Clinico-genetic heterogeneity in Pakistani families affected with muscular dystrophies
Riaz Ahmad, Muhammad Almas Hashmi, Asad Ullah, Ubaid Ur Rehman, Muhammad Naeem, Henry Houlden

TL;DR
This study identifies new genetic variants in Pakistani families with muscular dystrophies, highlighting the importance of genetic testing for diagnosis and treatment.
Contribution
The first report of MDC1A-associated phenotypes caused by LAMA2 gene variants in the Pakistani population.
Findings
Four novel LAMA2 gene variants were identified in families with MDC1A.
Two known DMD gene variants were found in families with BMD and DMD.
Next generation sequencing proved effective for diagnosing neuromuscular dystrophies.
Abstract
Muscular dystrophies are a group of inherited neuromuscular disorders characterized by degenerative and progressive muscle weakness. Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD) and merosin-deficient congenital muscular dystrophy type 1 (MDC1A) are frequent forms caused by DMD (NM_004006.3) and LAMA2 (NM_000426.4) mutations, respectively. Our study used whole exome sequencing to explore the molecular genetics of muscular dystrophies in six unrelated Pakistani families. We found four novel variants in the LAMA2; c.7470_7473del; p.(Lys2490AsnfsTer56) in family A, c.7807del; p.(Ala2603HisfsTer4) in family C, c.8651T > C; p.(Met2884Thr) in family D and c.4127T > A; p.(Leu1376Ter) in family E associated with MDC1A in these families. Furthermore, two already cited DMD variants were identified: c.10,801 C > T; p.(Gln872Ter) in family B and hemizygous genomic deletion in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
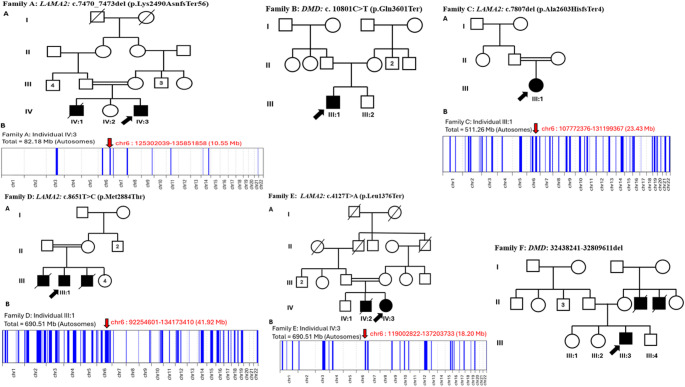 Figure 1
Figure 1- —https://doi.org/10.13039/501100010221Higher Education Commision, Pakistan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMuscle Physiology and Disorders · Cardiomyopathy and Myosin Studies · Ubiquitin and proteasome pathways
Introduction
Muscular dystrophies (MDs) are a group of inherited degenerative neuromuscular disorders that primarily manifest as progressive muscle weakness, leading to significant morbidity. Now, a total of nine specific classes of muscular dystrophy have been diagnosed because of mutations in 57 genes [1]. Duchenne muscular dystrophy (DMD) is the most prevalent and severe form of muscular dystrophy and is caused by mutations in the DMD (dystrophin) (ENST00000357033.9) gene, which is located on the X chromosome [2]. DMD affects approximately 1 in 3600 males [3]. DMD is clinically marked by muscle weakness and atrophy during early childhood that begins by the age of 3 years. Early manifestations include pain or difficulty with walking, delayed growth, a waddling gait and frequent falls [4]. Most affected individuals lose ambulation during late childhood or adolescence and die in early adulthood [5, 6]. The second common form of muscular dystrophy is Becker muscular dystrophy (BMD), which is also caused by variants in the DMD gene and has a global prevalence of approximately 1.5–3.6 in 100,000 males. The milder BMD form is investigated in patients around 11 to 25 years of age; however later age of onset is possible. BMD is characterized by progressive weakness in the shoulders, thighs, hips and pelvis muscles. Patients of BMD have nearly normal lifespans unless they experience cardiac arrest [7].
Merosin-deficient congenital muscular dystrophy type 1 (MDC1A), or laminin-α2chain-deficient congenital muscular dystrophy, caused by variants in the LAMA2 gene (ENST00000421865.3)and inherited in an autosomal recessive manner, is one of the most common subtypes of congenital muscular dystrophy (CMD). In MDC1A, the expression of laminin-α2chain is either reduced or completely absent, leading to early-onset clinical manifestations such as muscle weakness, profound hypotonia, skeletal deformities, inability to achieve ambulation and respiratory complications [8].
Congenital muscular dystrophies (CMDs) are rare disorders with an incidence of 0.82 per 100,000 live births [8]. The most frequent types are laminin-α2chain, collagen type VI and alpha-dystroglycan related types. As reported by Zambo and Muntoni, 37 genes have been associated with congenital muscular dystrophies [9]. The absence or deficiency of the laminin-α2 subunit results in the loss of laminin-211 and/or laminin-221, leading to compromised stability and strength of tissues in skeletal muscle. Depending on the degree of laminin-α2 deficiency, the clinical manifestations range vary from severe MDC1A (OMIM: 607855) to mild late onset LAMA2-MD (OMIM: 618138). In severe MDC1A, newborns are characterized by a weak cry, hypotonia and muscle weakness, which lead to delayed motor developmental milestones [10–11]. Only a few children can walk with support; otherwise, most are non-ambulatory [12]. Other complications include difficulty in feeding, swallowing and chewing because of restricted growth [13]. Several patients require enteral feeding and may also develop respiratory problems and occasionally need ventilatory support during their lifetimes [14]. In 30% of the patients, death is caused by respiratory tract infection (RTI) in the first decade of life with early onset of MDC1A [15]. In low- and middle-income countries (LMICs), mostly patients remain misdiagnosed or undiagnosed. The genetic architecture of muscular dystrophies varies across populations, and in regions with high consanguinity, such as Pakistan, autosomal recessive forms of inherited neurological disorders are more common [16]. Limited molecular data of muscular dystrophies from Pakistan have been published, leaving the population’s mutational spectrum poorly characterized, hindering accurate diagnosis, genetic counseling, and potential therapeutic interventions.
This study aimed to use exome sequencing as a first line of diagnostic approach to identify the cause of the disease. Through NGS technology we investigated six unrelated Pakistani families affected with mild to severe manifestations of muscular dystrophies. Disease-causing variants were identified in these families that strengthen the genotype-phenotype correlation and explore the disease burden around the globe. Early diagnosis and genetic counseling help in reducing the impact and mortality of the disorder.
Materials and methods
Ethical approval, recruitment and human subjects
Six unrelated human families were recruited from different regions of Pakistan including Khyber Pakhtunkhwa Province, Punjab Province and Islamabad Capital Territory. The study was approved by the Institutional Review Board of Quaid-I-Azam University, Islamabad, Pakistan (Approval No. QAU/DFBS/216). Written informed consent was obtained from the parents or legally authorized representatives of all minor subjects included in this study. Blood samples were collected from the participating normal and affected individuals of the families. Genomic DNA was extracted from peripheral blood lymphocytes by standard extraction protocol.
Exome sequencing and variants calling
Whole exome sequencing for six families (A, B, C, D, E and F) was performed at Macrogen, Korea through the Agilent SureSelect Human All Exome V6 Kit (Agilent Technologies, Santa Clara, CA, USA) as described by Efthymiou et al. [17] and Ahmad et al. [18]. Paired-end sequencing or PE150 was done by Illumina NovaSeq 6000 (Illumina, Santa Clara, CA, USA). The resultant read of sequencing against the reference genome of humans was aligned through Burrows-Wheeler Aligner v0.7.17 (BWA). Human genome assembly (hg19) was selected for our enrolled families, and hg38 assembly was used for sequencing reads. BAM files were sorted duplicated reads were identified via SAMtools (v1.8) and Picard (v2.18.9), respectively. Genome Analysis Toolkit (GATK) v4.0 was used for genotyping. Further, functional annotation and variant filtration were done by Annotate Variation (ANNOVAR) and FILTUS, respectively. After annotation, the obtained file was recovered in CSV format which was further filtered to identify the most accurate possible pathogenic variants. Our bioinformatic filtration strategy focused on searching for the exonic region (coding region), splice donor and acceptor site as described previously in supplementary Figure S1 [19]. Following pedigrees and clinical history, we chose the rare variants with MAF < 0.01% in databases including 1000 Genomes project, NHLBI Exome Variant Server, Exome Aggregation Consortium and Complete Genomics 6. Inheritance patterns and different in silico tools (shown in Table 1) were also considered for nonsense, missense, splice site and frameshift mutations. Pathogenic and likely pathogenic variants were prioritized based on ACMG guidelines. Our phenotypes are associated with muscular dystrophies so copy number variations (CNVs) analysis was also checked accordingly.
Table 1. Genetic findings of six unrelated families associated with muscular dystrophies in this studyFamilyID: (patient ID)GeneGenomic positioncDNAHGVSProtein HGVSModeType of variantAllelic FrequenciesCADDScoredbSNP IDMutation TasterPolyphen-2ReferenceACMGA: (IV:3) LAMA2 Chr6:129478708c.7470_7473delp.Lys2490AsnfsTer56ARFrameshiftNANANANANANovelLPB: (III:1) DMD ChrX:31,146,411c.10,801 C > Tp.Gln872TerX-linkedNonsenseNA45NADisease-causingNAReportedLPC: (III:1) LAMA2 Chr6:129486527c.7807delp.Ala2603HisfsTer4ARFrameshiftNANANANANANovelLPD: (III:1) LAMA2 Chr6:129505303c.8651T > Cp.Met2884ThrARMissenseExomes: ƒ =0.0000047919.35rs772282706Disease-causingPossibly damagingNovelVUSE: (IV:3) LAMA2 Chr6:129320606c.4127T > ALeu1376TerARStop gainedNA38NADisease-causingNANovelPF: (III:3) DMD ChrX: 32438241–32809611c.32438241-32809611delNAX-linkedGross deletionNANANANANAReportedPNA: Not available, LP: Likely pathogenic, P: Pathogenic, VUS: Variant of unknown significance, AR: Autosomal recessive
Homozygosity mapping
To confirm the homozygous region shared by ancestors, we performed homozygosity mapping through the AutoMap (https://automap.iob.ch/process), which was used with default settings (as shown in Fig. 1 for families A, C, D and E). VCF (Variant Call Format) files of WGS or WES could be used to generate homozygosity stretches [20].
Fig. 1. Pedigree and homozygosity mapping of six unrelated families (A, B, C, D, E and F). Probands subjected to whole exome sequencing are denoted by arrows. Squares represent males, while circles represent females. Unfilled symbols indicate healthy subjects, and filled symbols indicate affected individuals. Consanguinity is signified by a double line. Homozygosity mapping was performed for families A, C, D and E, with the target region was highlighted by a red arrow in the respective families
Prediction programs
The mutational effect was checked for all missense variants using the CADD score (Combined Annotation Dependent Depletion (CADD; https://cadd.gs.washington.edu), MutationTaster (www.mutationtaster.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2). Identified variants were further classified as likely pathogenic and pathogenic according to ACMG criteria using the Franklin variant assessment tool (https://franklin.genoox.com/clinical-db/home) and VarSome (https://varsome.com/).
Results
The genetic diagnoses and clinical findings of the patients are listed in Tables 1 and 2, respectively.
Table 2. Clinical findings of affected families with diverse muscular dystrophiesClinical featuresFamily AFamily BFamily CFamily DFamily EFamily FPatient or proband ID IV:3
III:1
III:1
III:1
IV:3
III:3 Likely OMIM syndrome: OMIM IDMDC1A: OMIM (607855)BMD: OMIM(300376)MDC1A: OMIM(607855)MDC1A: OMIM (607855)MDC1A: OMIM (607855)DMD: OMIM(310200)GenderMMFMFMCurrent age(year)17 years19 years13 years10 years5 years16–17 yearsDisease onsetBy birth, a weak cryLate-onset(5 years)3 months1 yearBy birth(delayed crying)(3 years)ConsanguinityYesNoYesYesYesNoProgressiveYesYesYesYesYesYesFacialdysmorphyElongated myopathic faceNoNoYesMilderMilderFeedingdifficultiesYesNoYesNAYesYesHypotoniaIn both legsYesYesYesYesYesMusculatureMuscle weakness in lower and upper limbsYesYesYesYesYesCreatine kinase levelHigh (1080 U/L)High (1255 U/L)NANAHigh (902 U/L)High (8200 U/L)ContractureYesYesYesYesYesYesScoliosisYesNANANAYesYesSpeechimpairmentYesMildYesNoYesYesRespiratory complicationYesYes (Repeated chest infections)NAYesYesYesLoss of Ambulation2-3 years15 years of ageYesNAYes8-9 yearsIntellectual disabilitiesYesMildYesModerateYesYesSeizuresFibrile seizuresNoNANoNoNo
Family A
Subject (IV:3) presented with congenital muscular dystrophy characterized by early onset with a weak cry. The patient was 17-year-old at the time of the study. Clinical features include an elongated, myopathic facial appearance, feeding difficulty, joint contractures, hypotonia of the lower limbs, spinal curvature or scoliosis and significant progressive muscle weakness of the proximal limbs. The patient has muscle wasting, joint immobility and developmental milestones delay and the child never walks on his own. The condition caused feeding difficulties because of hypotonia, with the possibility of dependency on enteral feeding. The patient has stunted growth while the overall motility of the patient is significantly affected. During winter, the proband’s respiratory complications tend to worsen, mostly leading to hospitalization. His 5-year-old brother had died because of the same condition. Clinical phenotype and molecular analysis confirmed our case correlated to MDC1A.
Family B
The patient (III:1) is a 19-year-old male who started developing slow progressive walking impairment at the age of 5 years. The patient started walking at an age of 1.5 years, neck holding at 9 months and sitting at 1 year of age. The subject exhibits a positive pseudohypertrophy and Gower sign – a clinical manifestation of proximal muscle weakness. Muscles of the pelvic girdle and lower limb are particularly weak. Over the last 11 years, the patient has shown a gradual deterioration in mobility during activities involving large muscle groups, such as climbing stairs or getting up from a seated position. A positive Gower sign indicates worsening muscle strength in the hip and thigh, which are vital for supporting the body and enabling movement. Weakness was gradual, progressive and bilateral. Other symptoms are summarized in Table 2. Clinicians suggested physical therapy, orthopaedic support and possible pharmacological intervention to reduce further functional decline. Family B phenotypes are closely associated with a mild type of muscular dystrophy, such as BMD.
Family C
Subject (III:1) is a female (13 years old) and the onset of congenital muscular dystrophy happened at 3 months of age; early clinical features included significant, persistent hypotonia, lower and upper limbs abnormalities and evidence of severe delay in motor development. Other clinical details are summarized in Table 1. All these manifestations are matched with MDC1A. Family history was unremarkable for this kind of muscular dystrophy and no evidence of this condition was observed in other members.
Family D
The proband (III:1) is a 10-year-old male (weight: 20 Kg; Height: 125 cm) born to healthy parents and presents with severe intellectual disabilities, progressive muscle weakness, low-set prominent ears, triangular face, deterioration in academic performance, and developmental delay. Cardiac and respiratory-related complications were observed. The family history is significant for two deceased siblings; one early neonatal death and another who died at 10 days of life due to prematurity. Elder male sibling with neuropsychiatric issues, while his four sisters are alive and healthy. Chromosomal or karyotyping analysis was observed normal. No speech impairment and seizures were observed in family D. All symptoms are consistent with MDC1A.
Family E
A 5-year-old female (IV:3) was born with delayed crying and hypotonia. Birth parameters and even neonatal adaptation were not normal. She has been presented with severe muscle atrophy and has developmental disabilities of an extremely severe degree. Her family history reveals the expiration of a brother to the same condition that implied. Intellectual functioning was abnormal with no seizures. During her last assessment at the age of 5 years, speech impairment and intellectual disabilities were predominantly noticed. Based on genetic and clinical features (Table 2) proband was diagnosed with MDC1A.
Family F
Subject (III:3) is a 14-year-old male who shows hypotonia, hyporeflexia, flexion contractures, increased lordosis, scoliosis, waddling gait, positive Gowers sign, short stature and mild mental retardation with developmental delay before 5 years of age. He was born normal to healthy parents following an unremarkable pregnancy. At a recent assessment, he is unable to stand or walk and is bound to a wheelchair. He could not speak a single word due to muscle weakness. The condition becomes worse because of the hip flexors and hamstrings. In the same family, two male patients have died because of the same condition within 20 years of life. Karyotyping analysis was done at the age of 5 years but confirmed normal for chromosomal abnormalities. Physical therapy has been conducted but no improvements have been noted.
Genetic analysis
We studied six unrelated families that exhibited overlapping features of muscular dystrophies, including MDCA1 (Family A, C, D and E), Becker muscular dystrophy (Family B) and Duchenne muscular dystrophy (Family F). These families harbor confirmed pathogenic and likely pathogenic variants in the LAMA2 (Human (GRCh38.p14) and the DMD genes. We performed WES for all families and found two novel frameshift variants c.7470_7473del; p.(Lys2490AsnfsTer56) in family A, c.7807del; p.(Ala2603HisfsTer4) in family C, one novel missense c.8651T > C; p.(Met2884Thr) in family D and one nonsense c.4127T > A; p. (Leu1376Ter) (in family E in the LAMA2 gene. Furthermore, a nonsense variant, c. 10801 C > T; p.(Gln872Ter), was detected in family B, whereas a large genomic deletion (chrX:32438241–32809611del; corresponding to the deletion of exons 7–29) involving the DMD gene was identified in family F by multiplex ligation-dependent probe amplification (MLPA).
Discussion
According to our knowledge, LAMA2 variants associated with congenital muscular dystrophy have not previously been reported in the literature from Pakistan. This suggests that our findings expand the clinical and mutation spectrum for the diagnosis of MDC1A. DMD gene-based variants are cited from Pakistan for DMD-related disorders [21–23].
As a diagnostic method, DNA sequencing or NGS technology is recommended by different groups for neuromuscular disorders such as MDC1A because of overlapping phenotypes and genetic heterogeneity. On the other hand, the muscle biopsy method has more complications than DNA sequencing. An autosomal recessive disorder MDC1A (MIM: 607855), is caused by homozygous or compound heterozygous variations in the LAMA2 gene [24]. The most common pathogenic mutations in LAMA2 are frameshifts with a frequency of 82.5% [25]. The LAMA2 gene comprises 65 exons located on chromosome 6q22-q23 [26]. Laminin alpha-2 is also known as merosin, an extracellular matrix protein that plays a crucial role in forming a connection between the extracellular matrix and cytoskeleton [27]. It is expressed in the striated muscle basement membrane, Schwann cells and basal lamina of the cerebral blood vessels [28, 29]. There is a significant variation in the clinical presentation of this disorder based on the age of onset, with most early-onset patients demonstrating severe phenotypes compared to late-onset cases. However, predicting the defined clinical manifestations based on the level of expressed laminin alpha-2 protein remains uncertain [30]. In early-onset cases, affected individuals typically present with muscle weakness, hypotonia, loss of independent ambulation, respiratory complications, proximal joint contractures and normal intellectual disabilities. In contrast, late-onset patients are characterized by proximal muscle weakness, delayed motor milestones and eventual loss of mobility [10].
In our study, we found two novel frameshift variants, c.7470_7473del; p.(Lys2490AsnfsTer56) and c.7807del; p.(Ala2603HisfsTer4), in family A and family C, respectively. Loss of function (LOF) mutations, such as frameshifts and nonsense, affect mRNA and resulting proteins that cause disease phenotype [25]. LAMA2-related LOF variants have pathogenic effects because of decreased levels of mRNA due to nonsense-mediated RNA decay [31]. Most reported variants in the LAMA2 gene create premature termination codons (PTCs), as in our patients [32]. Classic CMD1A or a more severe and early-onset form of congenital muscular dystrophy is caused by both defective alleles [33]. Mild and late-onset forms of the disorder have commonly been accompanied by missense variants, which are documented in a small number of cases. These cases are correlated with partial deficiency of laminin-α2 as confirmed with muscle biopsies-based immunohistochemical tests [34]. The phenotypes in family D are less severe than those with frameshift mutations (families A and C).
A brief comparison with published cohorts further contextualizes our results. In a large UK cohort of 51 MDC1A patients, complete merosin deficiency was associated with earlier onset, lack of independent ambulation, and increased need for ventilatory and enteral support [14], supporting the correlation between truncating variants and severe congenital phenotypes observed in our study. Similarly, an international MRI-based cohort (27 patients) demonstrated a clinical continuum from non-ambulant congenital cases to milder ambulant forms, with truncating and splice-site variants accounting for 65% of mutations [35]. In contrast, the Western Sicily Limb–Girdle muscular dystrophy (LGMD) cohort reported LAMA2 mutations in 12.5% of LGMD cases, typically with later onset, ambulant course, and lower CK levels [36]. Overall, these data highlight the broad phenotypic spectrum of LAMA2-related disorders and support the value of NGS in refining genotype-phenotype correlations among diverse populations.Biallelic nonsense variants exhibit variable clinical phenotypes and laminin-α2 absence can achieve independent ambulation [14]. We diagnosed family E with a novel homozygous c.4127T > A;p.(Leu1376Ter) nonsense variant with diverse clinical symptoms (Table 2).
Laminins are essential glycoproteins that play a key role in the architecture and function of the basement membrane, binding cell surface receptors via their laminin G-like (LG) domains [34]. Our variant is located in the LG5 domain together with the LG4 domain, these are the binding sites for α-dystroglycan and heparin. Therefore, this nonsense variant may affect the unions of molecules that bind the sarcolemmal cytoskeleton and extracellular matrix, ultimately disturbing the stability of laminin-α2 [14, 37]. The most common muscular disorder in childhood, Duchenne muscular dystrophy, is an X-linked recessive disorder mostly caused by deletions and duplications in the DMD gene. However, 10–15% of cases are caused by nonsense mutations [38]. Bitetti et al. (2021) reported a nonsense variant c.10,801 C > T; p.(Gln3601X) in the 76th exon of a 3-month-old baby from an Italian family who was a confirmed case of DMD. In family B, we report the same stop-codon point mutation as mentioned by Bitetti in a 19-year-old boy who clinically presented with hypotonia, musculature and mild speech impairment [39]. Our case was closely associated with documented cases of Duchenne muscular dystrophy. Furthermore, in family F, a gross deletion (chX: 32438241-32809611del) was observed, consistent with clinical symptoms of Duchenne muscular dystrophy. Thousands of different variations in DMD have been reported in BMD or DMD disorder [7, 40]. Around 60–70% of mutations in DMD patients are deletions, 5–15% are duplications and approximately 20% are point mutations, small insertions or deletions [7, 41]. By contrast, in BMD patients 60–70% of variations are deletions, 20% duplications, and 5–10% point mutations and small indels [7, 42, 43]. Several exons are considered hotspots, such as 3–19 (7%) and 45–55 (47%) for deletions in DMD [44, 45].
Conclusion
For the first time, we have elucidated LAMA2-related MDC1A phenotypes in the Pakistani population and underscored the diagnostic utility of next-generation sequencing in the clinical and molecular diagnosis of neuromuscular dystrophies, thereby facilitating targeted therapeutic strategies and informed prenatal interventions. In Pakistan like country, translating scientific breakthroughs into public health initiatives has been challenging despite the significant impact of neurological disorders on families and patients due to the lack of a national framework for incorporating genomics into healthcare. Medical infrastructure is insufficient for even infectious diseases, so genetic health services are ignored. Factors like religion, culture and society could shape the perceptions of genetic diagnosis in low and middle-income countries. Therefore, based on limited therapeutic options for inherited neurological disorders, prenatal diagnosis, carrier testing and genetic counseling should be encouraged in early diagnosis and prevention. Genomic research should be expanded for scientific interventions across the globe.