A systematic review of observational studies on long-term air pollution exposure and epigenetic alterations in adults
Lili Yu, Yuyuan Zhao, Wenxi Chen, Guirong Yu, Mark R Miller, Xue Li, Evropi Theodoratou

TL;DR
This study reviews how long-term air pollution exposure affects epigenetic changes in adults, finding some specific gene methylation patterns linked to pollutants.
Contribution
The paper provides a systematic review of observational studies linking long-term air pollution exposure to epigenetic alterations in adults.
Findings
Long-term exposure to PM2.5 and NO2 is associated with methylation changes at specific loci like cg08500171 and cg17629796.
SOX2 hypermethylation is linked to ambient PM2.5 exposure in candidate-gene studies.
Epigenome-wide studies show up to 189 methylation loci altered by specific ambient air pollutants.
Abstract
Evidence suggests that environmental exposures induce epigenetic modifications that can have long-lasting effects on multiple health outcomes, and an in-depth review of the epidemiological evidence is urgent. We aimed to comprehensively assess the associations between long-term exposure to air pollution and epigenetic changes in adults. We systematically searched EMBASE, MEDLINE, and Web of Science databases for relevant articles published in English from inception through 17 November 2023. We assessed and narratively synthesised eligible studies on ambient (i.e. non-occupational) and epigenetic alterations in adults. We separately documented relevant occupational studies identified by the search. We analysed 52 eligible articles, including 30 ambient air pollution and 22 occupational air pollution exposure studies. Long-term exposure to ambient particulate matter (PM) with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
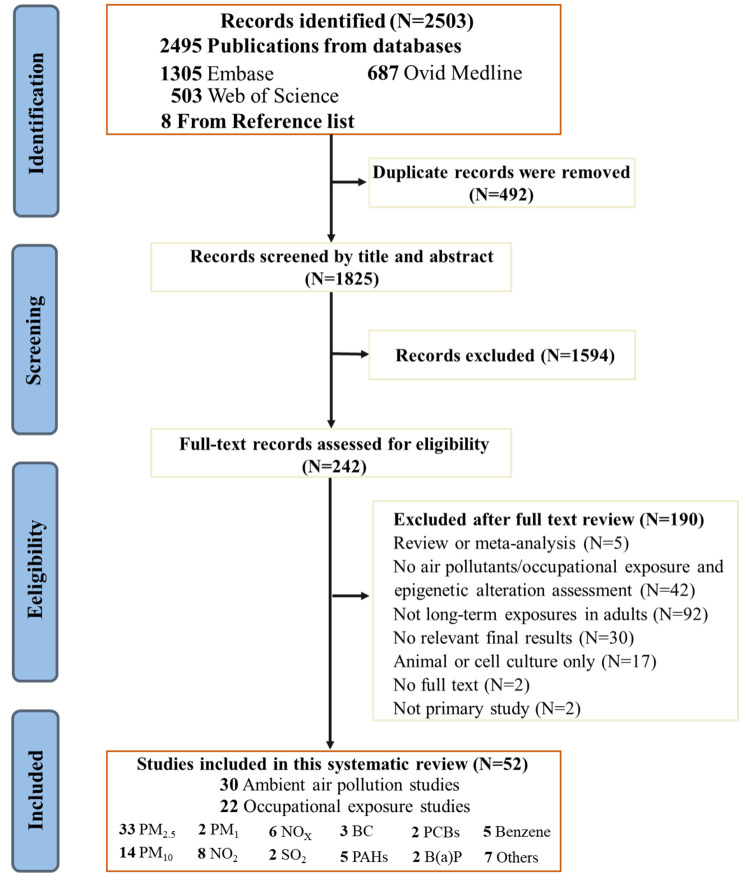 Figure 1
Figure 1| Author | Year | Country | Study population | Study design | Sample size | Age in years, x̄ (SD) | Pollutant type | Studied region or loci | Main findings |
|---|---|---|---|---|---|---|---|---|---|
| Cheng et al. [ | 2022 | China | NMU-LIFE study of the general population | Cross-sectional | 1554 | Range: 20–45; normal semen quality group: 30.6; abnormal semen quality group: 31.4 | PM10 | Global DNA Methylation: (5 mC) and Global DNA Hydroxymethylation (5-hmC) | PM10 exposure positively associated with 5-hmC levels ( |
| Chi | 2016 | USA | MESA in the general population | Cross-sectional | 1207 | 69.6 (9.4) | PM2.5, NOX | Global DNA Methylation: Alu and LINE-1 | Median DNA methylation of Alu and LINE-1 was not associated with PM2.5 (Alu, |
| Goobie | 2023 | USA | The University of Pittsburgh Simmons Centre for ILD Registry (Simmons) or the US-wide PFF Patient Registry | Cohort | 1059 | Simmons cohort: 68 (61–74); PFF cohort: 69 (64–74)* | PM2.5 | Global DNA methylation | Higher PM2.5 (1-y period exposures prior to blood collection) were associated with higher global DNA methylation percentage (%5 mC) in both Simmons ( |
| Plusquin | 2017 | Italy, Netherlands | Italian and Dutch components of the EPIC study of the general population | Cross-sectional | 613 | Italy: 54.2 (7.1); Netherlands: 58.8 (5.6) | NOX, NO2, PM10, PM2.5 | Global DNA methylation | NO2 (EPIC-Italy |
| Tao | 2014 | Poland | Residence of non-smoking women in Warsaw | Case-control | 42 | 59.4 (79.9) | Solid fuel | Global DNA methylation | The levels of LUMA methylation for women who had ever exposed to both coal and wood were reduced compared to women without exposure (6.70%; 95% CI = −13.36, −0.04) |
| Wang | 2020 | USA | Selected from the Sister Study | Cross-sectional | 1373 | Sub-cohort1:55.8 (8.9); sub-cohort2: 56.7 (8.8) | PM2.5 | Global DNA methylation: LINE-1 | There is no association between PM2.5 and LINE-1 |
| Xu | 2023 | Australia | AMDTSS female participants | Cross-sectional | 479 | 56.4 (7.9) | PM2.5 | Global DNA methylation: Alu, LINE-1, LTR | The 3-y average wildfire-related PM2.5 was negatively, but not significantly ( |
| Yadav | 2021 | India | A major research project of the general population | Cross-sectional | 512 | Low polluted: 47.9 (8.9); high polluted: 48.2 (10.2) | Total air pollution | Global DNA methylation | Air pollution showed a significant effect on global DNA methylation only among individuals with hyperhomocysteinemia ( |
| Author | Year | Country | Study population | Study design | Sample size | Age in years, x̄ (SD) | Pollutant type | Studied region or loci | Main findings | |
|---|---|---|---|---|---|---|---|---|---|---|
| Callahan | 2018 | USA | WEB Study | Case-control | 1170 | 35–79* | TSP | Gene-based: | Ambient air pollution was significantly associated with DNA methylation of the tumour suppressor genes SCGB3A1, SYK, and CCND2 in breast tumour tissue | |
| Cantone | 2020 | Italy | ED of Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, with a diagnosis of acute ischemic stroke | Cross-sectional | 55 | 74.6 (13.9) | PM2.5 | Gene-based: | PM2.5 exposure was not associated with | |
| Chou | 2020 | China | TWB database of the general population | Cross-sectional | 496 | No regular exercise: 45.832 (0.635), regular exercise: 55.185 (0.660) | PM2.5 | Gene-based: | PM2.5 was positively associated with | |
| Song | 2019 | China | A male prospective cohort study in China | Cohort | 157 | >29* | BPA | Gene-based: | Urine BPA concentration positively associated with 5hmC rate of the sperm | |
| Tantoh | 2019 | China | TWB data set of the general population | Cross-sectional | 708 | Men: 49.42 (11.76); women: 49.49 (10.97) | PM2.5 | Gene-based: | A unit increase in PM2.5 significantly associated with lower cg05575921 ( | |
| Tantoh | 2019 | China | TWB data set of the general population | Cross-sectional | 461 | Men: 48.86 (11.74); women: 48.78 (11.01) | PM2.5 | Gene-based: | x̄ PM2.5 level (2006–2011) associated with higher | |
| Su | 2020 | China | Taiwanese adults aged 30–70 who have no personal history of cancer | Cross-sectional | 948 | No exercise: 46.1 (0.46); exercise:54.4 (0.5) | PM2.5 | Gene-based: | PM2.5 significantly associated with higher levels of | |
| Wang | 2020 | USA | Selected from the Sister Study | Cross-sectional | 1373 | Sub-cohort1: 55.8 (8.9); subcohort2: 56.7 (8.8) | PM2.5 | Gene-based: | Estimated change of −6.5% (95% CI = −13.34%, 0.35%) in x̄ methylation of TNF-α per 5 μg/m3 increase in PM2.5 | |
| Author | Year | Country | Study population | Study design | Sample size | Age in years, x (SD) | Pollutant type | Main findings |
|---|---|---|---|---|---|---|---|---|
| Chi | 2016 | USA | MESA in the general population | Cross-sectional | 1207 | 69.6 (9.4) | PM2.5, NOX | Long-term PM2.5 exposure was significantly associated with 5 candidate CpGs methylation, including cg20455854 ( |
| Chi | 2022 | USA | MESA in the general population | Cross-sectional | 1207 | 69.6 (9.4) | PM2.5, NOX | Long-term PM2.5 exposure was significantly associated with 3 methylated CpGs, and NOx with cg11756214 within |
| de F C Lichtenfels | 2018 | Netherland | Lifeline cohort study participants of the general population | Cross-sectional | 1017 | 47.3 (11.0) | NO2, PM2.5, PM10 | 7 CpG sites were significantly associated with NO2 levels; no associations were found for PM10 and PM2.5 exposure at genome-wide level ( |
| Eze | 2020 | Switzerland | The SAPALDIA cohort of the general population | Cross-sectional | 1389 | SAPALDIA2: 50 (18), SAPALDIA3: 58 (18) | NO2, PM2.5 | Reported top 10 |
| Fiorito | 2018 | Italy | EPIC-Italy study of the general population | Case-control | 320 | CCVD: 54.8 (7.4); control: 54.9 (7.1) | NO2, NOx, PM2.5 | Identified enrichment of altered DNA methylation in ‘ROS/Glutathione/ Cytotoxic granules’ and ‘Cytokine signalling’ pathways related genes associated with NO2, NOx, PM2.5, respectively. |
| Gondalia | 2019 | USA | Participants of 2 cohorts (WHI, ARIC) | Cross-sectional | 8397 | 61.3 (7.4) | PM10, PM2.5 | 55 suggestively significant PM-associated CpG methylation were identified from trans-ethnic, fixed-effects inverse-variance-weighted meta-analyses |
| Honkova | 2022 | Czech Republic | Nonsmoking city policemen from 3 cities of Czech Republic | Cross-sectional | 125 | Prague: 40.4 (9.3); Ostrava: 39.3 (9.2); Ceske Budejovice: 38.0 (6.4) | PM2.5, B(a)P, NO2 | 13 643 DML were identified from a comparison between the Prague and Ostrava groups |
| Liu | 2021 | China | 3 panels of the healthy Chinese population (WHZH panel, COW panel, and Shiyan panel) | Cross-sectional | 304 | WHZH-Wuhan: 51.3 (13.9); WHZH-Zhuhai: 61.7 (11.7); SY: 40.0 (10.9); COW: 44.9 (8.4) | PAHs | Methylation level of cg09235308 was positively associated with urinary total OH-PAHs, but no significant epigenome-wide associations for the other 9 urinary OH-PAHs |
| Messingschlager | 2023 | Germany | General participants from Simmerath (Low PM10) and Stuttgart (Moderate PM10) | Cross-sectional | 60 | Stuttgart: 28 (20–36); Simmerath: 27 (20–39)† | PM10 | 231 DMRs between moderately and lowly PM10-exposed individuals |
| Mu | 2023 | China | Wuhan-Zhuhai cohort of the general population | Cross-sectional | 402 | 58.4 (7.89) | PM2.5 | 10 |
| Plusquin | 2017 | Italy, Netherlands | Italian and Dutch components of the EPIC study of the general population | Cross-sectional | 613 | EPIC-Italy: 54.2 (7.1); EPIC-Netherlands: 58.8 (5.6) | NOx, NO2, PM10, PM2.5 | NO2 and NOx were inversely associated with global DNA methylation, but not with PM2.5 and PM10 |
| Sayols-Baixeras | 2019 | Italy, Spain | General-based individuals from the REGICOR cohort and EPIC-Italy study | Cross-sectional | 1084 | REGICOR: 63.3 (11.7); EPIC-Italy: 54.2 (7.1) | PM10, PM2.5, NOx, NO2 | Did not identify any new genomic loci associated with long-term air pollution and did not replicate any previously identified loci |
| White | 2019 | USA | The Sister Study cohort of women participants who had a sister with breast cancer but no history of breast cancer themselves | Cross-sectional | 2747 | 45–65† | PM2.5, PM10, NO2 | NO2 was associated with methylation at 2 CpG sites |
| Wang | 2020 | China | Chinese Han general population-based cohort | Cross-sectional | 120 | HPR: 40.6 (2.2); LPR: 40.7 (3.5) | PM2.5, PM10, SO2, NO2, CO | Genome-wide methylation analysis revealed 191 hypomethylated and 180 hypermethylated DMRs ( |
| Xu | 2023 | Australian | Female participants in the AMDTSS | Cross-sectional | 479 | 56.4 (7.9) | PM2.5 | 26 CpGs and 33 DMRs associated with wildfire-related PM2.5 ( |
| Curtis | 2021 | USA | Those with the highest exposure to PBB selected from the Michigan PBB Registry | Cross-sectional | 641 | 54.3 (12.8) | PCBs | Current total PCBs level associated with the methylation proportion at 1345 CpGs |
| Pittman | 2020 | USA | Selected from the Anniston Community Health Survey | Cross-sectional | 817 | 57.6 (4.02) | PCBs | 28 significant CpGs were associated with PCBs, representing 17 unique genes |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · Air Quality and Health Impacts · Health, Environment, Cognitive Aging
Air pollution is considered to be one of the foremost environmental risk factors of disability-adjusted life-years worldwide [1]. According to a 2022 World Health Organization report, it is estimated that approximately 4.2 million premature deaths globally were caused by ambient outdoor air pollution alone in 2019, predominantly from respiratory disease, heart disease, and stroke [2]. Growing evidence links long-term exposure to air pollutants to many more adverse health outcomes in most major organ systems [3]. However, the underlying biological mechanisms of air pollution-induced adverse effects throughout the body have not been fully determined.
Findings from a large body of research demonstrate that environmental exposures may induce epigenetic alterations, including DNA methylation and histone modification. Epigenetic modifications can have profound effects on gene expression and cellular function [4,5], with de novo methylation associated with tumorigenesis [6]. DNA methylation, the most common epigenetic modification, involves adding a methyl group to cytosines at cytosine-phosphate-guanine (CpG) sites and is heritable and relatively stable over time [7]. Studies have shown that several small-scale alterations in DNA caused by environmental factors might accumulate over a long period [8,9]. In addition, evidence from epigenome-wide association studies (EWAS) shows that exposure to airborne particulate matter (PM) can affect DNA methylation in blood and tissues, identifying numerous air pollution-modified CpG sites and regions [10,11]. One systematic review showed that air pollution exposure was associated with locus-specific epigenome alterations or inverse global DNA hypomethylation [12]. Nonetheless, the effects of chronic exposure to air pollution on CpG sites or regions across different population settings, as well as the latest research on this topic, have not yet been comprehensively assessed.
Histone modification affects the interaction of histone with DNA and effector proteins, thus causing modifications in the structure and function of chromatin (i.e. an amalgamation of DNA and protein that forms chromosome structure) [13]. Environmental contaminants have been linked to histone acetylation with global modifications, including histone H3 lysine 9 di-methylation (H3K9me2), histone H3 tri-methylated lysine 4 (H3K4me2/3), tri-methylation at lysine 27 of histone H3 (H3K27me3), and histone H3 lysine 79 trimethylation (H3K79me3) [14–17]. Histone H3 modifications are the most studied [18], and some have been reported after exposure to heavy metals [19,20] and traffic-related PM [21].
We systematically reviewed the literature on associations between epigenetic alterations and long-term air pollution exposure to consolidate the available epidemiological evidence. Specifically, we focused on ambient air pollution in the general population; however, we also identified studies assessing epigenetic changes in response to ambient air pollutants in occupational cohorts (e.g. PM exposure in steel workers) or air pollutants found in ambient air, but more commonly associated with occupational exposures (e.g. benzene exposure in leather workers). We aimed to identify fundamental biological mechanisms underlying epigenetic alterations caused by air pollution exposure, and our findings may provide useful mechanistic insights.
METHODS
We registered the protocol in PROSPERO (CRD42023480771) on 20 November 2023 and followed the PRISMA guidelines [22].
Literature search and selection criteria
We systematically searched Embase, Medline, and Web of Science databases from inception through 17 November 2023 using a comprehensive search strategy (Table S1 in the Online Supplementary Document). The search terms comprised free text and medical subject headings for epigenetic alterations and air pollutant exposures (excluding tobacco smoke exposure). We reviewed each article that included a relevant abstract and those with uncertain relevance, and downloaded them to an EndNote 20 library (Clarivate, Philadelphia, Pennsylvania, USA). By following predefined inclusion and exclusion criteria, three authors (LY, WC, and YZ) conducted a two-step parallel review of the title and abstract, and full text to determine the eligibility of the retrieved publications.
The inclusion criteria were: participants are adults (>18 years), both in the general and occupational population; long-term (>1 year) ambient and occupational exposure; epigenetic alterations as a primary or secondary outcome (including DNA methylation and histone modifications based on approaches on global DNA methylation, candidate-gene, EWAS, and histone modification); and observational studies (including longitudinal, cross-sectional, cohort, and case-control).
We excluded studies that investigated prenatal air pollution exposure effects on epigenetic alterations in children (≤18 years); were based on the elderly (>65 years); did not report the effect of air pollution on epigenetic modifications; were abstract-only publications, comments, reviews, and interviews; and were non-human studies (including research experiments conducted in animals or animal/human cell lines).
When the same study population was investigated in multiple publications, we included only the most recent (larger sample size). All included studies were written in English and published in peer-reviewed journals. We checked the reference lists of all included articles to identify any additional eligible studies.
Data extraction
For each eligible study, two investigators (LY and WC) extracted the data independently using a predesigned data extraction form. We extracted the PubMed identification number, first author, year of publication, location, study design, number of participants, age of participants, exposure definition, type of pollutant, concentration of pollutant, exposure length, sample sites, studied region or loci, adjusted confounding factors, epigenetic alteration measurement technique, effect estimates for the major findings (coefficient of multiple linear regression or correlation coefficient with the 95% confidence intervals), and a brief summary of the reported main findings. Any discrepancies were resolved by consensus between the two data extractors. We attempted to contact the authors of the study when the exposure length for an eligible study could not be determined.
Quality assessment
Two reviewers (LY and GY) independently assessed all included studies using the ‘Risk of Bias in Non-randomised Studies - of Exposure’ (ROBINS-E) tool. With an emphasis on environmental exposures, the ROBINS-E tool is adapted from the original Risk of Bias in Non-randomised Studies of Interventions tool [23]. We evaluated seven main methodological aspects (i.e. domains) deduced from ROBINS-E, including bias due to confounding, bias due to exposure classification, bias due to missing data, bias in outcome measurement, bias due to departures from intended exposures, bias in the selection of reported results, and bias in selecting participants in the study, with the risk of bias judgments expressed as either ‘low’, ‘some’, or ‘high’ (Table S2 in the Online Supplementary Document). Any disagreements were resolved by consensus between the reviewers. We recorded the overall risk of bias for each study as the highest risk across any domain.
RESULTS
Literature review
We identified 2495 publications across Embase, Medline, and Web of Science databases (Figure 1). After removing 492 duplicates, we screened 1825 records by title and abstract, and selected the remaining 242 articles for full-text screening. Finally, 52 publications met the inclusion criteria, with 44 retrieved from the above three databases [17,24–65] and eight additional articles identified from the reference lists of other papers [66–73].
Flowchart of literature search. BC – black carbon, NO2 – nitrogen dioxide, NOx – nitrogen oxides, PAH – polycyclic aromatic hydrocarbon, PCB – polychlorinated biphenyl, PM – particulate matter, SO2 – sulfur dioxide.
Study characteristics
There were six cohort [26,37,45,54,69,73], three case-control [27,34,56], and 43 cross-sectional studies [17,24,25,28–33,35,36,38–43,45–52,54,55,57–68,70–72] (Tables 1–3??; Tables S3–5 in the Online Supplementary Document). Overall, 30 studies were classed as ‘ambient air pollution exposure’, with a median exposure window of three years (range = 1–29.6) [17,27,29–35,37,38,40,47,48,50,52–56,58–61,63,67,70,71], and 22 classed as ‘occupational exposure’, with a median of five years (range = 1–31) [17,24,25,35,40–45,49,52,57,62,64–66,68,69,72,73]. Some studies measured exposure to air pollutants using land-use regression models based on data from stationary monitoring networks [6,30,31,33,35,50,51,60,61]. Other studies used cyclone- or battery-operated channel optical particle counters as personal air pollution monitors during working hours at workplaces [17,25,40–42,44,52,56,57,62,68,69,73]. In particular, PM with aerodynamic diameters of ≤2.5 μm (PM_2.5_) and ≤10 μm (PM_10_), nitrogen oxides (NOx), nitrogen dioxide (NO_2_), polycyclic aromatic hydrocarbons (PAHs), solid fuel, benzo(a)pyrene (B(a)P), polychlorinated biphenyls (PCBs), sulphur dioxide (SO_2_), and bisphenol A (BPA) were analysed in ambient air pollution studies, whereas PM_1_, PM_2.5_, PM_10_, benzene, black carbon, B(a)P, PAHs, and formaldehyde were evaluated in occupational settings. PM (including PM_1_, PM_2.5_, PM_10_) was most frequently reported (n = 34) [17,26–34,36–44,47,48,50–52,54,55,57–61,71,74], followed by NO_2_ (n = 8) [6,32,34,39,50,51,59,60], NO_X_ (n = 6) [30–32,34,50,51], PAHs (n = 5) [24,38,46,52,65,66], benzene (n = 5) [45,68,69,72,73], black carbon (n = 3) [35,49,59], B(a)P (n = 2) [39,64], PCBs (n = 2) [67,70], SO_2_ (n = 2) [32,59], BPA (n = 1) [53], and formaldehyde (n = 1) [25]. Most studies used blood samples to extract DNA (n = 48) [17,24,25,28,30–52,54–73], whereas brain and breast tissue samples were used in two studies [26,27] and semen samples in two studies [29,53]. Of these, studies determined the occupational exposure by analysing biomarkers in the blood [43] or urine [24,64–66]. Eleven epigenetic alteration assessment techniques were applied, including pyrosequencing-based, array-based, high-throughput sequencing-based, analytical chemistry-based, and antibody-based enrichment approaches (Table S6 in the Online Supplementary Document), with Pyrosequencing [24,27,28,38,40,41,43,44,49,52,57,58,62,65,66,68,69] and Illumina Infinium Human Methylation 450K BeadChip Array [6,30,31,33,34,36,48,50,51,58,60,61] methods being the most used to estimate ambient air pollution-induced DNA methylation.
Risk of bias assessment
The domains with the highest risk of bias were confounding, exposure classification, missing data, outcome measurement, and exposure measurement (Table S7 in the Online Supplementary Document).
For confounding, we considered socioeconomic status, race and ethnicity, seasons, temperature, and smoking status to be the largest threats of bias. For studies where adjusted estimates were available (e.g. age, race/ethnicity, sex, study site, income, education, seasons, temperature, and smoking status), the bias was likely to be low [29-31,33,36,61], but when only crude estimates were available, we considered the study at high risk of bias [26,39,47,52,64]. For exposure classification, we considered studies that explored long-term exposure (>1 year) using models with good performance and/or estimated air pollution exposure using monitoring stations at low risk of bias [4,17,24,26,27,29-34,36-42,47–51,54,55,58–61,71,73]. We considered high risk when exposure levels were reported for long-term exposures using a model with poor performance or unclear exposure estimation [35]. For missing data, we considered studies that did not mention follow-up in cohort studies to be at high risk of bias [26,37,69,73]. For outcome measurement, studies with explicit methodologies for sample collection and technique controls in examining epigenetic alterations were considered low risk, whereas studies were at some risk of bias in assessing epigenetic alterations [24,25,27,35,37–39,41–45,49,52,53,56,63–69]. For the selection of reported results, studies reporting P-values with multiple test corrections (e.g. false discovery rate) were considered at low risk of bias; otherwise, at some risk of bias [17,28,40,42,43,45]. Regarding participant selection, the risk of bias for each study was assessed as low (Table S2 in the Online Supplementary Document). The overall risk of bias was considered ‘low’ for four studies [30,31,33,61], ‘some’ for 25 studies [17,24,27–29,32,34,36,46,48,50,51,54–60,63,65–67,70,71], and ‘high’ for 23 studies [6,25,26,34,37–45,47,49,52,53,62,64,68,69,72,73] (Figure S1 in the Online Supplementary Document).
Ambient air pollution studies
Overall, 30 studies measured exposure to long-term ambient air pollutants and epigenetic alterations [27-34,36,37,39,46–48,50,51,53–56,58–61,63,67,70,71]. Global DNA methylation approaches were reported in eight studies [29,30,37,50,56,58,61,63], candidate-gene approach in eight studies [27,28,32,54–56,59,72], the EWAS approach in 17 studies [6,30,31,34,35,37,40,47,48,50,51,59–61,67,70], and the histone modification approach in one study [26]. Among them, four studies used ≥2 approaches [30,50,58,61].
Global DNA methylation studies
We analysed eight studies [29,30,37,50,56,58,61,63] that assessed the association between ambient long-term exposure to specific air pollutants and global DNA methylation patterns (Table 1; Table S5 in the Online Supplementary Document). Seven studies reported a sample size of >100 participants [29,30,36,50,58,61,63]. There were seven global DNA methylation studies investigating DNA methylation levels in blood samples [30,37,50,56,58,61,63], and one in semen samples [29]. Three studies used methylation measurements of repetitive elements/regions (LINE-1 and Alu) as proxies for global DNA methylation levels [30,58,60]. Most of these studies (n = 7) [29,30,37,50,58,59,61] investigated changes in global DNA methylation following increased exposures to ambient PM_2.5_ or PM_10_. Among the ambient PM (PM_2.5_ or PM_10_) exposures reported in the included studies, two studies found that PM exposure was significantly associated with higher levels of the alternative epigenetic DNA modifications 5-methylcytosine and 5-hydroxymethylcytosine in blood and semen, respectively [29,37]. Nevertheless, four studies found no statistically significant association between long-term exposure to PM_2.5_ and PM_10_ and blood global DNA methylation [30,50,58,61].
Studies investigating exposure to NO_2_ (n = 1) [50], NO_X_ (n = 2) [30,50], solid fuel (n = 1) [56], and total air pollution (n = 1) [63] were also identified. Discrepant findings were found between studies exploring the association between global DNA methylation and exposure to ambient NOx. One study [50] showed an inverse association between NOx exposure and global DNA hypomethylation, whereas another study [30] suggested no significant association between NOx exposure and median DNA methylation of LINE-1.
Candidate-gene methylation studies
We identified eight studies investigating the association between air pollution and candidate-gene methylation (Table 2; Table S5 in the Online Supplementary Document), six of which assessed blood samples [28,32,54,55,58,71], whereas one in the breast tumour tissue sample [27] and one in the semen sample [53]. Air pollutants in these candidate-gene studies included PM (n = 6) [27,28,32,54,55,58,71] and BPA (n = 1) [53].
PM_2.5_ exposure was significantly and positively associated with higher levels of promoter methylation of blood sex-determining region Y-box 2 (SOX2) in two studies [55,71]. Long-term ambient PM_2.5_ exposure was significantly associated with decreased methylation of the deleted in lung and oesophageal cancer 1 (DLEC1) promoter and cg05575921 within the aryl hydrocarbon receptor repressor (AHRR) [32,54]. Two studies reported gene methylation in breast and sperm samples [27,53]. Total suspended ambient particulates were suggested to be significantly associated with DNA methylation of tumour suppressor genes (SCGB3A1, SYK, and CCND2) in breast tumour tissue [27]. Urine BPA concentration showed a positive association with sperm ACHE hydroxymethylation in men [53] (Table S8 in the Online Supplementary Document).
EWAS studies
Regarding the 17 EWAS studies, 16 were cross-sectional studies [6,30,31,33,36,39,46–48,50,51,59–61,67,70], and one was a case-control study [34] (Table 3; Table S9 in the Online Supplementary Document). Samples of >100 participants were analysed in the discovery phase of 16 studies [6,30,31,33,34,36,39,46,48,50,51,59–61]. All identified EWAS studies used blood samples, and the Infinium Human Methylation 450K BeadChip Array (Illumina) technique was the most commonly applied to assess the air pollution-induced CpG sites or regions [6,30,31,33,34,36,46,50,51,60,61], followed by Illumina Infinium 850K MethylationEPIC BeadChip [39,48,67,70], and whole-genome bisulfide sequencing [47,59]. Ambient PM exposure was assessed in 14 EWAS studies [6,30,31,33–35,39,47,48,50,51,59–61], NOx and NO_2_ in ten studies [6,30,31,33,34,39,50,51,59,60], PCBs in two studies [67,70], and PAHs in one study [48].
All the EWAS studies indicated that long-term exposure to ambient air pollution was associated with alterations in blood methylation of up to 189 unique loci (Table S9 in the Online Supplementary Document). The largest and most recent study, conducted by Eze and colleagues [33], included 1389 individuals and reported the top ten ambient NO_2_-associated and ten ambient PM_2.5_-associated CpGs in whole blood. One epigenome-wide study [46] found that the methylation level of cg09235308 in plectin 1 was positively associated with ten urinary mono-hydroxylated PAHs. In addition, the methylation in pescadillo ribosomal biogenesis factor 1 (PES1) was reported to be associated with both ambient PM_2.5_ and PM_10_ exposure in a multicentre prospective cohort [50]. Wang and colleagues [59] applied a multi-exposure model that included PM_2.5_, PM_10_, SO_2_, NO_2_, and CO, and reported that combined exposure to long-term ambient air pollution led to 371 differentially methylated regions (DMRs), located primarily in gene regulatory elements such as promoters and enhancers.
Four EWAS studies found significant signals in a discovery population and validated successfully in the external replication populations [6,50,51,70]. Pittman and colleagues [70] reported significant associations between exposures to PCBs and the methylation of 28 CpGs in whole blood in the discovery cohort. However, only one association was statistically significant in the replication external cohort: cg00475490 in the gene of serine protease 23 (PRSS23). Additionally, Eze and colleagues [33] replicated two previously identified air pollution-associated EWAS signals; increased methylation in cg08500171 in the gene of branched-chain-amino-acid transaminase 2 (BAT2) related to NO_2_ exposure and decreased methylation in cg17629796 related to PM_2.5_ exposure (Table S10 in the Online Supplementary Document). These three EWAS signals highlight strong evidence of the association between air pollutants and DNA methylation.
Histone modification studies
Ambient air pollution exposure and histone modification were assessed in one study [26]. It reported an inverse association between histone modification (mainly H3K9me2) and exposure to ambient PM_2.5_ in brain samples [26] (Tables S4 and S5 in the Online Supplementary Document).
Findings in occupational air pollution exposure studies
We identified 22 studies [17,24,25,35,38,40–45,49,52,57,62,64–66,68,69,72,73] that assessed the association between long-term occupational (including brick makers, boilermakers, shoe makers, electronic-waste recyclers, truck drivers, traffic policemen, welders, salon, steel, blue-collar, tanneries and coke oven plant workers) air pollution exposures and global DNA methylation patterns (n = 7) [24,25,35,43,44,49,65], candidate-gene studies (n = 13) [38,40–42,52,57,62,64,66,68,69,72,73], and histone modifications (n = 2) [17,45] (Tables S3–5 in the Online Supplementary Document).
Two studies found no statistically significant association between long-term occupational air pollution exposure (electronic-waste recyclers and boilermakers) and LINE-1/Alu methylation for PM_2.5_ and PM_10_ [43,44]. In candidate gene studies, occupation-related genes with epigenetic alterations were reported in our study. One study showed that methylation levels of adenomatous polyposis coli (APC) were positively associated with occupational exposure to PM_10_ or PM_1_ in steel plant workers, whereas tumour protein P53 (p53) and ras association domain family 1 isoform A (RASSF1A) were not [40]. Another study [57] reported that PM_10_ or PM_1_ was inversely associated with methylation of nitric oxide synthase 3 (NOS3) in steel workers, whereas methylation of endothelin 1 was not. Methylation of multiple tumour suppressor 1 (p16) was explored in two studies [40,72], with one [40] finding that methylation status in p16 in steel workers was not significantly associated with PM_1_ or PM_10_, while the other [72] indicated no association between the p16 gene methylation and benzene exposure in paint, shoe and printing workers. Two individual studies were conducted for ambient PAHs exposure, one found that 1-hydroxypyrene urine concentration was negatively associated with DNA hypomethylation of the interleukin 12 [24], and the other observed PAH metabolite concentrations were related to hypomethylation of coagulation factor II (thrombin) receptor-like 3 (F2RL3) and AHRR in chimney sweep workers [66]. Occupational exposure to PM_1_ and PM_10_ in steel workers was not associated with blood histone modification of the genes H3K4me2 and H3K9ac [17] (Tables S3, S4, and S8 in the Online Supplementary Document).
DISCUSSION
In this systematic review, we assessed the evidence for associations between long-term exposure to ambient and occupational air pollution with epigenetic modification in adults. More studies evaluated the association between air pollution exposure and locus-specific DNA methylation than those evaluating histone modifications. The studies identified showed high heterogeneity in exposure modelling and epigenetic alteration assessment. While most studies investigated ambient PM or NOx exposure, there was no consistent evidence of an association with global DNA methylation. Among candidate-gene studies, hypermethylation of the SOX2 gene was positively associated with ambient PM_2.5_ exposure, although this association was based on only two studies. There were 17 EWAS studies that found that long-term exposure to ambient air pollution was associated with methylation changes at up to 189 unique loci in blood. In EWAS studies successfully replicated in external validation cohorts, methylation was altered at loci such as cg00475490 (PRSS23) to PCBs, cg08500171 (BAT2) to NO_2_, and cg17629796 to PM_2.5_ exposure. In PM exposure in occupational settings, there was no significant effect on global DNA methylation. Although a greater number of potential associations were not significantly altered, PM exposure in steel workers was found to modify the methylation status of specific candidate genes. There was no change for histone modification of H3K genes.
DNA methylation patterns are disrupted in human malignant tumours and cell types, with genome-wide hypomethylation and gene-specific hypermethylation occurring simultaneously in the same cell, leading to gene expression and chromosome instability owing to large-scale alterations [75]. In addition, inhaled air pollutants are associated with adverse effects across many organ systems in adults via epigenetic changes [76,77]. While many air pollutants are harmful to health, associations with ambient PM exposure tend to be stronger and more consistent [78]. We found that PM_2.5_ and PM_10_ were the most investigated air pollutants in global DNA methylation studies. However, we observed discrepant findings regarding associations between PM_2.5_ and PM_10_ exposure and DNA methylation levels. Two studies reported a positive association between exposure to ambient PM_2.5_ or PM_10_ and global DNA methylation [29,37], while most studies reported no significant associations between these air pollutants and global DNA methylation [30,50,58,61]. We also found an inconsistency between ambient NOx exposure and global DNA methylation [30,50]. One potential explanation is that measurement error in DNA methylation (i.e. batch, chip, or cell composition effects) may result in differences in epigenetic alteration levels, thereby diminishing the statistical power required to identify significant associations. Additionally, global DNA methylation is not a very sensitive method for identifying or reflecting real biological changes, which might be one of the reasons findings from the included global methylation studies were inconsistent. In addition, future research should note combined exposure to different air pollutants, as indicated by the finding that joint exposure (PM_2.5_, PM_10_, SO_2_, NO_2_, and CO) could induce multiple DMRs.
We found that hypermethylation of SOX2 was influenced by ambient PM_2.5_ exposure in candidate-gene studies, although this was based on only two observational studies. The transcription factor SOX2 is essential for embryonic development, and plays a crucial role in controlling several features of cancer cells, such as proliferation, migration, invasion and metastasis, thus resulting in cancer pathogenesis [79]. SOX2 may present a potential molecular marker for lung cancer [80]. Methylation of the SOX2 promoter is thought to be useful for early prediction of lung cancer risk in non-smokers exposed to air pollution. Moreover, in oesophageal squamous cell carcinomas, SOX2 overexpression with p53 and p16 inactivation promotes chromatin remodelling by shaping the epigenome [81]. Yachida and colleagues also found that the SOX2 gene was overexpressed in neuroendocrine carcinomas of the gastrointestinal system due to hypermethylation of the promoter region [82]. These findings indicate that hypermethylation of SOX2 could serve as a biomarker for diseases (e.g. lung cancers and gastrointestinal diseases) and as a new avenue for exploring the mechanisms underlying the impact of environmental exposures on cancers. We also showed that total suspended particles were linked to epigenetic changes in tumour suppressor genes in breast tumours, such as SCGB3A1, SYK, and CCND2, suggesting that assessing methylation of these sites is warranted for cancer therapy.
EWAS is a commonly used strategy for discovering DNA methylation variations across conditions and phenotypes [83]. Methylation in PES1 was reported to be associated with both ambient PM_2.5_ and PM_10_ exposure [11]. The PES1 encodes a nuclear protein that contains the breast cancer-associated gene 1 C-terminal interaction domain, which plays a key role in the proliferation and carcinogenicity of breast cancer. PES1 is overexpressed in original breast tumours compared with normal mammary tissues, and knockdown of PES1 inhibits the growth of breast cancer cells [84]. We also identified that wildfire-related PM_2.5_-associated CpGs (mapped to genes) are enriched for pathways regarding inflammatory regulation and platelet activity [61]. Methylation of cg00475490 (PRSS23) by PCBs and cg08500171 (BAT2) by NO_2_ were successfully replicated in external validation cohorts. Originally discovered as an ovarian protease [85], PRSS23 has been demonstrated to be up-regulated by oestrogen receptor alpha and to stimulate the proliferation of breast cancer cells [86]. BAT2 encodes a mitochondrial protein that is expressed in all organs except liver cells and is essential for the branched-chain amino acid catabolic pathway [87]. Studies have shown that missense variants in BAT2 have been linked to lung cancer in multiple populations [88,89]. However, the identified CpGs/DMRs were heterogeneous across the included studies, because these studies are likely to report significant EWAS signals, making it difficult to identify overlapping EWAS findings across different studies. In addition, as noted with the global methylation studies, differences in the data sources and chemical composition of PM and NOx in different populations will contribute to the relative diversity of observations relating to PM and NOx, as suggested by the lack of common EWAS signals of the top PM_2.5_- and NO_2_-associated CpGs [33].
PM exposure can induce post-translational histone modifications, including H3K9me2, H3K4me3, H3K27me3, H3K79me3, and H3K4me2 [90]. While prenatal exposures were outside the scope of our search, it has been reported that air pollution-induced alterations in cord plasma histone H3 modifications during early life might be an indicator that circulating histones could be a risk factor in the development of air pollution-related diseases (e.g. lung cancer and stroke) in later life [91,92]. We found that ambient PM_2.5_ exposure in adults was inversely associated with histone modification (mainly H3K9me2). It has been shown that reactive oxygen species generated by PM_2.5_ alter H3K27 acetylation levels, leading to a global increase in enhancer-associated H3K27ac and further activating CXC-chemokine receptors, which may be related to changes in lung function through inflammatory pathways [93]. It should be noted that there are quite limited studies to elucidate the evidence between ambient air pollution and histone modifications. Given that DNA methylation and histone modification are key epigenetic alterations in cellular functions, prospective studies with larger sample sizes are warranted to assess the long-term consequences of these pathways.
We also identified occupational exposure studies related to pollutants in ambient air. Considering that the health impacts of exposures in ambient and occupational settings can differ in nature, including concentration, duration of exposure, composition, and co-exposure to other pollutants and stressors, we reported those occupational findings separately. The overall evidence from the identified studies suggests that long-term occupational exposure to inhaled pollutants could also affect epigenetic changes in a variety of genes (APC, p16, p53, RASSF1A, NOS3, EDN1, F2RL3, AHRR, H3K4me2, and H3K9ac). Aberrant DNA methylation of the APC could act as a strong predictor for cancer progression via the regulation of programmed cell death [94]. NOS3 encodes endothelial nitric oxide synthase, and nitric oxide plays an important and diverse signalling role in the cardiovascular system, contributing to the regulation of vascular tone. Evidence has reported that higher PM_2.5_ exposure was associated with DNA methylation of several CpG loci in the NOS3 gene, which indicates that these contaminants may change nitric oxide production through an epigenetic mechanism [95]. Accumulating studies consistently showed that methylation of F2RL3 and AHRR were the top-ranked signals associated with tobacco smoking, and the hypomethylation changes have been linked to increased risk of lung cancer and myocardial infarction [96–98]. One of the most common occupational exposures identified was PM exposure among steel workers, which did not cause histone alterations in H3K4me2 and H3K9ac. However, PM is rich in metals, which may instigate free radical generation, promoting pathophysiological changes driven by oxidative stress and inflammation [99]. Mechanistic studies together with new cohort studies exploring long-term health outcomes are required to further address these findings.
Current studies conducted on air pollution-induced epigenetic alterations have several challenges and limitations. In the context of air pollution, there will inevitably be some degree of measurement and attribution error in assigning individual air pollution concentrations. In line with this, among the 23 studies at overall high risk of bias assessed by the ROBINS-E tool, most of which are predominantly attributed to the high risk of measurement of the exposure bias. Land-use regression modelling based on actual measurements, combined with predictor variables such as nearby population/household density, can address this limitation to some extent [100,101]. In addition, people are exposed to mixtures of air pollution (such as differences in PM sources and sizes, and exposure to co-pollutants) [102], and it is difficult to separate the effects of closely correlated air pollutants, for example, PM and NO_2_ derived from traffic. Some studies assessed pollutant exposure by measuring the pollutant or a metabolite in a biological sample, such as blood or urine. There will be inherent differences in the biokinetics of the pollutant between individuals, but more so, the origin of the pollutant (i.e. PCBs) may not be certain and could have arisen from ingestion of the pollutant in water or food rather than inhalation. Regarding the measurement of epigenetic modifications, the accuracy of laboratory measurements, unmeasured batch effects, and the testing regions across different methylation arrays can vary widely, leading to inconsistencies in findings. In addition, four studies used tissue (brain and breast) and semen samples to measure epigenetic changes, indicating blood-specific (SOX2, DLEC1, and AHRR), breast tissue-specific (SCGB3A1, SYK, and CCND2), and sperm-specific (ACHE) gene methylation patterns related to different ambient air pollution exposures. Also, an inconsistency in histone modification of the gene H3K4me2 was observed in those participants exposed to PM, in both blood and brain tissue samples. Furthermore, a small number of observational studies were identified. In addition, in most studies, only significant EWAS signals or candidate genes were reported, which limited our ability to perform quantitative stratified meta-analysis and sensitivity analyses on overlapping CpG sites or genes associated with the same air pollutant. Most of the identified observational studies have relatively small sample sizes (range = 23–8397 participants), which likely contributed to the frequently observed trends that did not reach statistical significance. Lastly, most studies failed to control for season and temperature as confounders, and eight studies did not adjust for smoking status. DNA methylation at specific sites is robustly associated with environmental exposures, such as tobacco smoking and obesity [103]. Therefore, these should be considered as important sources of confounding in future studies.
The strengths of our review include its extensive coverage and its in-depth, reproducible evaluation of the existing evidence. Our synthesis of current evidence suggested certain patterns of epigenetic modifications altered by pollutants among public and occupation-based populations. Importantly, we highlighted locus-specific epigenome alterations that have been successfully validated in external data sets. These changes are significant, directing further studies to important mechanistic pathways and to those susceptible to the adverse effects of air pollution. We systematically assessed all included studies for risk of bias using the ROBINS-E tool, particularly for the evaluation of air pollution measurement, epigenetic alterations, and control of confounding factors. Meanwhile, several limitations of our review should also be considered. First, the search strategy was not designed to capture all occupational exposures. A purpose-designed review examining ambient air pollution and other occupational exposures may help identify patterns of epigenetic challenges associated with distinct characteristics of the pollutants in these settings, and potentially provide a mechanistic bridge between those exposures and diseases common to those occupations. Second, we only assessed participants aged <65 years to minimise bias due to ageing-associated epigenetic changes. Future research in the elderly is needed, as they are more vulnerable to air pollution exposure and may exhibit different patterns of epigenetic alterations [104]. Finally, we could not perform meta-analyses due to the highly heterogeneous study methodologies and the use of different epigenetic endpoints.
CONCLUSIONS
We found that exposures to ambient air pollution, notably PM, were associated with locus-specific DNA methylation changes in adults. However, these changes need to be balanced against many studies that did not consistently cause epigenetic alterations. Although the number of studies was small for specific air-pollutant-epigenetic pairs, the observed epigenetic changes can be linked to epigenetic changes in genes regulating pathways related to cancers. Different patterns of gene-specific DNA methylation were induced by ambient air pollution and occupational exposure, while commonalities emerged across broad classes of air pollutants in both ambient and occupational studies. Future studies with larger sample sizes are needed to identify the robustness of air pollution induced blood- and tissue-specific CpG sites or regions among different populations and assess the potential biological response of epigenetic alterations between air pollution and specific health outcomes, beyond cancers and cardiorespiratory traits, considering the growing links between air pollution and diseases in other organ systems.
Additional material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Global Burden of Disease 2021 Forecasting Collaborators Burden of disease scenarios for 204 countries and territories, 2022-2050: a forecasting analysis for the Global Burden of Disease Study 2021. Lancet. 2024;403:2204–56. 10.1016/S 0140-6736(24)00685-838762325 PMC 11121021 · doi ↗ · pubmed ↗
- 2World Health Organization. Ambient (outdoor) air pollution. 2024. Available: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health. Accessed: 12 December 2025.
- 3Schraufnagel DE Balmes JR Cowl CT De Matteis S Jung SH Mortimer K Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 2: Air Pollution and Organ Systems. Chest. 2019;155:417–26. 10.1016/j.chest.2018.10.04130419237 PMC 6904854 · doi ↗ · pubmed ↗
- 4Gref A Merid SK Gruzieva O Ballereau S Becker A Bellander T Genome-Wide Interaction Analysis of Air Pollution Exposure and Childhood Asthma with Functional Follow-up. Am J Respir Crit Care Med. 2017;195:1373–83. 10.1164/rccm.201605-1026 OC 27901618 PMC 5443897 · doi ↗ · pubmed ↗
- 5Lee HT Oh S Ro DH Yoo H Kwon YW The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J Lipid Atheroscler. 2020;9:419–34. 10.12997/jla.2020.9.3.41933024734 PMC 7521974 · doi ↗ · pubmed ↗
- 6de F C Lichtenfels A Jvan der Plaat D Ade Jong Kvan Diemen CC Postma DS Nedeljkovic I Long-term Air Pollution Exposure, Genome-wide DNA Methylation and Lung Function in the Life Lines Cohort Study. Environ Health Perspect. 2018;126:027004. 10.1289/EHP 204529410382 PMC 6047358 · doi ↗ · pubmed ↗
- 7Mattei AL Bailly N Meissner ADNA methylation: a historical perspective. Trends Genet. 2022;38:676–707. 10.1016/j.tig.2022.03.01035504755 · doi ↗ · pubmed ↗
- 8Baccarelli A Bollati V Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–51. 10.1097/MOP.0b 013e 32832925 cc 19663042 PMC 3035853 · doi ↗ · pubmed ↗