Bridging innate immunity and iron-dependent death: the interplay between cyclic GMP–AMP synthase–stimulator of interferon genes nexus and ferroptosis in cancer and inflammation
Xin-xin Chen, Yun-qing Hou, Xin-xu Chen, Qi Zhou, Xiang Wang

TL;DR
This review explores how the cGAS–STING immune pathway and ferroptosis, an iron-dependent cell death process, interact in cancer and inflammation.
Contribution
The paper systematically identifies key molecules linking cGAS–STING and ferroptosis and proposes therapeutic strategies targeting their crosstalk.
Findings
The cGAS–STING pathway detects DNA and activates immune responses through TBK1–IRF3 and nuclear factor-κB signaling.
Ferroptosis involves iron-dependent lipid peroxidation and is linked to disrupted antioxidant systems.
Therapeutic strategies targeting the crosstalk between these pathways show potential for cancer treatment.
Abstract
The cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway and ferroptosis have emerged as fundamental biological mechanisms that converge in regulating cellular homeostasis and disease pathogenesis. As a central component of innate immunity, the cGAS–STING axis detects cytosolic double-stranded DNA through cGAS-mediated synthesis of 2′3′-cyclic GMP–AMP, subsequently triggering STING-dependent activation of the TBK1–IRF3 and nuclear factor-κB signaling cascades. Ferroptosis, an iron-catalyzed form of regulated cell death, is characterized by the accumulation of phospholipid hydroperoxide due to compromised antioxidant activity and dysregulated iron metabolism. Accumulating evidence has revealed the intricate crosstalk between these pathways. This review systematically explores the structural and biochemical bases of both pathways, identifies key bridging molecules…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
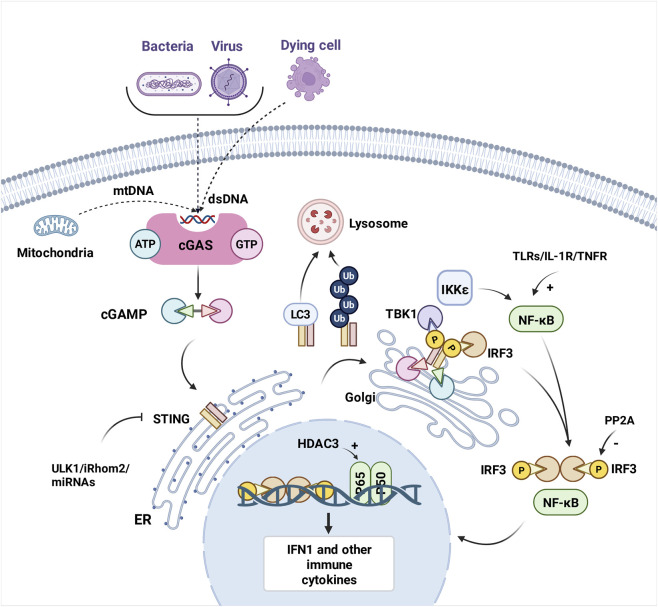 FIGURE 1
FIGURE 1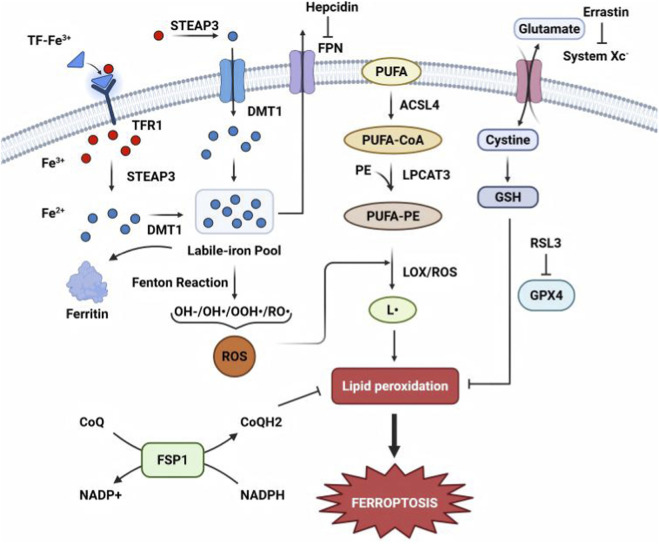 FIGURE 2
FIGURE 2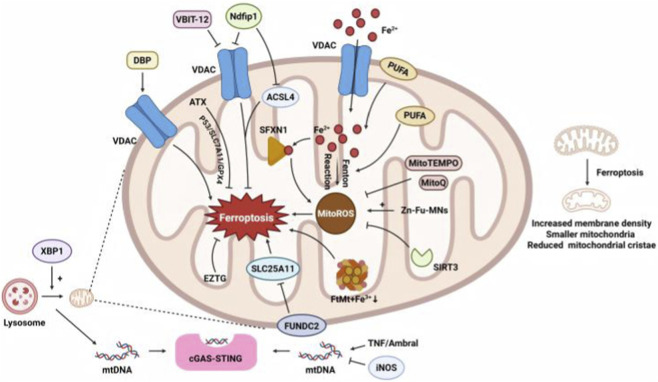 FIGURE 3
FIGURE 3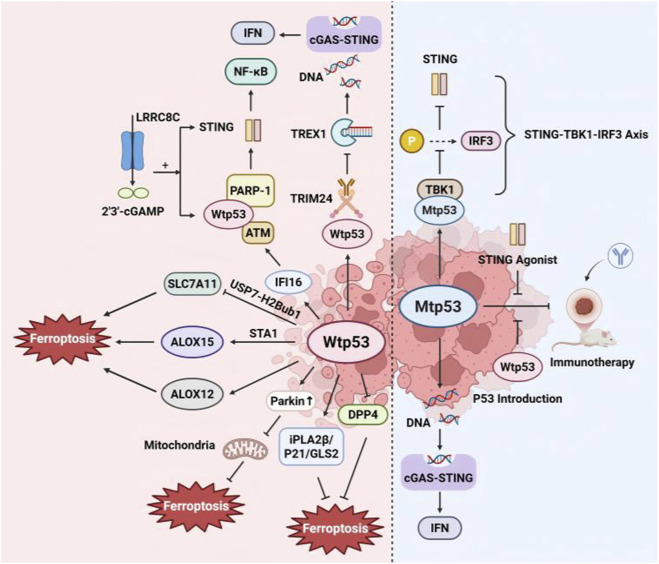 FIGURE 4
FIGURE 4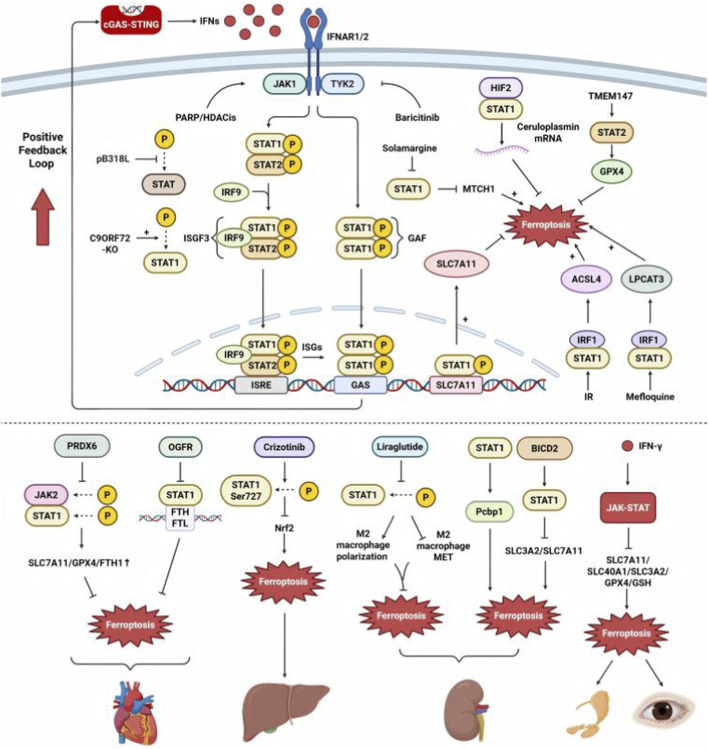 FIGURE 5
FIGURE 5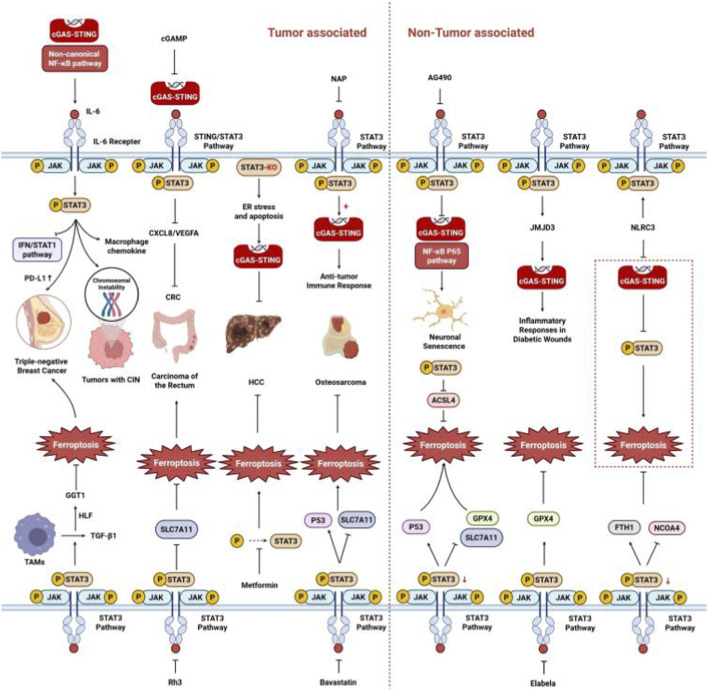 FIGURE 6
FIGURE 6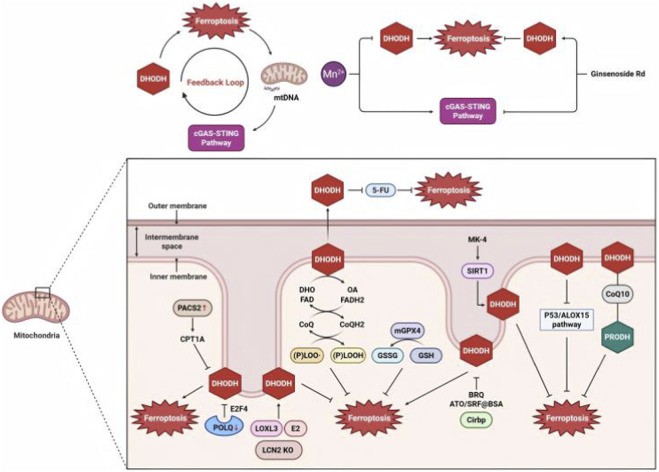 FIGURE 7
FIGURE 7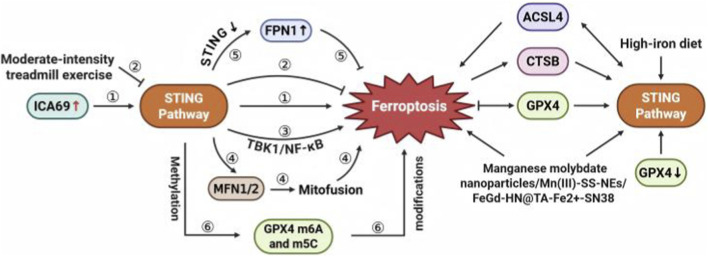 FIGURE 8
FIGURE 8- —Youth Science Fund Project10.13039/100014717
- —Natural Science Foundation of Jilin Province10.13039/100007847
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
Topicsinterferon and immune responses · Ferroptosis and cancer prognosis · Cancer Mechanisms and Therapy
Introduction
1
The cyclic GMP–AMP synthase (cGAS)–stimulator of interferon (IFN) genes (STING) pathway and ferroptosis are two critical biological processes that have garnered significant attention in recent years for their roles in immunity, cell death, and pathogenesis. The cGAS–STING pathway is a cornerstone of innate immunity, primarily recognized for its ability to detect double-stranded DNA and initiate type I IFN responses against viral infections and cellular damage (Sun et al., 2013). In addition, the cGAS–STING pathway has emerged as a promising therapeutic target for various diseases, including cancer and autoimmune disorders (Hopfner and Hornung, 2020). By contrast, ferroptosis is a unique form of regulated cell death characterized by iron-dependent lipid peroxidation (Dixon et al., 2012). Ferroptosis was initially identified as a mechanism of cell death in cancer cells; however, it is now recognized as a fundamental, conserved cell death pathway implicated in various pathological processes, including ischemia/reperfusion injury, neurodegenerative diseases, and chronic inflammation (Yang and Stockwell, 2016).
Emerging evidence reveals a critical interaction between the cGAS–STING pathway and ferroptosis, a mechanism that couples innate immune sensing with iron-dependent cell death (Wang Y. et al., 2025). STING activation can promote ferroptosis by enhancing lipid peroxidation, disrupting glutathione (GSH) metabolism (Yu et al., 2022), and altering iron homeostasis (Zhao J. et al., 2024). Ferroptotic cell death releases double-stranded DNA, which activates cGAS–STING signaling, amplifying inflammatory and antitumor responses (Kim et al., 2023). A recent review comprehensively outlines the roles and mechanisms of cGAS–STING signaling pathway in ferroptosis, emphasizing their close disease-level interconnection (Ding et al., 2025); however, key nodal regulators remain uncharacterized.
In this review, we aim to provide a comprehensive overview of the cGAS–STING pathway and ferroptosis, focusing on their individual roles, shared regulatory networks, and potential therapeutic targets.
Overview of the cGAS–STING pathway and ferroptosis
2
Activation and feedback regulation of the cGAS–STING pathway
2.1
The cGAS–STING pathway serves as a crucial component of the innate immune system, functioning as a key bridge between DNA sensing and immune response (Chen and Xu, 2023). The activation and feedback regulation of the cGAS–STING pathway are summarized in Figure 1.
Illustration of key molecular events in the cGAS–STING signaling pathway. The cascade is initiated when cGAS senses exogenous or endogenous DNA, leading to STING activation. Subsequently, TBK1 is recruited and phosphorylated, which activates IRF3 through phosphorylation. Activated IRF3 and NF-κB translocate to the nucleus to induce expression of type I interferons and various proinflammatory cytokines. The diagram highlights negative feedback mechanisms, including autophagy- and ubiquitination-mediated STING degradation, which regulate pathway activity to maintain immune homeostasis. cGAMP, 2′3′-cyclic GMP–AMP; cGAS, cyclic GMP–AMP synthase; dsDNA, double-stranded DNA; ER, endoplasmic reticulum; HDAC3, histone deacetylase 3; IRF3, interferon regulatory factor 3; IFN, interferon; mtDNA, mitochondrial DNA; NF-κB, nuclear factor-κB; PP2A, protein phosphatase 2A; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1.
The pathway is initiated when cytosolic DNA, resulting from genomic damage or microbial infection (Sun et al., 2013), activates cGAS, an enzyme that catalyzes the synthesis of 2′3′-cyclic GMP–AMP (cGAMP) from ATP and GTP. cGAMP acts as an endogenous second messenger by binding to and activating STING, a sensor predominantly localized in the endoplasmic reticulum (Ishikawa and Barber, 2008; Wu et al., 2013). Upon cGAMP binding, the STING–cGAMP complex translocates from the endoplasmic reticulum to the Golgi apparatus, where STING oligomerization triggers the recruitment and activation of TANK-binding kinase 1 (TBK1) (Liu et al., 2021). TBK1 phosphorylates the C-terminal tail of STING, enabling its interaction with both TBK1 and IFN regulatory factor 3 (IRF3). Phosphorylated IRF3 forms dimers and translocates to the nucleus to induce type I IFN production (Zhang et al., 2019). In parallel, STING oligomers recruit inhibitors of the nuclear factor-κB (NF-κB) kinase subunit ε and activate NF-κB (Ritchie et al., 2022). Although NF-κB signaling can be TBK1-dependent, it does not strictly require TBK1 (Balka et al., 2020). The combined actions of IRF3 and NF-κB drive the expression of type I IFNs and other proinflammatory cytokines, which collectively orchestrate antiviral defense and inflammatory responses (Hopfner and Hornung, 2020; Ritchie et al., 2022).
Following the activation of the STING signaling pathway, a tightly regulated termination process is actively or passively engaged to prevent excessive or prolonged immune activation and maintain homeostasis. The pivotal mechanism in this process involves autophagy-mediated degradation: LC3-associated autophagy induced upon STING activation primarily functions as a host defense mechanism for the direct clearance of intracellular pathogens, during which STING is degraded (Gui et al., 2019); ubiquitination-mediated degradation: E3 ubiquitin ligases such as RNF5 and TRIM29 mediate the ubiquitination of STING, promoting its degradation (Zhong et al., 2009); dephosphorylation: phosphatases such as protein phosphatase 2A dephosphorylate TBK1 and IRF3, thereby terminating signal transduction (Liu et al., 2015); regulation by inhibitory proteins: cyclic dinucleotide produced by cGAS initiates the activation of UNC-51-like kinase, and STING is phosphorylated and IRF3 function suppressed (Konno et al., 2013); and post-transcriptional regulation: microRNAs or RNA-binding proteins can suppress STING expression by regulating the stability or translation efficiency of STING mRNA (Shah et al., 2019).
Beyond IFN signaling, STING activation also leads to its function as a proton channel (Liu et al., 2023; Xun et al., 2024). STING-induced cell death and ferroptosis are context-dependent but mechanistically related. As a proton channel, STING primarily triggers PANoptosis—involving pyroptosis, apoptosis, and necroptosis—during acute stress, yet it also promotes lipid peroxidation, a hallmark of ferroptosis (Liu et al., 2023). In contrast, chronic STING activation directly drives ferroptosis by downregulating CDKN2A/p16 and increasing oxidizable lipids through Acyl-coenzyme A Synthetase Long-Chain Family Member 4 (ACSL4)-dependent lipid peroxidation (Xun et al., 2024). Therefore, while STING-induced death is not synonymous with ferroptosis, STING can either directly trigger ferroptosis under chronic acvtivation or create a permissive environment for its execution during acute stress, highlighting STING as a critical node linking innate immune sensing to oxidative cell death fates.
Drivers of ferroptosis
2.2
Ferroptosis is an iron-dependent, lipid peroxidation-driven form of programmed cell death. This primarily involves the key steps summarized in Figure 2. These processes ultimately result in cell membrane rupture and cell death (Dixon et al., 2012).
Mechanism of lipid peroxidation-mediated ferroptosis. Ferroptosis is initiated when labile iron (Fe2+) catalyzes Fenton reactions, generating reactive oxygen species (ROS) that oxidize polyunsaturated fatty acids (PUFAs) in membrane phospholipids, causing lethal lipid peroxidation. This process is exacerbated by dysregulated iron metabolism (e.g., hepcidin-mediated iron retention) and impaired antioxidant defenses, particularly inactivation of GPX4, which relies on glutathione to detoxify lipid peroxides, or the FSP1–CoQ10 system that scavenges lipid radicals. Accumulation of toxic lipid peroxidation products ultimately disrupts membrane integrity, leading to cell death. ACSL4, acyl-CoA synthetase long-chain family member 4; DMT1, divalent metal transporter 1; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; GPX4, glutathione peroxidase 4; GSH, glutathione; LPCAT3, lysophosphatidylcholine acyltransferase 3; PE, phosphatidylethanolamine; PUFA, polyunsaturated fatty acid; ROS, reactive oxygen species; STEAP3, six-transmembrane epithelial antigen of prostate 3; TF, transferrin; TFR1, transferrin receptor 1.
Iron plays a central role in ferroptosis, with its uptake, storage, and export being tightly regulated. Transferrin receptor 1 (TfR1) mediates iron uptake by internalizing transferrin-bound iron (Fe^3+^). Elevated TfR1 increases intracellular iron, thereby promoting ferroptosis. Ferritin, the primary iron storage protein, sequesters excess iron; its degradation (ferritinophagy) releases free iron, thereby exacerbating ferroptosis (Dixon et al., 2012). Iron regulatory proteins (IRP1/IRP2) control iron homeostasis by modulating TfR1 and ferritin expression. Ferroportin, the sole iron exporter, reduces intracellular iron. Its inhibition or degradation by hepcidin increases iron levels, thereby promoting ferroptosis (Ganz, 2013). Other molecules, such as Nrf2, counteract ferroptosis by upregulating ferritin and heme oxygenase 1 (Mancias et al., 2015), whereas p53 modulates iron metabolism genes (e.g., SAT1) to enhance lipid peroxidation (Jiang et al., 2015).
The Fenton reaction converts Fe^2+^ and H_2_O_2_ into hydroxyl radicals, which attack polyunsaturated fatty acids (PUFAs) in membranes, generating lipid radicals and peroxides (Su et al., 2019). Lipoxygenases directly oxidize PUFAs such as arachidonic acid and linoleic acid (Su et al., 2019). Additionally, oxidoreductases (NADPH-cytochrome P450 reductase and NADH-cytochrome b5 reductase) produce H_2_O_2_, further fueling the Fenton reaction (Yan et al., 2021). ACSL4 activates PUFAs by conjugating them to coenzyme A, whereas LPCAT3 incorporates them into phospholipids (e.g., phosphatidylethanolamine). These PUFA-rich lipids are highly susceptible to peroxidation, driving ferroptosis (Doll et al., 2017; Kagan et al., 2017).
The GSH–glutathione peroxidase 4 (GPX4) axis is the primary defense mechanism against ferroptosis. Erastin blocks cystine uptake via system xc^−^ (SLC7A11), depleting GSH and inactivating GPX4, thereby resulting in lethal lipid peroxide accumulation (Yang et al., 2014). GPX4 converts lipid hydroperoxides into harmless alcohols. Direct GPX4 inhibitors (e.g., RSL3) induce ferroptosis by allowing peroxide accumulation (Ursini and Maiorino, 2020). Ferroptosis suppressor protein one independently inhibits ferroptosis by reducing CoQ10 and vitamin K, which act as radical-trapping antioxidants (Bersuker et al., 2019; Mishima et al., 2022; Lv et al., 2023).
Crosstalk between the cGAS–STING pathway and ferroptosis: molecular mechanisms and functional consequences
3
STING, a molecule renowned for immune responses, has recently been found to exhibit an unexpected correlation with ferroptosis. Unraveling the molecules or pathways in this relationship may reveal novel therapeutic opportunities through immunogenic cell death mechanisms.
Link through mitochondria
3.1
Mitochondria and ferroptosis
3.1.1
Morphological and functional changes in mitochondria confirm their central role in ferroptosis (Dixon et al., 2012). Elevated mitochondrial ROS promote ferroptosis by enhancing lipid peroxidation (Gan, 2021). Mitochondria-targeted antioxidants (e.g., MitoQ) attenuate this process (Jelinek et al., 2018; Fang et al., 2019).
Various mitochondrial proteins are involved in ferroptosis; therefore, multiple therapeutic strategies targeting mitochondria to enhance or alleviate ferroptosis have been developed. Mitochondrial PUFA-rich membranes are prime lipid peroxidation targets, with mitochondrial ACSL4 serving as a key metabolic regulator (Tao L. et al., 2024). Mitochondrial ferritin deficiency exacerbates ferroptosis in brain cells by elevating labile iron during ischemia/reperfusion injury (Wang P. et al., 2021). Knockdown of SIRT3, a deacetylase localized in the mitochondrial matrix, exacerbates iron overload and reactive oxygen species (ROS) production, resulting in ferroptosis (Feng et al., 2020). SFXN1 is a mitochondrial inner membrane protein. Under lipopolysaccharide (LPS) induction, NCOA4 mediates ferritinophagy, and the released Fe^2+^ activates SFXN1, leading to mitochondrial ROS accumulation and ferroptosis (Li et al., 2020). Voltage-dependent anion channels (VDACs) are abundant mitochondrial outer membrane proteins that facilitate the transport of small molecules. The VDAC1 oligomerization inhibitor VBIT 12 protects mitochondria, elevates ceramide and cardiolipin levels, and attenuates acetaminophen overdose-induced ferroptosis in hepatocytes (Niu et al., 2022). Inhibition of ceramide kinase modulates VDAC-associated mitochondrial function, inducing ferroptosis in KRAS-mutant non-small cell lung-cancer cells (Vu et al., 2022). In Parkinson’s disease, Ndfip1 expression attenuates mitochondrial dysfunction by reducing VDAC levels and inhibiting ACSL4, thereby preventing neuronal ferroptosis (Fu et al., 2024). Dibutyl phthalate induces ferroptosis by upregulating and oligomerizing VDAC2, leading to mitochondria-dependent ferroptosis (Hou et al., 2024). FUNDC2, a mitochondrial outer membrane protein, interacts with SLC25A11 and plays a critical role in doxorubicin-induced ferroptosis in cardiomyocytes (Ta et al., 2022). Zinc-fluorouracil metallodrug networks induce mitochondrial ROS production, triggering ferroptosis in cancer cells (Lei et al., 2022). Polystyrene nanoplastics and cadmium inhibit the SIRT3–SOD2/GPX4 pathway, inducing oxidative stress and ferroptosis in ovarian mitochondria (Wu H. et al., 2025). ErZhiTianGui decoction maintains mitochondrial ROS levels and PINK1/Parkin-mediated mitochondrial homeostasis, preventing ferroptosis in aged mouse ovaries (Zhicheng et al., 2024). Astaxanthin stabilizes mitochondria membrane potential, mitigating interleukin (IL)-1β-induced ferroptosis in chondrocytes (Wang et al., 2022c).
Mitochondria and STING
3.1.2
Regarding the role of mitochondria in the cGAS–STING pathway, X-box binding protein one modulates mitophagy, leading to mitochondrial DNA (mtDNA) leakage from macrophages, which activates cGAS–STING (Wang Q. et al., 2022). In a chronic arthritis model, tumor necrosis factor inhibited PINK1-mediated mitophagy, resulting in mtDNA accumulation and subsequent activation of cGAS–STING (Willemsen et al., 2021). In pressure-overloaded hearts, inducible nitric oxide synthase (iNOS) controls the release of mtDNA into the cytosol, which activates the cGAS–STING pathway and triggers sterile inflammation, ultimately leading to cardiac dysfunction. (Guo Y. et al., 2023). Beclin1-regulated autophagy protein one on cardiomyocyte-derived extracellular vehicles is reportedly necessary for extracellular vehicle-mediated mtDNA transfer to fibroblasts, subsequent activation of the cGAS–STING pathway, and induction of cardiac remodeling after ischemia/reperfusion injury (Zhang C. et al., 2023).
Therefore, mitochondria serve as a crucial hub linking STING activation to ferroptosis through multiple mechanisms, including mtDNA leakage, mitochondrial ROS, and mitochondrial proteins (Figure 3). These interconnected pathways position mitochondria as both sensors of cellular stress (via STING) and executioners of ferroptosis, offering therapeutic targets for modulating inflammatory and cell death responses.
Role of mitochondria in ferroptosis and the cGAS–STING pathway. Mitochondrial dysfunction primes cells for ferroptosis by generating excessive ROS and disrupting iron homeostasis, leading to iron overload. Fe2+ catalyzes Fenton reactions that promote lipid peroxidation, particularly in mitochondrial membranes. Concurrently, mtDNA leakage activates the STING pathway, amplifying oxidative stress and inflammatory responses. The convergence of mitochondrial iron accumulation, uncontrolled lipid peroxidation, and STING-mediated inflammation drives ferroptotic cell death, as toxic lipid peroxides compromise membrane integrity and induce irreversible damage. ACSL4, acyl-CoA synthetase long-chain family member 4; DBP, dibutyl phthalate; EZTG, ErZhiTianGui; FUNDC2, FUN14 domain-containing 2; FtMt, mitochondrial ferritin; iNOS, inducible NO synthase; MitoQ, mitochondrial-targeted coenzyme Q10; mtDNA, mitochondrial DNA; Ndfip1, Nedd4 family interacting protein 1; PUFA, polyunsaturated fatty acid; SFXN1, siderofexin; SIRT3, sirtuin 3; TNF, tumor necrosis factor; VDACs, voltage-dependent anion channels; XBP1, X-box binding protein 1; Zn-Fu MNs, zinc-fluorouracil metallodrug networks.
Link through p53
3.2
p53 and STING
3.2.1
STING and p53 synergistically suppress tumors through their interconnected roles in innate immunity and tumor suppression. Wild-type p53 collaborates with the ubiquitin ligase TRIM24 to degrade DNA exonuclease TREX1, resulting in the accumulation of cytoplasmic DNA and activation of the cGAS/STING pathway (Ghosh et al., 2023). Additionally, upon activation of the DNA-binding protein IFI16, p53 —in conjunction with the DNA damage response factors ATM and poly (ADP-ribose) polymerase (PARP) 1—participates in STING signaling complex assembly, facilitating non-canonical STING activation (Dunphy et al., 2018). LRRC8C mediates cyclic dinucleotide transport via T-cell anion channels, activating STING–p53 signaling to suppress T-cell immunity (Concepcion et al., 2022).
However, mutations in p53 can reverse its immune regulatory effects. p53 mutations confer immunotherapy resistance in high-mutation tumors, reversible by STING agonists or p53 restoration (Zhu et al., 2023a). Mechanistically, mutant p53 binds to TBK1, preventing IRF3 activation and disrupting downstream signaling, thereby promoting tumor cell survival (Ghosh et al., 2021). In addition, p53 mutations impair cGAS–STING signaling but may lead to chromosomal instability and DNA fragment release, resulting in chronic low-level IFN-I inflammation (Qin et al., 2023). This further illuminates the complexity of upstream regulation within the STING pathway.
p53 and ferroptosis
3.2.2
Recent research has revealed that p53-mediated regulation of ferroptosis is highly context-dependent, exhibiting bidirectional effects.
p53 as a promoter of ferroptosis
3.2.2.1
The p53–SAT1 axis drives ALOX15-dependent ferroptosis via polyamine catabolism, inhibiting tumor growth (Ou et al., 2016). Another lipoxygenase family member, ALOX12, reportedly mediates ferroptosis via p53 in an ACSL4-independent manner (Chu et al., 2019). In the context of ferroptosis induced by erastin or RSL3, p53 targets ferredoxin reductase, leading to ferroptosis in a p53-dependent manner (Zhang et al., 2017). iPLA2β acts as a key repressor of p53-mediated ferroptosis, functioning independently of GPX4 in tumors (Chen et al., 2021).
p53 as an inhibitor of ferroptosis
3.2.2.2
In addition, p53 acts to restrain ferroptosis; specifically, it limits erastin-induced ferroptosis via inhibiting dipeptidyl peptidase-4-dependent lipid peroxidation (Xie et al., 2017). The p53–p21 axis inhibits ferroptosis by preventing GSH depletion and reducing ROS accumulation (Tarangelo et al., 2018). Overexpression of Parkin, a p53 target gene, significantly eliminates mitochondria through mitophagy, thereby inhibiting ferroptosis (Zhang et al., 2011). p53 increases the expression of glutaminase 2, which plays a pivotal role in antioxidant defense; however, its precise role in ferroptosis requires further investigation (Hu et al., 2010).
Therefore, p53 does not directly regulate STING-induced or STING-inhibited ferroptosis pathways but acts as an upstream modulator that fine-tunes immune responses through STING and governs cell-fate decisions via ferroptosis (Figure 4).
Role of p53 in ferroptosis and the cGAS–STING pathway. STING and p53 exert synergistic antitumor effects by coordinating immune activation and cell death pathways. p53 directly binds promoter regions of cGAS–STING genes, dynamically modulating their expression to enhance STING signaling. p53 regulates cytosolic DNA accumulation, the primary trigger for STING activation. Additionally, p53 controls ferroptosis via downstream effectors, including lipid peroxidation enzymes (e.g., ALOX15), ROS generation systems, and oxidative stress defense proteins (e.g., SLC7A11). This dual regulation promotes a therapeutic synergy: p53-mediated STING activation boosts antitumor immunity, whereas p53-driven ferroptosis eliminates cancer cells through iron-dependent lipid peroxidation. ALOX12, arachidonate 12-lipoxygenase; ALOX15, arachidonate 15-lipoxygenase; ATM, ataxia telangiectasia mutated; DPP4, dipeptidyl peptidase 4; GLS2, glutaminase 2; IFI16, interferon-γ-inducible factor 16; IRF3, interferon regulatory factor 3; LRRC8C, leucine-rich repeat-containing protein 8; Mtp53, mutant p53; PARP 1, poly (ADP-ribose) polymerase 1; SAT1, spermidine/spermine N1-acetyltransferase 1; TBK1, TANK-binding kinase 1; SLC7A11, solute carrier family 7 member 11; Wtp53, wild-type p53.
Link through the JAK–STAT pathway
3.3
STAT1/2 and STING
3.3.1
STING activation triggers type I IFN production, which binds IFNAR1/2 to activate Janus kinase (JAK)1/TYK2 and initiate JAK–STAT signaling. Specifically, STAT2 forms a complex with STAT1 and IFN regulatory factor 9, also known as IFN-stimulated gene factor 3. This complex translocates to the nucleus, binds to IFN-stimulated response elements, and promotes the transcription of numerous IFN-stimulated genes. Additionally, STAT1 homodimers can form the IFN-γ-activated factor, which activates gene expression via the GAS promoter element. This process is integral to STING-initiated immune signaling and plays a critical role in the host’s defense against various immune challenges (Katze et al., 2002). Furthermore, type I IFNs induce STING expression through the STAT1 promoter binding site, creating a positive feedback loop that amplifies the immune response (Ma et al., 2015). Moreover, IFNs trigger the release of mtDNA into the cytoplasm, and JAK1-mediated phosphorylation of cGAS further activates the STING pathway (Tong et al., 2024).
The STING–JAK–STAT axis is regulated by multiple factors. The African swine fever virus pB318L protein suppresses host immunity by blocking STING Golgi translocation and STAT1/2 phosphorylation, inhibiting IFN-I/ISG production to enhance viral virulence (Liu et al., 2024). Loss of C9ORF72 results in increased STAT1 phosphorylation levels, which act upstream to further induce STING activation (Pang and Hu, 2023). Regarding therapeutic applications, dual PARP and histone deacetylase inhibitors activate the cGAS–STING and JAK–STAT pathways, boosting type I IFN responses in triple-negative breast cancer (TNBC) (Zhu Q. et al., 2024). Combined treatment with the farnesyltransferase inhibitor lonafarnib and the JAK–STAT inhibitor baricitinib suppresses the cGAS–STING–STAT1 pathway, reducing cytotoxicity and improving symptoms of Hutchinson–Gilford progeria syndrome (Arnold et al., 2021).
STAT1/2 and ferroptosis
3.3.2
STAT1/2 regulates ferroptosis by modulating its key regulators. In tumors, hypoxia-induced HIF-2α/STAT1 upregulates ceruloplasmin mRNA in tumor-associated macrophages, which is transferred via extracellular vesicles to tumor cells, reducing iron accumulation and lipid peroxidation to confer ferroptosis resistance (Schwantes et al., 2024). Epigallocatechin-3-gallate inhibits the procancer effects of leptin by reducing STAT1 binding to the SLC7A11 promoter, thus facilitating ferroptosis (Li F. et al., 2023). Solamargine reduces STAT1 expression, downregulating mitochondrial carrier one and promoting ferroptosis in hepatocellular carcinoma (HCC) (Zhang et al., 2025). In tumor treatment, ionizing radiation stimulates ACSL4 and ferroptosis in intestinal cells via the STAT1/IRF1 axis (Kong et al., 2023). In cancer immunotherapy, mefloquine enhances PD-1 immunotherapy efficacy by upregulating LPCAT3 via the IFN-γ/STAT1–IRF1 axis, thereby promoting ferroptosis (Tao Q. et al., 2024). Conversely, CircPIAS1 inhibits STAT1 phosphorylation, activating the SLC7A11/GPX4 pathway and preventing IFN-induced ferroptosis in melanoma cells, revealing the mechanisms of immunotherapy resistance (Zang et al., 2024). Moreover, STAT2 mediates ferroptosis. In colorectal cancer, multilayer analysis has identified STAT2 as a potential upstream transcription factor that regulates ferroptosis (Zhong et al., 2022). In HCC, TMEM147 enhances STAT2-mediated DHCR7 expression, increasing 27-hydroxycholesterol levels to upregulate GPX4 and suppress ferroptosis (Huang et al., 2023).
In addition to tumor pathogenesis, STAT1 affects various organs by modulating ferroptosis. In the heart, peroxiredoxin 6 inhibits adriamycin-induced ferroptosis and heart failure by suppressing JAK2 and STAT1 phosphorylation, thus increasing SLC7A11, GPX4, and ferritin heavy chain levels (Xiong et al., 2024). STAT1 stimulates ferritin gene transcription, whereas opioid growth factor receptor inhibits STAT1 binding to ferritin promoters, preventing ferroptosis and providing cardioprotection to doxorubicin-treated patients (Chen X. et al., 2024). Crizotinib promotes STAT1 Ser727 phosphorylation, inhibiting Nrf2 transcription and inducing ferroptosis, thereby exacerbating liver injury (Guo et al., 2024). In the kidney, STAT1 induces macrophage ferroptosis by regulating the iron chaperone protein Pcbp1, promoting fibrosis in senescent kidneys (Wu et al., 2024). Liraglutide reduces STAT1 phosphorylation while increasing STAT3 and STAT6 phosphorylation, promoting M2 macrophage polarization and protecting against ferroptosis-induced renal dysfunction (Sun et al., 2024). Bicaudal D2 promotes IFN-γ signaling by facilitating STAT1 nuclear translocation, inhibiting SLC3A2 and SLC7A11 expression, and enhancing ferroptosis. Conversely, VHL degrades Bicaudal D2, mitigating renal injury (Hao et al., 2023). In Sjögren’s syndrome—a chronic autoimmune disorder in which the immune system attacks the body’s moisture-producing glands—elevated IFN-γ levels activate the JAK/STAT1 pathway, downregulating system xc^−^ components (e.g., SLC3A2, glutathione, and GPX4), leading to ferroptosis in salivary gland epithelial cells (Cao et al., 2023). In eye diseases, IFN-γ activates the JAK1-2/STAT1 pathway, which inhibits Fe^2+^ efflux protein, GPX4, and GSH, ultimately causing ferroptosis in human retinal pigment epithelial cells in age-related macular degeneration (Wei et al., 2022).
Therefore, STAT1/2 serve as critical molecular bridges connecting STING-mediated immune responses with ferroptosis (Figure 5). Following STING activation, STAT1/2 regulate ferroptosis by modulating key regulators of ferroptosis, including SLC7A11, GPX4, ACSL4, ferritin, and LPCAT3. The STING-STAT1/2–ferroptosis nexus represents a therapeutic target, in which precise modulation could either enhance tumor immunogenicity or protect against tissue damage, depending on disease context.
Mechanistic regulation of ferroptosis via the cGAS–STING–JAK–STAT1/2 pathway. The cGAS–STING pathway induces interferon (IFN) production, activating IFN receptors (IFNAR1/2) to initiate JAK–STAT signaling. Upon IFN binding, JAK1 and TYK2 phosphorylate STAT1 and STAT2, promoting transcription factor complex formation that binds interferon-stimulated response elements (ISREs) and gamma-activated sequence motifs. These complexes regulate key ferroptosis-related genes, thereby modulating lipid metabolism (ACSL4, LPCAT3), antioxidant defenses (SLC7A11, GPX4), and iron homeostasis/redox balance. This cascade culminates in ferroptotic cell death in multiple organs (heart, liver, kidney, eye), underscoring its physiological and pathological significance. BICD2, bicaudal D2; FTH, ferritin heavy chain; FTL, ferritin light chain; GSH, glutathione; HDAC, histone deacetylase; IFNAR1/2, IFNα/β receptor; IRF9, IFN regulatory factor 9; IR, ionizing radiation; ISGF3, IFN-stimulated gene factor 3; ISGs, interferon-stimulated genes; ISRE, interferon-stimulated response element; JAK, Janus kinase; LPCAT3, lysophosphatidylcholine acyltransferase 3; METs, macrophage extracellular traps; MTCH1, mitochondrial carrier 1; Nrf2, nuclear factor erythroid 2-related factor 2; OGFR, opioid growth factor receptor; PRDX6, peroxiredoxin 6; STAT, signal transducer and activator of transcription; TAK, tyrosine kinase; TMEM147, transmembrane protein 147.
STAT3 and STING
3.3.3
STING and STAT3 exhibit reciprocal regulation, with STAT3 acting as a downstream effector of STING. In tumors with chromosomal instability, activation of cGAS–STING and non-canonical NF-κB pathway induces IL-6/STAT3 signaling, which paradoxically promotes tumor cell survival (Hong et al., 2022; Vasiyani et al., 2022), contrasting with the conventional understanding of STING as a tumor suppressor. Mechanistically, chemotherapy-induced DNA damage activates the STING–NF-κB pathway, leading to IL-6 production and STAT3 activation. This upregulates PD-L1 expression, mediating immunosuppression and enabling tumor cell survival (Vasiyani et al., 2022). All of these mechanisms coincide with the IL-6/JAK/STAT3 axis being a well-established pathway that suppresses antitumor immune responses (Johnson et al., 2018). STING-mediated STAT3 activation also plays a role in balancing cell survival and death, particularly in mitigating IFN/STAT1-associated cell death (Bakhoum, 2022). In addition to tumor biology, STING promotes macrophage chemokine production and migration via STAT3 activation, which reduces inflammation (Chen et al., 2025).
Regarding the feedback modulation of STAT3 on STING, STAT3 inhibition enhances STING signaling and antitumor immunity, suggesting a synergistic strategy in immunotherapy (Pei et al., 2019; Suter et al., 2021). In HCC, STAT3 deficiency potentiates the efficacy of sorafenib by activating the STING–IFN axis via apoptosis-induced DNA release (Wang et al., 2022b). Napabucasin, a STAT3 inhibitor, boosts cGAS–STING signaling via mitochondrial modulation to overcome tumor immune resistance (Wang et al., 2025). Additionally, AG490, a JAK2/STAT3 inhibitor, suppresses the cGAS/STING/NF-κB/p65 pathway, exacerbating neuronal senescence and inflammation post-stroke (Zhang W. et al., 2023). In metabolic disorders, STAT3 in pre-senescent adipocytes negatively regulates STAT1/cGAS–STING signaling, thus inhibiting cellular senescence and inflammation (Madani et al., 2021). IL-6 activates the JAK/STAT3 pathway, upregulating Jumonji domain-containing protein 3, which regulates STING gene expression and mediates inflammatory responses in diabetic wounds (Audu et al., 2022).
STAT3 and ferroptosis
3.3.4
The regulatory relationship between STAT3 and ferroptosis is closely related to the expression of key ferroptosis-related proteins, including p53, ACSL4, GPX4, and SLC7A11. STAT3 inhibition promotes ferroptosis, suppresses tumor growth, and reduces chemotherapy resistance by upregulating p53 and directly downregulating GPX4 and SLC7A11 at the transcriptional level (Ouyang et al., 2022). STAT3 inhibits ACSL4 expression, thus suppressing ferroptosis (Wang Y. et al., 2021). Auranofin upregulates hepcidin via the NF-κB/IL-6/STAT3 pathway, thereby reducing ferroptosis (Yang et al., 2020). Chronic exposure of osteosarcoma cells to cisplatin hyperactivates the STAT3/Nrf2 signaling pathway, increasing GPX4 activity and inhibiting ferroptosis (Liu and Wang, 2019). The IL-6/JAK/STAT3 axis triggers tumor growth factor-β1 secretion from tumor-associated macrophages, activating GGT1 through the hepatic leukemia factor oncoprotein to suppress ferroptosis and drive TNBC progression (Li et al., 2022).
Considering the potential role of STAT3 in ferroptosis, several therapeutic strategies have been developed for cancer treatment. Thiostrepton, a STAT3 inhibitor, blocks GPX4 expression, inducing ferroptosis in pancreatic cancer cells (Zhang et al., 2022b). Ginsenoside Rh3 induces ferroptosis in rectal cancer cells by downregulating SLC7A11 via the STAT3/p53/NRF2 pathway (Wu et al., 2023). Bavastatin inhibits STAT3, upregulates p53, and downregulates SLC7A11, thereby promoting ferroptosis in osteosarcoma cells (Luo et al., 2021). In HCC, metformin downregulates ATF4 and inhibits STAT3 phosphorylation, promoting ferroptosis and enhancing sorafenib efficacy (Hu et al., 2023).
In contrast to its role in cancer, STAT3 exhibits a positive correlation with ferroptosis in noncancerous diseases. Inhibition of the IL-6/STAT3 pathway by Elabela activates xCT/GPX4, inhibiting ferroptosis in endothelial cells and offering therapeutic potential for hypertensive heart disease (Zhang Z. et al., 2022). STAT3 inhibition reduces NCOA4 and increases ferritin, protecting cardiomyocytes from high-fat diet-induced ferroptosis (Zhu et al., 2023b). In the liver, the inhibition of the AhR-STAT3-HO1/COX-2 pathway alleviates hepatocyte ferroptosis and improves human mesenchymal stem cell survival (Han et al., 2024). Lipocalin-2 activates the NF-κB/STAT3 pathway, promoting ferroptosis in hypoxic–ischemic brain damage (Luo et al., 2023). Additionally, STING drives ferroptosis by inhibiting STAT3 phosphorylation; STING knockdown mitigates LPS-induced ferroptosis via STAT3 suppression in lung epithelial cells (Gu et al., 2024).
Therefore, STAT3 serves as a pivotal molecular switch that connects STING signaling to ferroptosis, exhibiting context-dependent dual functions (Figure 6).
Regulatory interplay between cGAS–STING and ferroptosis via JAK/STAT3 signaling. The cGAS–STING pathway produces the second messenger cGAMP, which directly activates STAT3. STAT3, in turn, modulates cGAS–STING activity through multiple mechanisms, establishing a bidirectional regulatory loop. Within the nucleus, STAT3 regulates transcription of key ferroptosis-related genes, including tumor suppressor p53, GPX4, cystine/glutamate antiporter (SLC7A11 and ACSL4). Notably, the IL-6/JAK2/STAT3 axis, which is functionally linked to both cGAS–STING activation and ferroptosis regulation, is implicated in various pathological conditions such as neuropsychiatric disorders (depression), cerebrovascular disease (stroke), and metabolic disorders (diabetes). ACSL4, acyl-CoA synthetase long-chain family member 4; CRC, colorectal cancer; CIN, chromosomal instability; CXCL8, chemokine ligand 8; ER, endoplasmic reticulum; FTH1, ferritin heavy chain 1; GPX4, glutathione peroxidase 4; HCC, hepatocellular carcinoma; HLF, hepatic leukemia factor; JMJD3, Jumonji domain-containing protein 3; NAP, napabucasin; NCOA4, nuclear receptor coactivator 4; NLRC3, NLR family CARD domain-containing 3; Rh3, ginsenoside Rh3; TAMs, tumor-associated macrophages; TGF-β1, transforming growth factor beta 1; VEGFA, vascular endothelial growth factor A.
Link through dihydroorotate dehydrogenase (DHODH)
3.4
DHODH and ferroptosis
3.4.1
DHODH is a mitochondrial enzyme that is critical for maintaining mitochondrial membrane potential, regulating ROS production, and inhibiting ferroptosis in tumors. DHODH inactivation induces mitochondrial lipid peroxidation, particularly in cancer cells with low GPX4 expression. DHODH catalyzes the oxidation of dihydroorotic acid to orotate, while reducing coenzyme Q (CoQ) to CoQH2. The DHODH–CoQH2 system plays a key role in suppressing ferroptosis in cancer cells (Mao et al., 2021). In glioblastoma, deletion of proline-rich protein 11 increases lipid peroxidation and promotes ferroptosis in a DHODH-dependent manner (Miao et al., 2024). Inhibition of polymerase theta suppresses DHODH expression via the E2F4 transcription factor, increasing ferroptosis sensitivity and reducing stemness in gastric cancer cells (Peng et al., 2024).
Therapeutic strategies targeting DHODH in ferroptosis have been developed. Inhibition of DHODH synergizes with cisplatin and induces ferroptosis in cervical cancer cells (Jiang et al., 2023). Brequinar, a DHODH inhibitor, is modified with a mitochondrial targeting motif to enhance its anticancer activity by inducing ferroptosis (Hai et al., 2024). The nanomedicine ATO/SRF@BSA promotes mitochondrial lipid peroxidation by inhibiting GPX4 and downregulating the DHODH–CoQH2 system in TNBC (Zhou et al., 2024). SOD2 depletion and DHODH inhibition enhance the sensitivity of nasopharyngeal carcinoma cells to radiotherapy (Amos et al., 2023).
In a noncancer context, 17β-estradiol (E2) increases DHODH expression in the hippocampus, exerting neuroprotective effects against ferroptosis and addressing memory decline (Tian et al., 2023). In heart failure, E2 upregulates DHODH to inhibit ferroptosis, alleviating pathological hypertrophy and fibrosis (Wang et al., 2024). In aged donor hearts, reduced expression of cold-inducible RNA-binding protein leads to decreased DHODH levels, exacerbating ferroptosis in transplanted hearts (Zhu Y. et al., 2024). In diabetic cardiomyopathy, PACS2 upregulation leads to increased CPT1A expression, which inhibits DHODH and promotes cardiomyocyte ferroptosis (Xiang et al., 2024). In subarachnoid hemorrhage, menaquinone-4 activates SIRT1, which upregulates DHODH to attenuate ferroptosis (Zhang et al., 2024). DHODH inhibits the p53/ALOX15 pathway, ameliorating neuronal ferroptosis and spinal cord injury (Li D. et al., 2023). Under hypoxia, DHODH promotes cell survival through cooperation with GPX4 in corneal epithelial cell (Wu M. F. et al., 2025). In benzene-induced inflammatory anemia, DHODH interacts with IRP1 and ALOX12 to regulate ferroptosis (Zhang et al., 2022c). In septic mice, the absence of Lipocalin-2 increases DHODH expression, reducing ferroptosis and liver injury (Li et al., 2025).
DHODH and STING
3.4.2
DHODH-induced ferroptosis promotes mitochondrial lipid peroxidation, leading to the release of mtDNA, which activates the cGAS–STING pathway, creating a feedback loop between ferroptosis and STING signaling. Notably, Mn^2+^ activates cGAS–STING to boost type I IFN, while inhibiting DHODH to trigger IFN-dependent ferroptosis via mitochondrial ROS and lipid peroxidation (Zhao et al., 2020; Zhang S. et al., 2022). In subarachnoid hemorrhage models, ginsenoside Rd inhibits the cGAS/STING pathway while activating DHODH, thereby suppressing neuronal ferroptosis (Jiang et al., 2025).
DHODH serves as a crucial mitochondrial regulator linking STING signaling to ferroptosis (Figure 7). By maintaining CoQH2 levels, DHODH suppresses lipid peroxidation and ferroptosis, while its inhibition triggers mitochondrial ROS accumulation and mtDNA release, activating cGAS–STING.
Mitochondrial DHODH-mediated ferroptosis regulation and its crosstalk with cGAS–STING signaling. In mitochondria, dihydroorotate dehydrogenase (DHODH) catalyzes the redox-dependent conversion of dihydroorotate (DHO) to orotate (OA) via FAD/FADH2 and CoQ/CoQH2 cycling. This metabolic process generates lipid peroxides [(P)LOO• and (P)LOOH], which induce ferroptosis. In cancer, DHODH interacts functionally with LCN2-mediated iron metabolism and the p53/ALOX15 lipid peroxidation pathway. In noncancer contexts, DHODH activity is modulated by metabolic regulators (CPT1A) and organelle trafficking factors (PACS2). Importantly, mtDNA released during ferroptosis links mitochondrial dysfunction to cGAS–STING activation. This network is further regulated by endogenous modulators like Mn2+ and SIRT1, as well as pharmacological agents such as ginsenoside Rd, brequinar (BRQ), arsenic trioxide/BSA nanocomplexes (ATO/SRF@BSA), and menaquinone-4 (MK-4). ATO, arsenic trioxide; BRQ, brequinar; Cirbp, cold-inducible RNA-binding protein; CoQ, coenzyme Q; CPT1A, carnitine palmitoyltransferase 1A; DHODH, dihydroorotate dehydrogenase; DHO, dihydroorotic acid; E2, 17β-estradiol; LOXL3, lysyl oxidase-like 3; mtDNA, mitochondrial DNA; KM-4, menaquinone-4; OA, orotate; PACS2, phosphofurin acidic cluster sorting protein 2; POLQ, polymerase theta; PRODH, proline dehydrogenase.
Other associations between STING and ferroptosis
3.5
STING reportedly promotes ferroptosis. Increased ICA69 activates the STING pathway, leading to cardiomyocyte ferroptosis (Kong et al., 2022). Moderate-intensity treadmill exercise reduces neuronal ferroptosis after traumatic brain injury by inhibiting STING (Chen J. et al., 2023).
STING promotes ferroptosis through its interaction with key regulators of mitochondrial dynamics and ferroptosis. STING promotes ferroptosis by binding to mitofusin 1/2, mediating mitofusion, which increases mitochondrial ROS and enhances ferroptosis (Li et al., 2021). In lupus nephritis, STING induces ferroptosis and inflammatory responses through TBK1/NF-κB-mediated downregulation of ACSL4, GPX4, and SLC7A11 (Chen J. et al., 2024). In diabetic nephropathy, STING inhibition stabilizes ferroportin 1, thereby attenuating ferroptosis (Zhao Q. X. et al., 2024). Hypertension induces mtDNA release from renal epithelial cells, activating STING, which interacts with ACSL4 to promote ferroptosis, renal inflammation, and fibrosis (Gao et al., 2023).
A complex bidirectional interaction has been demonstrated between key regulatory molecules of ferroptosis and the cGAS–STING pathway. GPX4 is essential for cGAS–STING signaling pathway activation, which is critical for the innate immune response against DNA viruses (Jia et al., 2020). In patients with chronic hepatitis B, reduced GPX4 expression in peripheral blood mononuclear cells during the immune tolerance period is associated with increased STING mRNA levels, suggesting a potential relationship between GPX4 and the cGAS–STING pathway (Su et al., 2024). GPX4 methylation modifications, activated by the cGAS–STING pathway, facilitate cancer immunotherapy (Chen B. et al., 2023). A high-iron diet or GPX4 depletion activates the STING pathway, leading to macrophage activation in pancreatic cancer (Dai et al., 2020). Ferroptosis increases cathepsin B expression, causing DNA damage and activating STING, creating a feedback loop in human pancreatic cancer cells (Kuang et al., 2020).
Therapies have been developed to evaluate STING activation and ferroptosis induction in various diseases. Manganese molybdate nanoparticles, containing MoO_4_ ^2–^ and Mn^2+^, activate the cGAS–STING pathway, promote IFN-γ secretion, and inhibit GPX4, thereby inducing ferroptosis in tumor cells (Lei et al., 2024). Mn(III)-SS-NEs activate the cGAS–STING pathway, secrete IFN-γ, and induce ferroptosis, creating an antitumor loop through manganese-induced immunogenic cell death (He et al., 2023). FeGd-HN@TA-Fe^2+^-SN38 nanoparticles activate STING and release Fe^2+^, inducing ferroptosis in tumor cells (Guo S. et al., 2023).
Therefore, the cGAS–STING pathway and ferroptosis engage in a complex bidirectional regulatory relationship with significant therapeutic implications (Figure 8).
Additional pathways linking the cGAS–STING pathway and ferroptosis regulation. ICA69 protein upregulation activates STING signaling, promoting ferroptotic cell death (①). Moderate-intensity treadmill exercise suppresses STING activation, thereby inhibiting ferroptosis (②). STING-mediated activation of the TBK1/NF-κB cascade enhances ferroptosis execution by modulating key ferroptosis regulators (③). The STING pathway interacts with mitofusins (MFN1/2) to modulate mitochondrial fusion, resulting in ferroptosis (④). STING downregulation increases ferroportin 1 (FPN1) expression, inhibiting ferroptosis via iron efflux (⑤). STING activity influences glutathione peroxidase 4 (GPX4) modifications, altering its ferroptosis-suppressive function (⑥). Moreover, lipid metabolism enzymes (ACSL4), lysosomal proteases (CTSB), and antioxidant systems (GPX4) connect STING activity to ferroptosis regulation. CTSB, cathepsin B; FPN1, ferroportin 1; ICA69, islet cell autoantigen 69; MFN1/2, mitofusin 1/2; NF-κB, nuclear factor-κB; P62/SQSTM1, sequestosome 1; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1.
Discussion
4
The intricate and context-dependent interplay between the cGAS–STING pathway and ferroptosis represents a sophisticated regulatory network that significantly affects cellular fate in pathological and physiological settings. Our synthesis of shared molecular mediators—such as mitochondrial dysfunction—highlights nodes of convergence that are not merely bystanders but active regulators of both pathways. These converging molecules present promising, yet complex, therapeutic targets for diseases where immune response and iron-driven cell death collide, such as in cancer, neurodegenerative disorders, and sterile inflammatory conditions. Consequently, our findings extend beyond the effects of individual pathways, revealing a novel intersection of immune and metabolic cell death.
While this review consolidates evidence linking key molecules to both pathways, the precision with which these shared elements coordinate cGAS–STING activation and ferroptosis remains incompletely mapped. For instance, it is still unclear whether STING activation preferentially sensitizes cells to ferroptosis in a spatiotemporally controlled manner. A key future direction involves the employment of live-cell imaging techniques to track the real-time transport of STING between organelles (such as the endoplasmic reticulum and mitochondria) and the generation of ferroptosis markers (such as GPX4 activity and lipid ROS), thereby elucidating the spatiotemporal coupling mechanisms between these processes. Furthermore, we observed the paradoxical phenomenon whereby p53 may both promote and inhibit ferroptosis. The present study hypothesizes an integrated model in which the intensity of intracellular stress signals and the cellular microenvironment jointly determine p53’s post-translational modification status and downstream pathways. Future research should systematically map p53 expression patterns under diverse stress conditions to decipher the key code governing this cell fate decision.
Looking forward, we propose that moving beyond observational linkage to mechanistic engineering of this interplay will open transformative therapeutic avenues. For example, in cancer therapy, designing combination regimens that sequentially activate STING and then trigger ferroptosis could maximize immunogenic cell death and antitumor immunity. Conversely, in inflammatory diseases, targeted disruption of specific branches of this network (e.g., by inhibiting lipid peroxidation at mitochondrial-ER contact sites) may mitigate pathological cGAS–STING activation without compromising overall immune function. The development of dual-acting agents or context-dependent modulators that can toggle between pro-death and pro-survival outcomes holds particular promise for precision medicine. Thus, unraveling the dynamic crosstalk between STING and ferroptosis will not only improve our understanding of cell death and immune signaling but also accelerate the translation of these insights into novel, pathway-aware therapeutics. It is noteworthy that, despite the promising mechanistic insights described here, key translational parameters for many of these agents remain poorly characterized. A substantial portion of the listed interventions is primarily supported by in vitro evidence, with limited in vivo data available on efficacy, toxicity, pharmacokinetics, or pharmacodynamics and minimal clinical validation to date. Therefore, further development of such therapeutics will require extensive additional preclinical and clinical research for robust validation.
In conclusion, this review presents the interplay between STING and ferroptosis as a dynamic and multifaceted relationship with profound implications for cancers and noncancerous diseases. By unraveling the molecular mechanisms and exploring innovative therapeutic approaches, researchers can harness the potential of this interplay to develop effective treatments and improve patient care.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Amos A. Jiang N. Zong D. Gu J. Zhou J. Yin L. (2023). Depletion of SOD 2 enhances nasopharyngeal carcinoma cell radiosensitivity via ferroptosis induction modulated by DHODH inhibition. BMC Cancer 23 (1), 117. 10.1186/s 12885-022-10465-y 36737723 PMC 9896811 · doi ↗ · pubmed ↗
- 2Arnold R. Vehns E. Randl H. Djabali K. (2021). Baricitinib, a JAK-STAT inhibitor, reduces the cellular toxicity of the farnesyltransferase inhibitor lonafarnib in progeria cells. Int. J. Mol. Sci. 22 (14), 7474. 10.3390/ijms 22147474 34299092 PMC 8307450 · doi ↗ · pubmed ↗
- 3Audu C. O. Melvin W. J. Joshi A. D. Wolf S. J. Moon J. Y. Davis F. M. (2022). Macrophage-specific inhibition of the histone demethylase JMJD 3 decreases STING and pathologic inflammation in diabetic wound repair. Cell Mol. Immunol. 19 (11), 1251–1262. 10.1038/s 41423-022-00919-5 36127466 PMC 9622909 · doi ↗ · pubmed ↗
- 4Bakhoum S. F. (2022). c GAS-STING and the deadly CIN: how chronic inflammation represents a therapeutic vulnerability in chromosomally unstable cancers. Trends Cancer 8 (10), 788–789. 10.1016/j.trecan.2022.07.006 35915014 · doi ↗ · pubmed ↗
- 5Balka K. R. Louis C. Saunders T. L. Smith A. M. Calleja D. J. D’Silva D. B. (2020). TBK 1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. 31 (1), 107492. 10.1016/j.celrep.2020.03.056 32268090 · doi ↗ · pubmed ↗
- 6Bersuker K. Hendricks J. M. Li Z. Magtanong L. Ford B. Tang P. H. (2019). The Co Q oxidoreductase FSP 1 acts parallel to GPX 4 to inhibit ferroptosis. Nature 575 (7784), 688–692. 10.1038/s 41586-019-1705-2 31634900 PMC 6883167 · doi ↗ · pubmed ↗
- 7Cao T. Zhou J. Liu Q. Mao T. Chen B. Wu Q. (2023). Interferon-γ induces salivary gland epithelial cell ferroptosis in Sjogren’s syndrome via JAK/STAT 1-mediated inhibition of system Xc. Free Radic. Biol. Med. 205, 116–128. 10.1016/j.freeradbiomed.2023.05.027 37286044 · doi ↗ · pubmed ↗
- 8Chen C. Xu P. (2023). Cellular functions of c GAS-STING signaling. Trends Cell Biol. 33 (8), 630–648. 10.1016/j.tcb.2022.11.001 36437149 · doi ↗ · pubmed ↗