper, Hussain Dahodwala, Marianna Fleischman, Julia C. Frederick, Haley Fuller, Jason Gall, Isaac Godfroy, Daniel Gowetski, Krishana Gulla, Vera Ivleva, Lisa Kueltzo, Q. Paula Lei, Yile Li, Venkata Mangalampalli, Sarah O’Connell, Aakash Patel, Erwin Rosales-Zavala, Elizabeth Scheideman, Nicole A. Schneck, Zachary Schneiderman, Andrew Shaddeau, William Shadrick, Alison Vinitsky, Sara Witter, Yanhong Yang, and Yaqiu Zhang.
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Macaque study
Fifteen outbred male and female rhesus macaques of Indian origin (Macaca mulatta) aged 3–5 years old were assigned to three study groups (n = 5/group) with even sex and weight distributions. All macaque were housed and maintained at Alpha Genesis Inc. (Yemasee, SC), and all macaque studies were conducted in compliance with all relevant local, state, and federal regulations and were approved by the Alpha Genesis Institutional Animal Care and Use Committee (IACUC). Group 1 received three bimonthly immunizations of rTTHC.FP8.v1 (100μg) mixed with 459C repair and stabilized (RnS) wild-type (WT) gp140 Env (100μg) at weeks 0, 8 and 16 followed by six bi-monthly immunizations of 459C RnS WT gp140 Env at weeks 24, 32, 40, 48, 56 and 64. After a resting interval of 48 weeks, animals received an additional five bimonthly booster immunizations of rTTHC.FP8.v1 (100μg) combined with 459C RnS WT gp140 Env (100μg) at weeks 112, 120, 128, 136, 142 with a sixth and final boosting immunization of 459C RnS WT gp140 only at week 152. Group 2 received the same immunization regimen as group 1 with 459C RnS WT gp140 Env replaced with trivalent 459C V2-SET gp140 Env (100μg total). Group 3 received the same regimen as groups 1 and 2 using trivalent 459C V2-SET (100μg total) only. All immunizations were administered intramuscularly and bilaterally in quadriceps muscles using Adjuplex adjuvant (Sigma-Aldrich Inc, MO or equivalent).
Mice immunization and sample collections
All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Beth Israel Deaconess Medical Center (BIDMC). A total of 72 eight week-old female Balb/cJ mice (strain #000651, The Jackson Laboratory) were randomly assigned to four experimental groups (n = 18). Mice were housed in pathogen-free conditions on a 12-h light/dark cycle at 21 ± 1°C with ad libitum access to food and water. Mice were anesthetized and immunized intramuscularly with 50 μg of Alexa Fluor 647-conjugated immunogens (WT-A647, OPT-A647, or ALT-A647) formulated in a 1:1 ratio with Adju-Phos adjuvant (InvivoGen, vac-phos-250), administered in a total volume of 100 μL (50 μL per hind leg). A group of unimmunized animals served as naive controls. Draining lymph nodes (dLNs) and spleens were harvested at days 1, 3, 5, 7, 9, and 12 post-immunization following euthanasia and placed into RPMI-1640 medium supplemented with 2% FBS for further processing.
Cell lines
Expi293F cells (cat# A14257) and FreeStyle 293-F cells (cat# R79007) were purchased from ThermoFisher Scientific Inc. Cells were maintained in FreeStyle 293 Expression Medium. The cell line was used directly from commercial sources and cultured following the manufacturer’s suggestions, as described in the method details section below. Cell lines were authenticated by the manufacturer and were tested for mycoplasma contamination.
METHOD DETAILS
Antigen labeling and characterization
HIV Env trimer protein antigens (WT, OPT, and ALT) were conjugated to Alexa Fluor 647 (A647) using the Alexa Fluor 647 Protein Labeling Kit (Invitrogen, A20173), following the manufacturer’s protocol. Env trimers were first diluted to 2 mg/mL in 0.5 mL of PBS. To this, 50 μL of 1 M sodium bicarbonate buffer (pH 8.4) was added, and the mixture was maintained on ice. The buffered protein solution was then added to the vial containing the A647 dye. Labeling reactions were carried out at RT for 2 h with gentle stirring. Following conjugation, excess dye was removed by passing the samples through Zeba Spin Desalting Columns pre-equilibrated with PBS, with two rounds of purification. Labeled proteins were sterile-filtered using 0.22 μm Spin-X centrifuge tube filters (Millipore Sigma, CLS8160) and stored at 4°C until further use. The degree of labeling was determined by measuring absorbance at 280 nm and 650 nm, corresponding to total protein and A647 dye, respectively. Protein and dye concentrations were calculated using extinction coefficients of 118,000 M^−1^ cm^−1^ for each trimer subunit (WT, OPT, and ALT) and 239,000 M^−1^ cm^−1^ for the A647-NHS ester. The labeling ratio was calculated as the molar ratio of A647 dye to trimer protein.
Flow cytometry analysis of draining lymph nodes and spleen
Following tissue collection, dLNs were enzymatically digested in 1 mL of digestion buffer containing 0.8 U/mL Dispase (STEMCELL), 0.1 mg/mL Collagenase D (Worthington Biochemicals), and 0.1 mg/mL DNase I (Sigma) at 37°C for 20 min. Tissues were subsequently dissociated into single-cell suspensions by mechanical disruption through a 70-μm cell strainer (BD Biosciences). Spleens were processed without enzymatic digestion by mechanical disruption through a 70-μm filter, followed by red blood cell lysis using ACK lysis (Gibco) buffer for 3 min at RT. Single-cell suspensions from dLNs and spleens were washed with PBS and stained with Live/Dead Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific, L34957) for 10 min at 25°C. After two washes with MACS buffer supplemented with 2% BSA (Miltenyi) and 1.5% penicillin-Streptomycin (Fisher Scientific), cells were blocked with anti-mouse CD16/CD32 Fc receptor antibody (BioLegend, 101319) for 10 min at 4°C, followed by surface staining with fluorophore-conjugated antibodies for 30 min at 4°C. Flow cytometric analysis was performed on a BD LSR II flow cytometer (BD Biosciences). To identify immune cell populations associated with Alexa Fluor 647-labeled immunogens, the cells were stained with the CD3 BUV737 (clone 17A2, BD Biosciences), CD21/CD35 PE (clone 7E9, BioLegend), CD169 BV605 (clone 3D6.112, BioLegend), and B220 BUV395 (clone RA3–6B2, BD Biosciences).
Mannose-binding lectin (MBL) deposition ELISA
High-binding 96-well ELISA plates were coated overnight at 4°C with WT, OPT, or ALT SOSIP protein at 1μg/mL diluted in 1X DPBS. After incubation, plates were washed once with wash buffer (0.05% Tween 20 in 1X DPBS) and then blocked with 350 μL/well of a casein-based blocking solution for 2–3 h at room temperature (RT). After incubation, the blocking solution was discarded, and the plates were gently blotted dry. Serum was freshly collected from naive BALB/c mice using Sarstedt serum gel tubes and kept on ice. Initial serum dilutions were prepared at 30% (v/v) in blocking buffer and further serially diluted 2-fold. Diluted samples were added to the antigen-coated wells and incubated for 1 h at 37°C. Following incubation, plates were washed three times with wash buffer and incubated with 2 μg/mL of rat anti-mouse MBL monoclonal antibody (clone 14D12, Abcam, cat# ab106046) prepared in blocking buffer and incubated for 1h at RT. After three washes, HRP-conjugated anti-rat IgG secondary antibody (1:5000 dilution; Invitrogen, cat# 31470) was added, and plates were incubated for 1h at RT. After three washes, 100 μL of SeraCare KPL TMB SureBlue substrate was added to each well and incubated for 11 min at RT. The reaction was stopped by adding 100 μL of SeraCare KPL TMB stop solution. Absorbance was measured at 450 nm with a reference reading at 650 nm using a VersaMax microplate reader (Molecular Devices). Endpoint titers were determined by fitting the data to a four-parameter logistic (4PL) regression model. The reciprocal serum dilution corresponding to a corrected absorbance value (A450–A650) of 0.1 was reported as the endpoint titer.
Expression of 459C V2-SET gp140 Env constructs
The 459 V2-SET gp140 Env proteins were generated as previously described.^43^ Constructs were synthesized and codon-optimized (GenScript) and cloned into pCDNA2004 or generated by standard methods involving site-directed mutagenesis and PCR. HEK-Expi293 cells were transiently transfected with 90% of pCDNA2004 plasmid with the Env insert and 10% of Furin-pCDNA2004, according to the manufacturer’s instructions and cultured for 5 days at 37°C and 10% CO2. Culture supernatants were spun for 10 min at 1,250 × g. For expressions in 96-well format, the cells were cultured for 3 days at 37°C and 10% CO2. 4 μL of Opti-MEM was mixed with 4 μL 100 ng/μL DNA, and 8 μL Expi293F mix (54 μL/mL Opti-MEM) as added and incubated for 20 min. Subsequently, 200 μL/well Expi293F cells were added at 2.5 × 10E6 cells/mL. The culture supernatant was harvested and spun for 10 min at 200 × g to remove cells and cellular debris. The clarified supernatant was subsequently sterile-filtered using a 0.22 μm vacuum filter and stored at 4°C until use. For crystallization, the protein was produced in HEK293 GnTI−/− cells via transient transfection with 10% of Furin-pCDNA2004.
Purification of 459 V2-SET gp140 trimers using lectin and SEC
The recombinant 459C V2-SET Envs were purified by a 2-step purification protocol applying a Galantus nivalis-lectin column (Vectorlabs) for the initial purification and subsequently a Superdex200 Increase column (GE) for the polishing step to remove residual contaminants. For the lectin step, the culture supernatant was diluted with 40 mM Tris, 500 mM NaCl pH7.5, and passed over a 4 mL CV Tricorn 10–50 lectin agarose column at 300 cm/h. Subsequently, the column was washed with 4 column volumes (CV) of 40 mM Tris, 500 mM NaCl pH7.5, and eluted with 4 CV of 40 mM Tris, 500 mM NaCl, 1 M mannopyranoside pH 7.5 with an upflow of 120 cm/h. The eluate was concentrated using a spin concentrator (50 K, Amicon Ultra, Millipore) and the protein was further purified using a Superdex200 Increase 10/300 column using 20 mM citrate, 75 mM NaCl, 5% sucrose, and 0.03% Tween 80 pH 6.0 as running buffer. The second peak contained the HIV gp140 trimer. The fractions containing this peak were pooled, and the protein concentration was determined using OD280 and stored a 4°C until use.
Size exclusion chromatography and multi-angle light scattering analysis
Purity and mass were confirmed by size exclusion chromatography (SEC) and multi-angle light scattering (MALS) using a high-performance liquid chromatography system (Agilent Technologies) and miniDAWN TREOS (Wyatt) instrument coupled to an Optilab T-rEX Refractive Index Detector (Wyatt). In total, 40 μg of purified protein was applied to a TSK-Gel G3000SWxl column (Tosoh Bioscience) equilibrated in running buffer (150 mM sodium phosphate, 50 mM sodium chloride, pH 7.0) at 1 mL/min. In other experiments, analysis was performed on supernatants instead of purified Env. The data were analyzed using the Astra 6 software package, and molecular weight calculations were derived from the refractive index signal.
Bio-Layer interferometry (Octet)
Antibodies were immobilized on anti-hIgG (AHC) sensors (FortéBio) at a concentration of 1 μg/mL in 10× kinetics buffer (FortéBio) in 96-half well black flat bottom polypylene microplates (Forté-Bio). Experiments were performed on an Octet RED384 instrument (Pall-FortéBio) at 30°C, shaking speed 1,000 rpm. Activation was 300 s, immobilization of antibodies 600 s, washing 300 s, and binding the Envs 1,200 s, followed by a dissociation of 1,200 s, all shaking at 1,000 rpm. The data analysis was performed using the FortéBio Data Analysis 8.1 software (FortéBio).
459C trimer probe and H703, CT184, T250.4 DS SOSIP trimer probe production
AVI-tagged H703, CT184, T250.4 repaired and stabilized (RnS) DS SOSIP trimers, and 459C SET trimers, were produced using transient transfection in FreeStyle 293F cells (Thermo Fisher Scientific). In brief, pre-mixed 600 mg constructs encoding N terminus HRV3C cleavable single-chain Fc-tagged and C terminus AVI-tagged trimers and 150 mg furin plasmid in 2.25 mL Turbo293 transfection reagent (Speed BioSystems) were added to 0.8 L of cells at a cell density of 2 × 106 viable cells/ml. Transfected cells were cultured for 6 days in an orbital shaker at 125 rpm at 37°C in a humidified 9% CO2 incubator before the supernatant was harvested. Subsequently, the supernatant was incubated with 5 mL of PBS-equilibrated protein A resin for 1–2 h. The trimer-bound resin was then washed with PBS, collected, and then incubated at 4°C overnight in a 3-mL mixture comprised of 200 μg of HRV3C and BirA biotin-protein ligase mixture (Avidity) according to manufacturer instructions. The HRV3C-liberated and biotinylated trimers were applied to a Superdex 200 16/600 gel filtration column equilibrated with PBS the next day. Peak fractions corresponding to trimers were pooled and negatively selected using a V3 cocktail column containing six V3-directed antibodies (1006–15D, 2219, 2557, 2558, 3074, and 50.1).
Trimer ELISA for serum samples and monoclonal antibodies
The ELISA using the autologous 459C trimer was performed by coating 96-well Costar half plates (Costar High Binding Half-Area; Corning, Kennebunk, ME) with 2 μg/ml streptavidin overnight at 4°C. The plates were then washed and coated with biotinylated 459C WT, OPT and ALT trimers. For heterologous BG505 DS-SOSIP trimer binding, the method was modified based on previously reported method with lectin captured trimer. ^15^ Briefly, 96-well Costar half plates were coated with 2 mg/mL Lectin and after a one-hour blocking step with 5% skim milk/PBS, the BG505 trimer or glycan-base trimer was incubated on the plates for two hours at RT. After washing the plates 5x with PBS-T, serially diluted sera (at various starting dilutions) or monoclonal antibodies at 2 mg/mL was added to the plates and further incubated for one hour at RT. The plates were then washed 5x with PBS-T and goat anti-rhesus HRP-conjugated secondary antibody was added to the plates at a 1:5000 dilution for one hour at RT. The plates were then developed for 10 min using tetramethylbenzidine (TMB) substrate (SureBlue; KPL, Gaithersburg, MD). To stop the reaction, 1 N H2SO4 sulfuric acid was added. The plates were then read at OD450 nm and values were documented. EC50 for serum binding was calculated when all of the bindings reached the peak OD value, otherwise AUC or endpoint titers were calculated if not all samples reached the peak values.
Anti-Fusion-Peptide (FP8) ELISA
The ELISA for assessing the response to Fusion-Peptide (FP8) was conducted according to a previously described method.^15^ Biotinylated eight-residue fusion peptide (FP8-PEG-biotin) was coated in 96-well streptavidin-coated plates (Thermo Fisher) overnight. Subsequently, the plates were blocked with B3T buffer (comprising 150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 3.3% fetal bovine serum, 2% bovine albumin, 0.07% Tween 20, and 0.02% thimerosal). The serum was serially diluted at 7-point 5-fold dilution and incubated for 1hr. Goat anti-rhesus HRP conjugated secondary was added and incubated for an hour. The plates were then developed for 10 min using tetramethylbenzidine (TMB) substrate (SureBlue; KPL, Gaithersburg, MD). To stop the reaction, 1 N H2SO4 sulfuric acid was added. Finally, the plates were read on a microplate spectrophotometer (Biotek Epoch, Winooski, VT) to determine the endpoint titer for each sample.
Neutralization assays
Neutralization assays using a single round of infection Env-pseudovirus were performed using TZM-Bl target cells and heat-in-activated sera.^42^ Briefly, 293T cells were cotransfected with an Env expression plasmid and a sPG3ΔEnv backbone to generate the Env-pseudovirus stocks used in neutralization assays. The sera were assessed at various dilutions, using an 8-point 4-fold dilution method which began at a dilution factor of 1:20. The 50% inhibitory dilutions (ID50) were used to assess breadth across the 10 FP-sensitive HIV-1 strains and to test for any correlative relationship with the plasma and B cell parameters tested with Prism.
HIV-1 envelope trimer immunogens
All HIV-1 envelope trimer immunogens were prepared using transiently transfected 293F cells as previously described.^26^ Broadly neutralizing antibodies 2G12 or VRC01 were used in affinity chromatography to purify the trimers, along with gel filtration (Superdex200 16/60HL column) and a 447–52D affinity column functioning as a negative selection to remove V3-exposed trimers. Antigenicity tests were performed on the trimers with or without mixing with adjuvant using a Meso Scale Discovery (MSD) platform as previously described.
Isolation of rhesus monoclonal antibodies
Cryopreserved rhesus macaque PBMCs were thawed and stained with LIVE/DEAD fixable violet dead cell stain (Life Technologies). After washing, cells were stained with a cocktail of anti-human antibodies, including CD3 (clone SP34–2; BD Biosciences), CD4 (clone OKT4; BioLegend), CD8 (clone RPA-T8; BioLegend), CD14 (clone M5E2; BioLegend), CD20 (clone 2H7; BioLegend), IgG (G18–145;BD Biosciences), and IgM (clone G20–127; BD Biosciences), and with fluorescently labeled trimer probe (BG505 DS-SO-SIP.avi) and peptide probe (FP9-PEG12-biotin).^42^ Vivid-CD3^−^CD4^−^CD8^−^CD14^−^CD20+IgG+IgM-memory B cells that are positively stained with both trimer and peptide probes were sorted into 96-well plates containing lysis solution as previously described.^42^ Nested PCR was performed using published primers.^65,66^ Heavy and light chain sequences were cloned into expression vectors containing rhesus macaque immunoglobulin constant regions. IgG was expressed by cotransfecting Expi293FTM cells with equal amounts of paired heavy and light chain plasmids and purified using protein A Fast Flow (GE Healthcare) according to the manufacturer’s instructions.
Rapid assembly, transfection, and production of immunoglobulins (RATP-Ig)
BG505+BG505 glycan-base trimer+ or Cap256 + K169E-sorted memory B cells were subjected to RATP-Ig analysis as previously described.^67^ Single-cell RNA was purified with RNAclean beads (Beckman Coulter). cDNA was then synthesized using 5′ RACE reverse-transcription and amplified by PCR. An aliquot of enriched cDNA was sequenced using 2×150 paired-end reads on an Illumina MiSeq. For immunoglobulin production, enriched variable regions were assembled into expression cassettes that include CMV, and HC/LC-TBGH polyA fragments. Assembled cassettes were amplified by PCR and transfected into Expi293 cells in 96-well deep-well plates using the Expi293 Transfection Kit (ThermoFisher Scientific). Cell cultures were grown at 37C and 8% CO2, with 1100 RPM shaking for 5–7 days. Cell culture supernatants were harvested by centrifugation.
Antibody gene assignment, nomenclature and genetic analysis
Antibody sequences were annotated using IMGT V/Quest^59^ and IgBlast^58^ gene database. Antibodies sharing both heavy and light chain V and J gene assignments and junction region were grouped into one lineage. Monoclonal antibodies were named to include information from the donor and the sorting plate and well containing the single B-cell. For example, the designation mAb J601–1B2, indicates a mAb isolated from well B2 on sorting plate 1 from animal J601. For both heavy chain and kappa chains, germline V genes from a public database^68^ and genes predicted to be used by FP antibodies but specific to an animal were combined to construct a phylogenetic tree using MEGA6 with GTR+G substitution model.^69^
Antibody preparation
The antibody heavy- and light-chain variable region DNA sequences were synthesized and separately cloned into an mammalian cell expression plasmid and sequenced. Plasmids encoding heavy and light chain pairs were co-transfected into Expi293F cells (Thermo Fisher) as described previously. The cells were grown for six days, and the culture supernatants were harvested and loaded on protein A columns. After washing the columns with PBS, the IgG proteins were eluted with a low pH buffer. For antibody Fab preparation, an HRV3C cleavage site was inserted in the heavy-chain hinge region, and the purified IgG protein was digested with HRV3C. The Fab was separated from Fc by passing the digestion mixture through a protein A column and then further purified through a Superdex 200 column (Cytiva).
Negative-stain EM analysis
The purified antibody Fab was mixed with HIV-1 Env trimer at a 3:1 molar ratio. The mixture was diluted with a negative-stain buffer (10 mM HEPES, pH 7.0, and 150 mM NaCl) to approximately 0.02 mg/mL, and the diluted Fab-Env mixture was transferred to a freshly glow-discharged carbon-coated grid and incubated for a few seconds, and the liquid was removed with a piece of filter paper. The grid was rinsed 3 times with the negative-stain buffer and then stained with 0.75% uranyl formate for 30 s. Data were collected using a Talos F200C transmission electron microscope (ThermoFisher Scientific) operated at 200 kV. Particle picking, 2D classification, and 3D reconstruction were carried out using CryoSparc 4.6.^57^ The 3D volumes of the Fab-Env complex density were visualized and analyzed, and figures were prepared using UCSF ChimeraX.^55^ For EMPEM analysis, polyclonal Fabs were prepared with Pierce Fab Preparation Kit (ThermoFisher Scientific) and then mixed with Env trimer. The Fab-Env complexes were separated by SEC Superose 6 column (Cytiva, Wilmington, DE) in PBS, and the Fab-Env complex peak was collected for negative-stain EM data collection.
Cryo-EM structure determination
To prepare cryo-EM grids, purified Env trimers at 1–4 mg/mL were mixed with antibody Fab; n-Dodecyl β-D-maltoside (DDM) was added to a final concentration of 0.1mM, and the mixture was applied to a freshly glow discharged carbon-coated copper grid (CF 1.2/1.3 300 mesh) or a Quantifoil-gold 2/2 holey carbon grid. The sample was vitrified in liquid ethane using a Vitrobot Mark IV. Cryo-EM data were collected using Leginon software^60^ on a Titan Krios electron microscope operating at 300 kV, equipped with a Gatan K3-BioQuantum direct detection device. Exposures were taken with a total electron fluence of 58.06 e−/Å2. The total dose was fractionated for 2.5 s over 50 raw frames. Some cryo-EM data were acquired on a Titan Krios operating at 300 kV, equipped with a K2 Summit detector (Gatan) operating in counting mode and using SerialEM 4.0.^64^ The dose was fractionated over 40 raw frames. The movie frames were aligned and dose-weighted, and CTF estimation, particle picking, 2D classifications, ab initio model generation, heterogeneous refinements, homogeneous 3D refinements, non-uniform refinement, and local resolution calculations were carried out using cryoSPARC 4.6.^57^
A model of the antibody Fab was generated using AlphaFold2 or AlphaFold3. The Fab model and the structure of BG505 DS-SOSIP (PDB: 6cdi)^26^ were docked into the cryo-EM map using UCSF ChimeraX.^55^ This initial model was then manually rebuilt to fit into the density and match the sequence of the Env trimer of the specific HIV-1 strain using Coot.^56^ The structural models were refined using Phenix.^63^ Overall structures were evaluated using MolProbity.^61^ Protein interface calculations were performed using PISA.^62^ Structural figures were generated using PyMOL (Schrodinger; http://www.pymol.org) and UCSF ChimeraX.
QUANTIFICATION AND STATISTICAL ANALYSES
A two-tailed Mann-Whitney t test comparing the mean and standard error of the mean (SEM) was performed when comparing two groups in serologic and B-cell studies. For multiple comparison analysis, a Kruskal-Wallis test followed by the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli tests was performed to assess differences in multiple groups. A two-tailed Pearson correlation coefficient test was used to assess correlations between plasma and B cell parameters and neutralization titers and breadth, when data obeyed normality assumptions. When the normality assumption failed, a 2-tailed Spearman correlation coefficient test was performed. *: p < 0.05, **: p < 0.01, ***: p < 0.0001.
Supplementary Material
1
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2025.116905.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bbosa N, Kaleebu P, and Ssemwanga D (2019). HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 14, 153–160. 10.1097/coh.0000000000000534.30882484 · doi ↗ · pubmed ↗
- 2Ferguson MR, Rojo DR, von Lindern JJ, and O’Brien WA (2002). HIV-1 replication cycle. Clin. Lab. Med. 22, 611–635. 10.1016/s 0272-2712(02)00015-x.12244589 · doi ↗ · pubmed ↗
- 3Cale EM, Driscoll JI, Lee M, Gorman J, Zhou T, Lu M, Geng H, Lai YT, Chuang GY, Doria-Rose NA, (2022). Antigenic analysis of the HIV-1 envelope trimer implies small differences between structural states 1 and 2. J. Biol. Chem. 298, 101819. 10.1016/j.jbc.2022.101819.35283191 PMC 9006658 · doi ↗ · pubmed ↗
- 4Hodge EA, Naika GS, Kephart SM, Nguyen A, Zhu R, Benhaim MA, Guo W, Moore JP, Hu SL, Sanders RW, and Lee KK (2022). Structural dynamics reveal isolate-specific differences at neutralization epitopes on HIV Env. i Science 25, 104449. 10.1016/j.isci.2022.104449.35677643 PMC 9167985 · doi ↗ · pubmed ↗
- 5Munro JB, and Lee KK (2018). Probing Structural Variation and Dynamics in the HIV-1 Env Fusion Glycoprotein. Curr. HIV Res. 16, 5–12. 10.2174/1570162×16666171222110025.29268688 · doi ↗ · pubmed ↗
- 6Stewart-Jones GBE, Soto C, Lemmin T, Chuang GY, Druz A, Kong R, Thomas PV, Wagh K, Zhou T, Behrens AJ, (2016). Trimeric HIV-1-Env Structures Define Glycan Shields from Clades A, B, and G. Cell 165, 813–826. 10.1016/j.cell.2016.04.010.27114034 PMC 5543418 · doi ↗ · pubmed ↗
- 7Sanders RW, Derking R, Cupo A, Julien JP, Yasmeen A, de Val N, Kim HJ, Blattner C, de la Peña AT, Korzun J, (2013). A next-generation cleaved, soluble HIV-1 Env trimer, BG 505 SOSIP.664 gp 140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. P Lo S Pathog. 9, e 1003618. 10.1371/journal.ppat.1003618.24068931 PMC 3777863 · doi ↗ · pubmed ↗
- 8Bianchi M, Turner HL, Nogal B, Cottrell CA, Oyen D, Pauthner M, Bastidas R, Nedellec R, Mc Coy LE, Wilson IA, (2018). Electron-Microscopy-Based Epitope Mapping Defines Specificities of Polyclonal Antibodies Elicited during HIV-1 BG 505 Envelope Trimer Immunization. Immunity 49, 288–300.e 8. 10.1016/j.immuni.2018.07.009.30097292 PMC 6104742 · doi ↗ · pubmed ↗