Neurodevelopmental Disorder and Cortical Myoclonus in ZMYM2 Deficiency
Luca Pollini, Maria Novelli, Lorena Travaglini, Fabiola Panvino, Vincenzo Leuzzi, Francesco Pisani, Serena Galosi

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
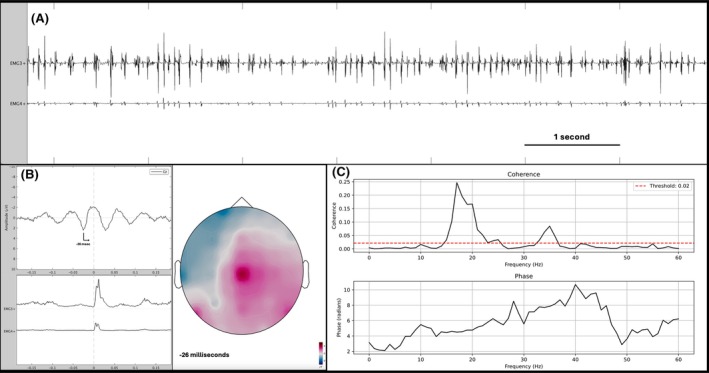 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetics and Neurodevelopmental Disorders · Congenital heart defects research · Genomics and Rare Diseases
ZMYM2 encodes a regulatory protein that functions as a transcriptional corepressor by interacting with several nuclear receptors. ZMYM2 autosomal dominant pathogenic variants have been associated with a neurodevelopmental‐craniofacial syndrome with variable renal and cardiac abnormalities.1, 2
In this letter, we describe the first case of ZMYM2 deficiency associated with prominent neurological phenotype and cortical myoclonus.
Our patient presented with developmental delay and stereotypies, prompting an initial diagnosis of autism spectrum disorder. Since the age of 3 years he suffered from generalized tonic‐clonic seizure and focal motor seizure. A diagnosis of mild intellectual disability was made at the age of 5 years.
He exhibited bilateral hand jerky movements since childhood. At age 14, he came to our attention because of a progressive spastic gait and sensitivity deficits in the lower limbs and abdomen caused by a thoracic spinal lesion from severe kyphoscoliosis, which required surgical intervention.
On examination at age 18, he had an asymmetrical spastic paraplegia and a high‐frequency jerky movement of the upper and lower limbs that were aggravated by outstretching and opening and closing his hands, and on feet dorsiflexion (Video S1). Jerks could be triggered by tactile stimulation of the hands.
A neurophysiological examination helped classify its involuntary movement as myoclonus because of the presence of short and arrhythmic electromyographic (EMG) bursts, synchronous between agonist and antagonist muscles. Jerk‐locked back‐averaging, corticomuscular coherence and phase analysis showed the cortical origin of myoclonus (Fig. 1).
A whole‐exome sequencing disclosed the presence of a novel de novo likely pathogenic truncating variant c.421C>T; p.Arg141Ter in ZMYM2.
The neurological phenotype associated with ZMYM2 deficiency is expanding rapidly, with new cases reported in the literature suggesting links with epilepsy, Rett‐like phenotypes, cerebral palsy mimics, and spastic diplegia.1, 2, 3 Unlike several previously reported cases, this patient showed no renal or cardiac involvement, but rather a predominantly neurological phenotype characterized by epilepsy and cortical myoclonus. Cortical myoclonus has not yet been associated with ZMYM2 deficiency, further broadening its neurological spectrum.
Cortical myoclonus is defined as a brief and sudden muscular activation arising from cortical areas. Although in many cases the distinction is straightforward, high‐frequency myoclonus can mimic a tremor, and neurophysiological analysis can aid the diagnosis,4 as observed in our patient. The distinction is crucial because myoclonus and tremor underpin different pathophysiological mechanisms and require different treatments.
Before the association of ZMYM2 deficiency with human disease was elucidated, tremor was reported in a boy with a neurodevelopmental disorder. This boy had a 2.1‐Mb deletion on chromosome 13q12.11 that included the ZMYM2 gene.5
However, no further neurophysiological study was performed to elucidate the nature of his movement disorders, and the deletion contained other genes in addition to ZMYM2.
In summary, we present a novel phenotype characterized by neurodevelopmental disorder, epilepsy, and movement disorder in ZMYM2 deficiency, suggesting that cortical myoclonus may be part of the ZMYM2‐related neurological spectrum.
Further characterization of additional patients is required to definitively include ZMYM2 on the list of genes responsible for myoclonic syndromes associated with neurodevelopmental disorders.
Ethical Compliance Statement
Written informed consent for offline and online video distribution of the video material was obtained from the patient and his caregiver and is available upon request.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution;
(2) Data Acquisition and Analysis: A. Acquisition, B. Analysis;
(3) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
L.P.: 1A, 1B, 1C, 2A, 2B, 3A, 3B.
M.N.: 1B, 1C, 2A, 3B.
L.T.: 2B, 3B.
F. Panvino: 2B, 3B.
V.L.: 2B, 3B.
F. Pisani: 2B, 3B.
S.G.: 1A, 1B, 1C, 3A, 3B.
Supporting information
Data S1. Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Connaughton DM , Dai R , Owen DJ , et al. Mutations of the transcriptional corepressor ZMYM 2 cause syndromic urinary tract malformations. Am J Hum Genet 2020;107(4):727–742. 10.1016/j.ajhg.2020.08.013 32891193 PMC 7536580 · doi ↗ · pubmed ↗
- 2Politano D , Marazzi F , Scognamillo I , et al. A de novo ZMYM 2 gene variant associated to a Rett‐like phenotype: case report of a new phenotype and review of the literature. Brain Dev 2025;47:104351. 10.1016/j.braindev.2025.104351 40112685 · doi ↗ · pubmed ↗
- 3De Wachter M , Schoonjans AS , Weckhuysen S , et al. From diagnosis to treatment in genetic epilepsies: implementation of precision medicine in real‐world clinical practice. Eur J Paediatr Neurol 2024;48:46–60. 10.1016/j.ejpn.2023.11.003 38039826 · doi ↗ · pubmed ↗
- 4Schwingenschuh P , Van der Stouwe M , Pandey S , et al. Clinical neurophysiology for tremor: common questions in clinical practice. Parkinsonism Relat Disord 2025;130:107196. 10.1016/j.parkreldis.2024.107196 39627925 · doi ↗ · pubmed ↗
- 5Der Kaloustian VM , Russell L , Aradhya S , Richard G , Rosenblatt B , Melançon S . A de novo 2.1‐Mb deletion of 13q 12.11 in a child with developmental delay and minor dysmorphic features. Am J Med Genet A 2011;155A(10):2538–2542. 10.1002/ajmg.a.34198 22043489 · doi ↗ · pubmed ↗