Type I Thanatophoric Dysplasia: Clinical Outcome in the Absence of Molecular Genetic Confirmation: A Case Report
Khadija Mesbah, Kaoutar Ettoini, Kawtar Khabbach, Yousra El Boussaadni, Abdallah Oulmaati

TL;DR
This case report describes a newborn diagnosed with thanatophoric dysplasia before birth, a rare and severe skeletal disorder that is often fatal shortly after birth.
Contribution
The paper contributes a clinical case of TD without molecular genetic confirmation, highlighting diagnostic challenges.
Findings
The newborn exhibited typical features of thanatophoric dysplasia, including severe micromelia and frontal bossing.
The diagnosis was made antenatally based on clinical and imaging findings, without molecular confirmation.
The case underscores the importance of clinical evaluation in the absence of genetic testing.
Abstract
Thanatophoric dysplasia (TD) is the most common lethal congenital skeletal dysplasia, characterized by severe micromelia, a narrow thorax, and frontal bossing due to activating mutations in the fibroblast growth factor receptor 3 (FGFR3) gene on chromosome 4p16.3. First described by Maroteaux and Lamy in 1967, it carries a near-uniform perinatal mortality from respiratory insufficiency. We present the case of a male newborn in whom the diagnosis of thanatophoric dysplasia was established during the antenatal period.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
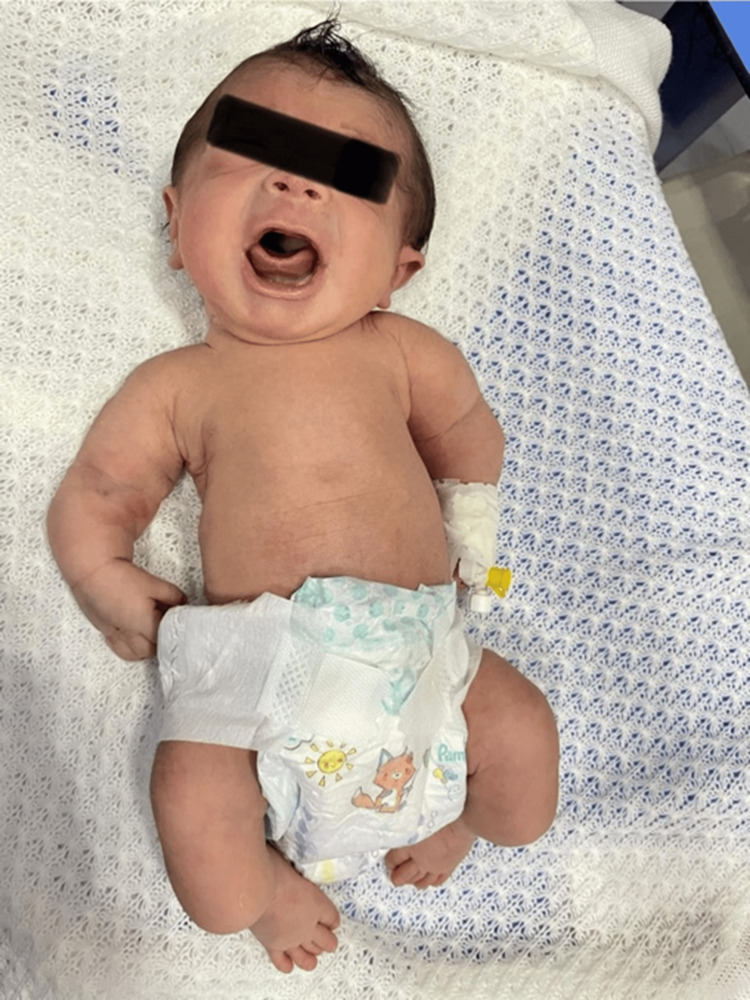 Figure 1
Figure 1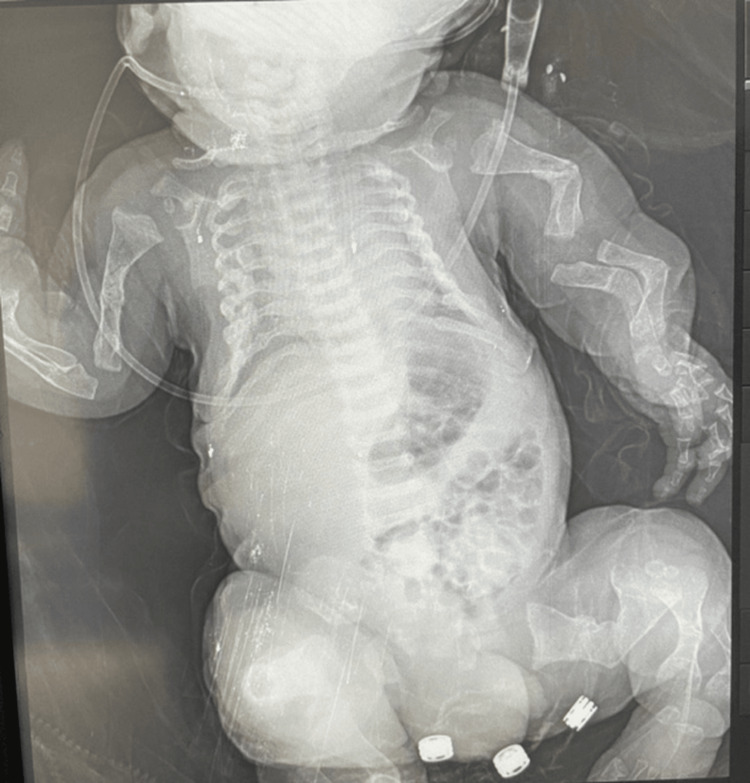 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective tissue disorders research · Protein Tyrosine Phosphatases · Craniofacial Disorders and Treatments