The metallophosphoesterase Rv0805 regulates carbon flux and cell envelope homeostasis during growth of mycobacteria in propionate
Priyanka Biswas, Nishad Matange, Sintu Samanta, Vishwas Mishra, Gerald Larrouy-Maumus, Sandhya S. Visweswariah

TL;DR
This study shows that the enzyme Rv0805 helps mycobacteria process propionate, a byproduct of cholesterol, and maintain cell structure, which is crucial for their survival in the host.
Contribution
The study identifies Rv0805 as a key regulator linking propionate metabolism and cell envelope integrity in mycobacteria.
Findings
Loss of Rv0805 impairs propionate uptake and alters cell envelope lipid composition.
Rv0805 is essential for carbon flux through the methylcitrate cycle during propionate metabolism.
Vitamin B12 supplementation rescues growth by restoring metabolic balance in Rv0805-deficient mycobacteria.
Abstract
Mycobacterium tuberculosis relies on host-derived lipids, including cholesterol, for intracellular survival, generating propionyl-CoA—a metabolite that must be efficiently assimilated to prevent toxicity. The metallophosphoesterase Rv0805 is required for optimal growth on cholesterol, and an Rv0805 knockout strain exhibits impaired ability to colonize the murine lung. However, the mechanisms underlying the essential role of Rv0805 under host-relevant conditions remain unclear. The deletion of the rv0805 ortholog (bcg_0857) in Mycobacterium bovis BCG reveals that both its catalytic activity and membrane localization are essential for growth on propionate, a by-product of cholesterol metabolism. Loss of Rv0805 impaired propionate uptake, altered cell envelope lipid composition with an accumulation of methyl-branched lipids, and reduced carbon flux through the methylcitrate cycle,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
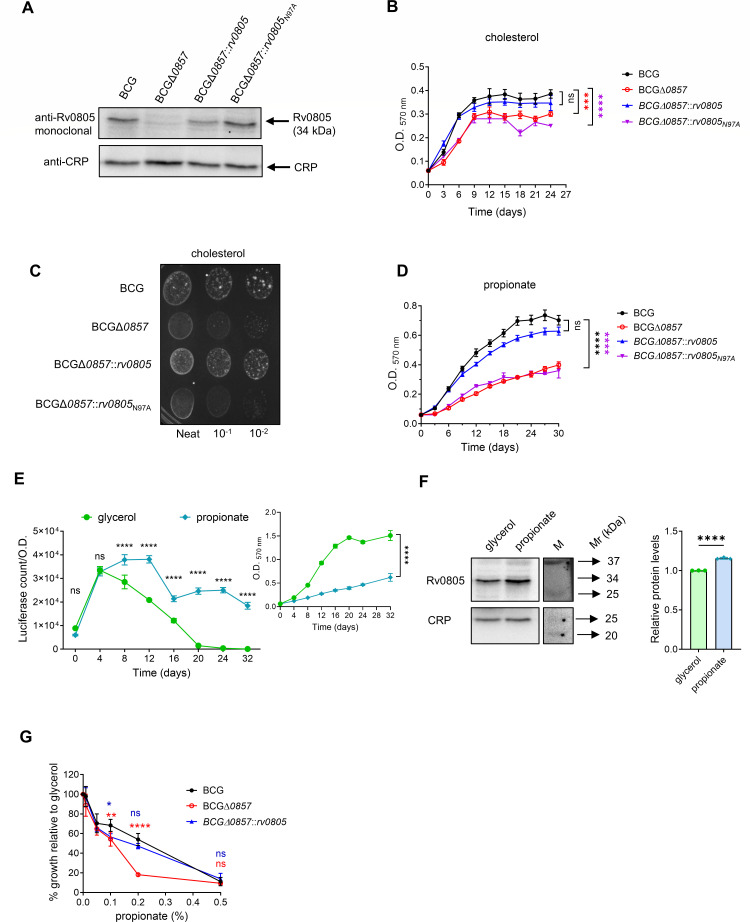 Fig 1
Fig 1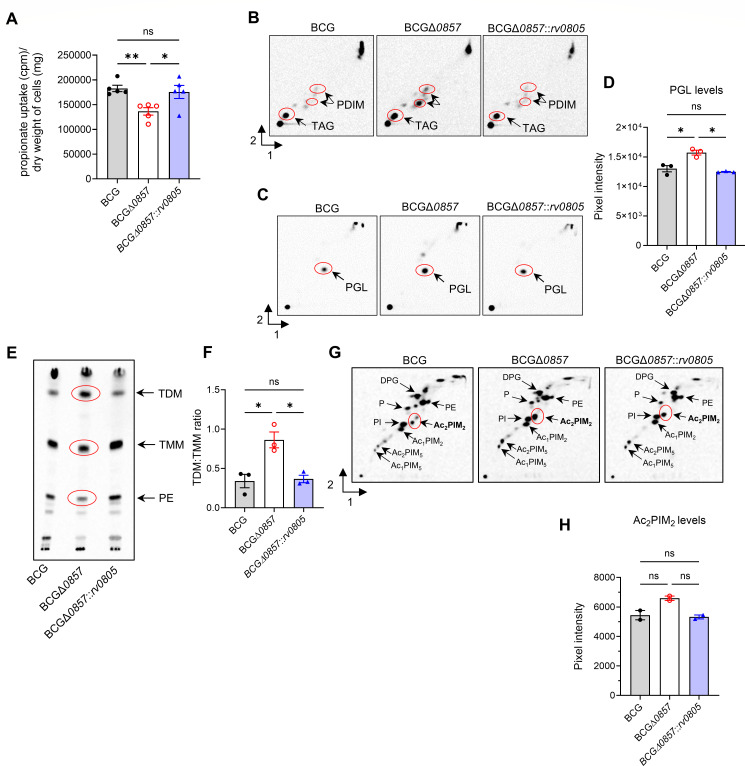 Fig 2
Fig 2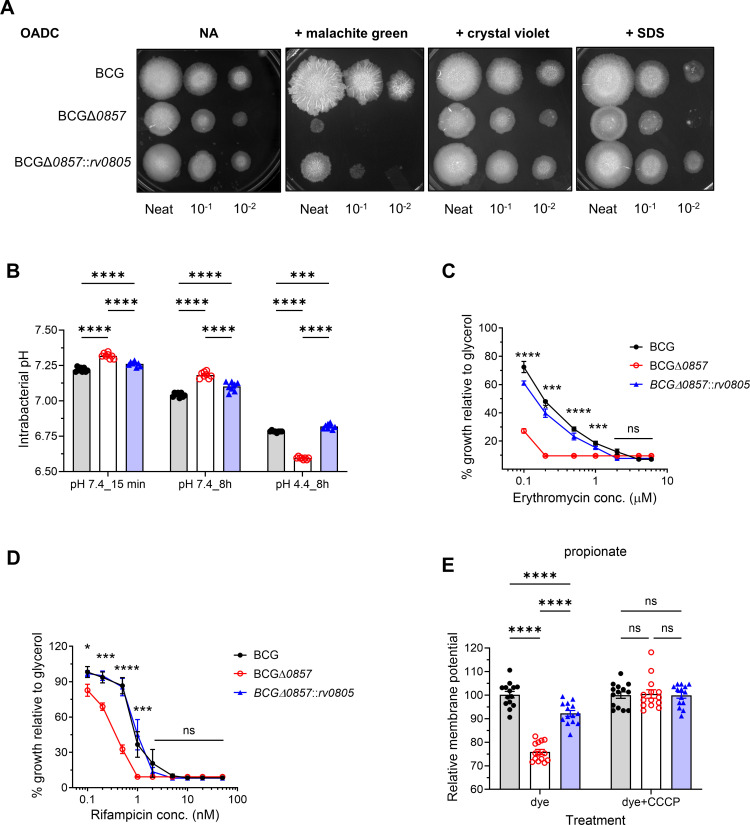 Fig 3
Fig 3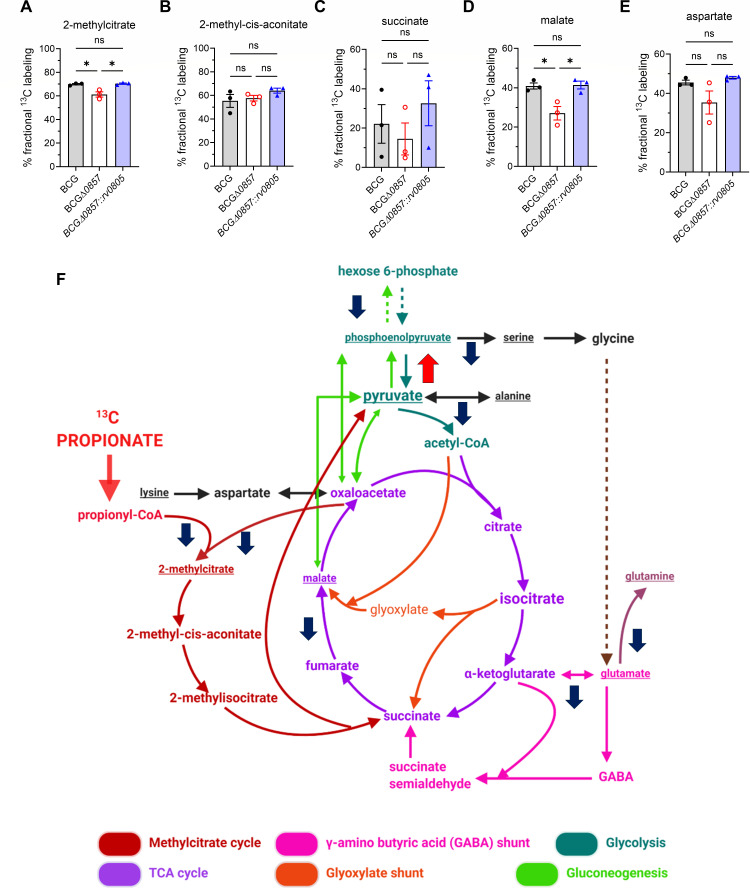 Fig 4
Fig 4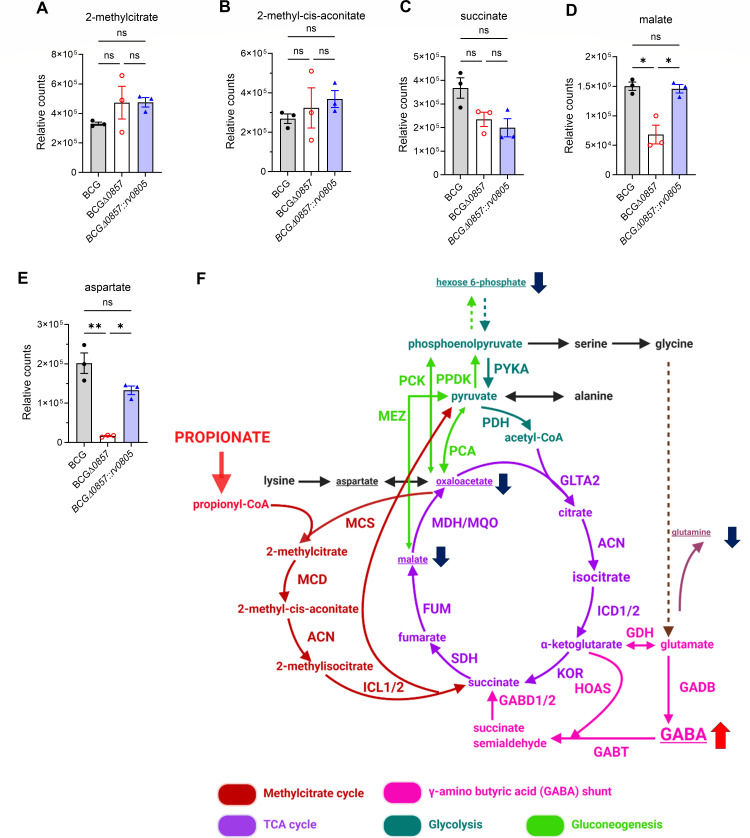 Fig 5
Fig 5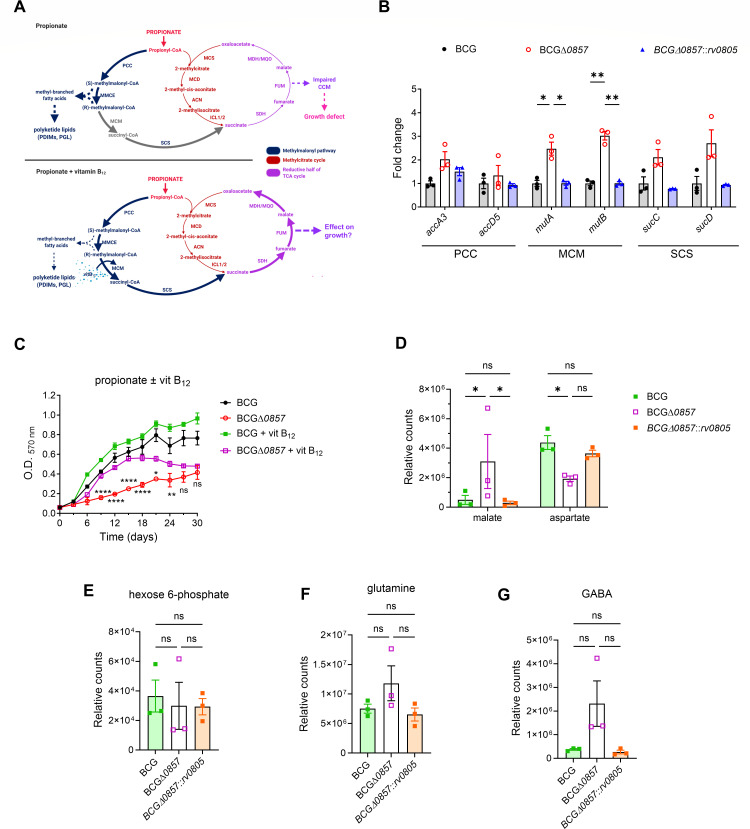 Fig 6
Fig 6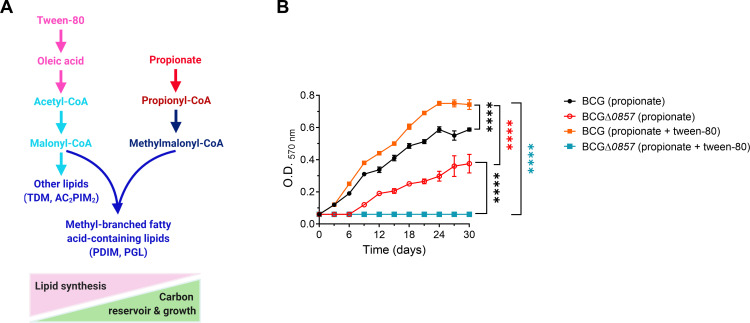 Fig 7
Fig 7| Strain | Description | Reference |
|---|---|---|
| BCG/BCG wild type | This study | |
| BCGΔ | ||
| BCGΔ | BCGΔ | |
| BCGΔ | BCGΔ | |
| BCGΔ | BCGΔ | |
| BCG_WT_ | ||
| BCG_WT_ |
| Medium | Composition |
|---|---|
| OADC | 7H9 base medium supplemented with 10% OADC (oleic acid-albumin-dextrose-catalase), 0.2% glycerol, and 0.05% Tween-80 |
| No carbon | 7H9 base medium supplemented with 0.025% Tyloxapol |
| Cholesterol | 7H9 base media supplemented with 250 μM cholesterol and 0.125% Tyloxapol as a vehicle of cholesterol |
| Glycerol | 7H9 base medium supplemented with 0.2% glycerol and 0.025% Tyloxapol |
| Propionate | 7H9 base medium supplemented with 0.2% sodium propionate and 0.025% Tyloxapol |
| OADC agar | 7H10 agar supplemented with 0.5% glycerol and 10% OADC (oleic acid-albumin-dextrose-catalase) |
| Cholesterol agar | 7H10 agar supplemented with 250 μM cholesterol and 0.125% Tyloxapol as a vehicle of cholesterol |
| Primer name | 5' to 3' sequence | Purpose |
|---|---|---|
|
| Screening of BCG and BCGΔ | |
|
| ||
| BCG |
| Screening of BCGΔ |
|
| Amplification of flanking region of ~1 kbp at the 5′ end of | |
|
| ||
|
| Amplification of flanking region of ~1 kbp at the 3′ end of | |
|
|
| Primer name | 5' to 3' sequence | Gene | BCG ortholog |
|---|---|---|---|
| 16S RT FWD |
|
|
|
| 16S RT REV |
| ||
| accA3 RT FWD |
|
|
|
| accA3 RT REV |
| ||
| accD5 RT FWD |
|
|
|
| accD5 RT REV |
| ||
| bcg_1605 RT FWD |
|
|
|
| bcg_1605 RT REV |
| ||
| frdA RT FWD |
|
|
|
| frdA RT REV |
| ||
| frdD RT FWD |
|
|
|
| frdD RT REV |
| ||
| fum RT FWD |
|
|
|
| fum RT REV |
| ||
| icl1 RT FWD |
|
|
|
| icl1 RT REV |
| ||
| icl2 RT FWD |
|
|
|
| icl2 RT REV |
| ||
| mdh RT FWD |
|
|
|
| mdh RT REV |
| ||
| mutA RT FWD |
|
|
|
| mutA RT REV |
| ||
| mutB RT FWD |
|
|
|
| mutB RT REV |
| ||
| mqo RT FWD |
|
|
|
| mqo RT REV |
| ||
| mez RT FWD |
|
|
|
| mez RT REV |
| ||
| pca_RT FWD |
|
|
|
| pca_RT REV |
| ||
| pckA_RT FWD |
|
|
|
| pckA_RT REV |
| ||
| ppdk_RT FWD |
|
|
|
| ppdk_RT REV |
| ||
| prpC RT FWD |
|
|
|
| prpC RT REV |
| ||
| prpD RT FWD |
|
|
|
| prpD RT REV |
| ||
| prpR RT FWD |
|
|
|
| prpR RT REV |
| ||
| rv0247c RT FWD |
|
|
|
| rv0247c RT REV |
| ||
| rv0248c RT FWD |
|
|
|
| rv0248c RT REV |
| ||
| rv0249c RT FWD |
|
|
|
| rv0249c RT REV |
| ||
| sdhA RT FWD |
|
|
|
| sdhA RT REV |
| ||
| sdhB RT FWD |
|
|
|
| sdhB RT REV |
| ||
| sdhC RT FWD |
|
|
|
| sdhC RT REV |
| ||
| sdhD RT FWD |
|
|
|
| sdhD RT REV |
| ||
| sucC RT FWD |
|
|
|
| sucC RT REV |
| ||
| sucD RT FWD |
|
|
|
| sucD RT REV |
| ||
| rv0805 RT FWD |
|
|
|
| rv0805 RT REV |
|
| Solvent system | Run direction | Components |
|---|---|---|
| A | 1 | Petroleum ether:ethyl acetate = 96:4 (*3) |
| 2 | Petroleum ether:acetone = 80:20 | |
| B | 1 | Petroleum ether:ethyl acetate = 85:15 (*3) |
| 2 | Toluene:acetone = 85:15 | |
| D | 1 | Chloroform:methanol:water = 100:20:2 |
| 2 | Chloroform:acetone:methanol:water = 50:60:2.5:3 | |
| E | 1 | Chloroform:methanol:water = 60:30:6 |
| 2 | Chloroform:acetic acid:methanol:water = 40:25:3:6 |
- —Department of Biotechnology, Ministry of Science and Technology, Indiahttp://dx.doi.org/10.13039/501100001407
- —Anusandhan National Research Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTuberculosis Research and Epidemiology · Infectious Diseases and Tuberculosis · Phosphodiesterase function and regulation
INTRODUCTION
Mycobacterium tuberculosis (Mtb) infection causes human tuberculosis which remains the leading cause of death with an estimated 1.25 million deaths and 10.8 million new infections in 2023 (1). Mtb inhabits the hostile environment of macrophage phagosomes, exposing them to low pH, hypoxia, reactive oxygen and nitrogen species, and nutrient limitation (2, 3). However, Mtb undergoes metabolic adaptations crucial for its survival and pathogenesis (4–6). Deciphering the metabolic strategies employed by Mtb is, therefore, imperative for identifying new antimycobacterial strategies.
Mycobacteria possess remarkable metabolic flexibility, which facilitates survival in intracellular environments by co-utilizing multiple carbon sources in a compartmentalized manner to support efficient growth (5, 7, 8). Importantly, Mtb shifts toward host-derived lipids, including fatty acids and cholesterol as major carbon substrates during infection (9–11). These substrates not only fuel central carbon metabolism (CCM) but also provide building blocks for virulence-associated cell envelope lipids, which modulate host immunity and support pathogenic persistence (9, 12).
Mtb utilizes cholesterol as a carbon source, which confers an evolutionary advantage in mammalian hosts, where an abundant supply of cholesterol is available (13). Cholesterol is broken down to yield four propionyl-CoA, four acetyl-CoA, one pyruvate, and one succinyl-CoA (14), which feed into CCM, fueling energy production, biosynthesis, and anaplerosis (10, 15–17). Mtb prominently produces propionyl-CoA from odd-chain fatty acids and branched-chain amino acids during infection. Propionyl-CoA is converted to succinate either via the vitamin B_12_-independent methylcitrate cycle (MCC) (13, 18, 19) or the vitamin B_12_-dependent methylmalonyl (MM) pathways (17, 20). In the absence of vitamin B_12_, MCC becomes the predominant route for anaplerosis from propionate as mycobacteria are unable to synthesize this cofactor (18, 21). However, recent evidence from murine and human infection models indicates that sufficient host-derived vitamin B_12_ is available in vivo to activate the MM pathway, rendering MCC largely dispensable during infection (22). Propionyl-CoA detoxification is also possible via conversion to methylmalonyl-CoA (MM-CoA), followed by incorporation of MM-CoA precursor molecules into the methyl-branched lipids. Thus, during infection, mycobacteria utilizing cholesterol harbor elevated levels of phthiocerol dimycocerosates (PDIMs), sulfolipid (SL), and phenolic glycolipids (PGLs) as excess propionyl-CoA derived from cholesterol is shunted toward the biogenesis of these methyl-branched lipids (20). Similarly, the methyl-branched acyl chains of PDIMs and SL become longer as they assimilate cholesterol or propionate. This metabolic flexibility around propionyl-CoA is central to Mtb’s ability to maintain efficient CCM and mitigate cholesterol-derived metabolic stress (9, 19).
In our earlier studies on the role of cAMP in mycobacteria, we identified Rv0805 as a cyclic nucleotide phosphodiesterase (23). Rv0805 is a member of the metallophosphoesterase family (24), and structural studies elucidated the conservation of the metallophosphoesterase fold and identified critical residues required for catalytic activity (25). Rv0805 orthologs are only found in the genomes of slow-growing and pathogenic mycobacteria (23). While Rv0805 does hydrolyze cAMP, it does so with low affinity and can exhibit phosphodiesterase (PDE) activity against other cyclic and linear substrates in vitro (23, 25, 26). The overexpression in Mtb leads to only modest (~30%) reduction in intracellular cAMP levels and triggers phenotypes that appear independent of its cAMP hydrolase activity (27). This functional promiscuity suggests broader physiological roles beyond the canonical cAMP signaling pathway.
Rv0805 adopts the conserved αββα-MPE fold with a binuclear Mn^2+^/Mg^2+^ active site coordinated by residues including Asp97 (N97), which is essential for PDE activity (23, 25, 26). Substitution of Asp97 with alanine (N97A) abolishes catalytic activity without affecting folding or localization. A unique C-terminal cap domain regulates access to the active site, affecting enzymatic stability, oligomerization, and membrane localization (23, 25, 26). The deletion of the final 40 residues (Rv0805Δ40) disrupts envelope localization and attenuates the enzyme’s physiological functions without affecting catalytic activity (26, 28). When expressed in Mycobacterium smegmatis (M. smegmatis), the cap domain modulates cell envelope permeability, altering susceptibility to hydrophobic cytotoxic compounds (26, 28).
In a study by Griffin et al., rv0805 was identified as one of the genes essential for optimum growth of Mtb on cholesterol (16, 29). An independent study showed that the deletion of rv0805 from Mtb reduced growth in cholesterol and colonization of the lungs in mice (30). Supplementation with vitamin B_12_ could rescue the growth defect in BCGΔrv0805 observed in propionate-containing media*,* and the report suggested that these changes were due to alterations in intracellular cAMP levels (30). However, the mechanistic link between Rv0805 and propionate metabolism, coupled with cell wall localization, remains unanswered.
Mycobacterium bovis bacille Calmette-Guérin (BCG), the only licensed vaccine against tuberculosis, is a live attenuated member of the M. tuberculosis complex (MTBC) and was derived from M. bovis through prolonged in vitro passage (31). BCG lacks several virulence-associated regions, including RD1 encoding the ESX-1 secretion system (32, 33). Despite these differences, BCG shares >99% genome sequence identity with Mtb and conserves core metabolic pathways essential for intracellular survival, making it a widely used and genetically tractable model for investigating mycobacterial physiology under non-Biosafety Level 3 (BSL-3) conditions (33, 34).
Here, we elucidate the mechanism underlying the role of Rv0805 in propionate metabolism. We show that Rv0805 promotes efficient propionate utilization by maintaining cell envelope lipid homeostasis, which is associated with altered carbon allocation at the propionyl-CoA “node” that intersects the MCC and MM pathways. These findings demonstrate that the loss of function of Rv0805 creates a metabolic vulnerability that could be exploited for the treatment of tuberculosis.
RESULTS
Rv0805 is critical for growth in propionate
To validate earlier reports demonstrating a requirement of Rv0805 during mycobacterial growth on propionate (30), we deleted bcg-0857 from wild-type Mycobacterium bovis BCG. Rv0805 from Mtb and its BCG ortholog BCG_0857 are 100% identical at both the nucleotide and amino acid levels, supporting functional conservation between two members of the M. tuberculosis complex. We generated complementation strains with wild-type rv0805, the catalytically inactive N97A, or the membrane localization-defective Rv0805Δ40 mutant expressed from the rv0805 native promoter integrated at the att site (Fig. S1A). Western blot analysis confirmed the deletion and the presence of the expected 34 kDa Rv0805/BCG_0857 protein in the complemented strains BCGΔ0857::rv0805 and BCGΔ0857::rv0805_N97A_ (Fig. 1A).
We assessed the growth of BCG strains on cholesterol and propionate as the sole carbon source, and as reported earlier (30), BCGΔ0857 displayed slower growth when either cholesterol or propionate was used as a carbon source (Fig. 1B through D). In contrast to the earlier published report (30), however, a significant growth defect was also observed when glycerol was used as a carbon source (Fig. S2A). Bcg_0857 promoter activity and protein expression were significantly higher in propionate-grown cells compared to glycerol-grown cells (Fig. 1E and F), indicating a requirement for Rv0805 protein in propionate-containing media. Propionate supplementation to glycerol-containing medium significantly impaired the growth of BCGΔ0857 (Fig. 1G), suggesting that toxicity is a result of the presence of propionate in the medium.
The BCGΔ0857::rv0805_N97A_ showed phenotypes similar to those of BCGΔ0857 (Fig. 1B through D; Fig. S2A) in cholesterol, glycerol, or propionate-containing media, confirming that catalytic activity is essential for Rv0805 function during growth in propionate. Complementation of BCGΔ0857 with a C-terminal truncation mutant lacking the final 40 residues (Rv0805Δ40) that are required for envelope association (28) did not restore growth (Fig. S2B), confirming earlier observations (30). In contrast, however, no significant differences in intracellular cAMP levels were detected, indicating that the growth defect was not dependent on cAMP signaling (Fig. S2C and D).
*Rv0805 is crucial for growth in propionate. (A) Representative Western blot showing the 34 kDa band for Rv0805/BCG_0857 present in BCG, BCGΔ0857::rv0805, BCGΔ0857::rv0805N97A, but not BCGΔ0857, grown in propionate (n = 2). C-reactive protein (CRP) was used as a normalization control. (B) Growth curves in cholesterol minimal media (n ≥ 4). (C) Growth analysis on cholesterol agar (n ≥ 4). (D) Growth curves in propionate minimal medium (n ≥ 4). (E) bcg_0857 promoter activity in modified wild-type BCG (BCG_WT_rv0805prom_luxA) grown in glycerol or propionate minimal media (n = 4). The inset shows the growth of the same strain in glycerol or propionate. (F) Representative Western blot showing the 34 kDa BCG_0857 protein in exponential phase BCG cultures grown in glycerol or propionate minimal media (n = 3) and its quantification. (G) Determination of sensitivity to propionate-induced toxicity: growth analysis in glycerol minimal media supplemented with increasing concentrations of propionate (n = 4). For B, D–G, results show mean ± SEM from biological replicates. Two-way ANOVA determined statistical significance with Dunnett’s multiple comparison test for B and D; by two-way ANOVA with Sidak’s multiple comparison test for E and G; and by two-tailed unpaired t-test for F. *P < 0.05; **P < 0.01; ***P < 0.001; ***P < 0.0001; ns, not significant.
Together, these data suggest that Rv0805 plays a crucial role during the growth of Mycobacterium during cholesterol metabolism and propionate detoxification.
Deletion of bcg_0857 affects propionate uptake via altering cell wall lipid levels
Rv0805 localizes and interacts with the cell membrane and cell wall of mycobacteria (26, 28). We, therefore, asked if the growth defect observed was a direct consequence of altered cell membrane properties and nutrient uptake. Indeed, ^14^C-propionate uptake was significantly lower in BCGΔ0857 than in BCG or BCGΔ0857::rv0805 (Fig. 2A). We next analyzed polar and apolar lipids in propionate-grown cells. Despite reduced propionate uptake, BCGΔ0857 accumulated higher levels of PDIM and PGL that are derivatives of propionyl-CoA and methylmalonyl-CoA than the wild type or complemented strain (Fig. 2B through D). BCGΔ0857 also displayed an altered TDM/TMM ratio and elevated Ac_2_PIM_2_ levels (Fig. 2E through H), indicating envelope lipid remodeling.
*Loss of Rv085 impairs propionate uptake and is associated with altered cell envelope composition. (A) 14C-propionate uptake in exponential phase propionate-grown cultures (n ≥ 4). Representative TLC shows polar/apolar lipid analysis in propionate-grown and 1,2-14C sodium acetate radiolabeled exponential-phase cultures (n = 3). Accumulation of (B) PDIM and (C) PGL levels, both derived from propionyl-/methylmalonyl-CoA (n = 3). (D) Quantification of PGL levels. Levels of (E) apolar TDM, TMM, and PE, and (G) polar lipids (n = 3). Quantification of (F) TDM: TMM ratio, and (H) Ac2PIM2 levels. Marked in red are lipids of different intensity among strains. For A, D, F, and H, results show mean ± SEM from biological replicates, and statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. *P < 0.05; *P < 0.01; ns, not significant. TLC, thin-layer chromatography; PDIM, phthiocerol dimycocerosate; PGL, phenolic glycolipid; TDM, trehalose dimycolate; TMM, trehalose monomycolate; PE, phosphatidylethanolamine; DPG, diphosphatidyl glycerol; PIM, phosphatidylinositol mannosides (integers denote number of mannoside or acyl [Ac] groups); PI, phosphatidylinositol; P, phospholipid.
To assess the functional consequences of altered lipid composition, we tested the susceptibility of these strains to cell wall-perturbing agents, investigated intracellular pH as an indicator of membrane homeostasis, analyzed their sensitivity to lipophilic antibiotics, and estimated the membrane potential in these strains. BCGΔ0857 showed a higher sensitivity to cell wall-perturbing agents than BCG and BCGΔ0857::rv0805 (Fig. 3A). Furthermore, BCGΔ0857 was poorer in maintaining intracellular pH (Fig. 3B). Interestingly, BCGΔ0857 was more sensitive to the lipophilic antibiotics, erythromycin and rifampicin (Fig. 3C and D). Finally, BCGΔ0857 exhibited a reduced membrane potential when grown in propionate medium (Fig. 3E). Together, these results suggest a role for Rv0805 in maintaining an optimal lipid profile that preserves membrane integrity, a role that has been previously underappreciated.
*Loss of Rv0805 induces cell envelope defects. (A) Representative images of spotting assays on OADC agar ± cell wall-perturbing agents (malachite green, crystal violet, SDS) (n ≥ 4). (B) Intrabacterial pH of glycerol-grown exponential phase cultures exposed to neutral (pH 7.4) or acidic (pH 4.4) conditions for indicated timepoints (n = 8). (C and D) Strains were grown in glycerol ± lipophilic antibiotics (erythromycin, rifampicin) (n ≥ 2). (E) Membrane potential of propionate-grown exponential phase cultures (n ≥ 7). CCCP was used as a control. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparison test for B–D, and by one-way ANOVA with Tukey’s multiple comparison test for F. *P < 0.05; ***P < 0.001; ***P < 0.0001; ns, not significant. CCCP, carbonyl cyanide m-chlorophenyl hydrazone.
Deletion of bcg_0857 affects CCM via disrupting carbon flux from propionate
We hypothesized that the reduced propionate uptake and higher PDIM and PGL of the BCGΔ0857 strain could be due to an altered metabolic flux of propionate. Propionate catabolism converges on CCM via the MCC and MM pathway. ^13^C tracing with [U-^13^C] propionate revealed lower incorporation of ^13^C into 2-methylcitrate and malate in BCGΔ0857, while the labeling of 2-methyl-cis-aconitate, succinate, and aspartate remained unchanged (Fig. 4A through F). Similarly, lower ^13^C enrichment was also observed in phosphoenolpyruvate, serine, alanine, glutamate, and glutamine with no differences in glycine, trans-aconitate, isocitrate, and α-ketoglutarate (Fig. S3A through D, F through J). Interestingly, a modest increase in pyruvate labeling, which remained at very low fractional abundance (~1%–3%) (Fig. S3E; Fig. 4F) was seen. Consistent with the increased ^13^C labeling of pyruvate, the anaplerotic node genes (35) pca, ppdk, and pckA were upregulated in BCGΔ0857 compared to BCG, while mez expression remained unchanged (Fig. S3K). These data indicate that the deletion of bcg_0857 reduces the flux of carbon from propionate into the MCC, impairing CCM.
*Absence of Rv0805 alters carbon flux into CCM. Fractional 13C-labeling in (A) 2-methylcitrate, (B) 2-methyl-cis-aconitate, (C) succinate, (D) malate, and (E) aspartate from [U-13C] propionate (n = 3). Results show mean ± SEM from biological replicates. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test, P < 0.05; ns, not significant. (F) Visual representation of impaired 13C incorporation into key CCM intermediates. Blue-downward and red-upward arrows indicate lower and higher 13C fractional labeling, respectively.
To determine whether the reduced flux to MCC was a consequence of dysregulated expression of enzymes involved in the pathway, we performed quantitative real-time PCR (qRT-PCR) analysis of the MCC pathway genes. As expected, MCC pathway genes (prpC, prpD, icl1, icl2) and their regulator prpR (36) were upregulated in both BCG and BCGΔ0857 in propionate compared to glycerol (Fig. S4A through E). No significant differences in the expression levels of prpC, icl1, and prpR were observed across the strains (Fig. S4A, C and E). However, prpD was significantly downregulated, and icl2 was upregulated in BCGΔ0857 (Fig. S4B and D). Similarly, other MCC-associated genes encoding SDH1 subunits (rv0247c, rv0248c, and rv0249c), fumarate reductase subunits (frdABCD), SDH subunits (sdhABCD), fumarase (fum), malate: quinone oxidoreductase (mqo), and malate dehydrogenase (mdh) were expressed at similar or higher levels in BCGΔ0857 than in BCG (Fig. S4F). Although prpD expression was reduced and icl2 expression increased in BCGΔ0857, these opposing transcriptional changes are unlikely to explain the reduced carbon flux through the MCC. Reduced prpD expression would be expected to constrain early steps of the pathway, while increased icl2 expression may reflect a compensatory response. Consistent with reduced ^13^C incorporation into MCC intermediates, the data indicate that impaired propionate assimilation is not primarily driven by altered MCC gene expression.
To further understand the consequences of the reduced flux into CCM, we profiled metabolites of the MCC. Levels of 2-methylcitrate, 2-methyl-cis-aconitate, and succinate were comparable across strains (Fig. 5A through C); however, BCGΔ0857 exhibited ~7-fold lower malate and ~36-fold lower aspartate (a proxy for oxaloacetate) (Fig. 5D through F) relative to BCG and BCGΔ0857::rv0805. Moreover, while no differences were observed in the levels of other central carbon metabolites—glycine, pyruvate, alanine, aconitate, α-ketoglutarate, and glutamate—BCGΔ0857 exhibited significantly lower hexose 6-phosphate and glutamine levels, and higher GABA levels than BCG (Fig. S5A through I; Fig. 5F), indicating an altered and impaired CCM.
*Loss of Rv0805 alters CCM metabolite levels. Steady-state metabolite levels of (A) 2-methylcitrate, (B) 2-methyl-cis-aconitate, (C) succinate, (D) malate, and (E) aspartate in propionate-grown cultures (n = 3). (F) Visual representation of impaired CCM in propionate-grown BCGΔ0857. Blue down arrows indicate metabolites (underlined) with lower levels, and red up arrows indicate metabolites (underlined) with increased levels in BCGΔ0857. For A–E, results show mean ± SEM from biological replicates, and statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test, *P < 0.05; *P < 0.01; ns, not significant.
Vitamin B12 rescues the growth defect by activating the MM pathway
The results shown above suggested that propionate is shuttled into lipid anabolism in the BCGΔ0857 strain. We, therefore, hypothesized that the activation of the MM pathway may shuttle propionate back into the MCC pathway via the enzymatic activity of MCM and SCS (Fig. 6A). To confirm this, we first verified the expression of key enzymes involved in the MM pathway. Genes encoding propionyl-CoA carboxylase (PCC genes accA3 and accD5) and succinyl-CoA synthetase (SCS genes sucC and sucD) subunits were expressed at similar levels in all strains, whereas the expression of mutA and mutB encoding methylmalonyl-CoA mutase (MCM) subunits was higher in BCGΔ0857 than BCG and BCGΔ0857::rv0805 (Fig. 6B). Supplementation of the growth media with Vitamin B_12_ rescued the growth defect of BCGΔ0857 in propionate medium (Fig. 6C), and levels of the malate (Fig. 6D) and normalized the levels of other central carbon metabolites (Fig. 6E through G). However, aspartate levels were still lower in BCGΔ0857 than in BCG (Fig. 6D). Together, this suggests that the absence of Rv0805 results in improper carbon rationing at the propionyl-CoA node, which can be rescued on vitamin B_12_ supplementation by redirecting the carbon back from MM-CoA to CCM.
*Vitamin B12 supplementation rescues CCM metabolite levels and growth defect in propionate. (A) Propionate metabolism with or without vitamin B12 supplementation. (B) QRT-PCR analysis of MM pathway genes in exponential phase cultures grown in propionate minimal medium (n = 3). 16s was used as the reference gene. (C) Growth curves in propionate minimal medium with or without 10 μg/mL vitamin (vit) B12 supplementation (n = 4). Rescued steady-state metabolite levels of (D) malate and aspartate, (E) hexose 6-phosphate, (F) glutamine, and (G) GABA in propionate media supplemented with vitamin B12 (n = 3). For B–G, results are presented as mean ± SEM from biological replicates. For B, D–G, statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. In C, statistical significance for the growth of BCGΔ0857 between propionate and propionate ± vit B12 was determined by two-way ANOVA with Tukey’s multiple comparison test. In B, asterisks are used only for comparisons where the P-value is less than 0.05. *P < 0.05; **P < 0.01; ***P < 0.0001; ns, not significant.
Loss of bcg_0857-mediated modulation of methyl-branched lipid synthesis diminishes carbon flow toward CCM
A higher conversion of propionyl-CoA to (S)- and (R)-methylmalonyl-CoA could lead to increased synthesis of methyl-branched fatty acid-containing polyketide lipids, such as PDIMs and PGLs, potentially compromising CCM and impairing the growth of strains lacking Rv0805 (Fig. 6A). To test this, we sought to perturb carbon allocation further and analyze its effect on growth. We grew the BCGΔ0857 strain in propionate-containing medium, supplemented with 0.05% Tween-80. Tween-80 is catabolized to form oleic acid, ultimately generating acetyl-CoA that can be converted to malonyl-CoA (37) (Fig. 7A). The addition of Tween-80 and consequent elevation of malonyl-CoA pools could increase the conversion of propionyl-CoA to methylmalonyl-CoA and deplete the flux of carbon toward MCC and, thus, restrict the growth of BCGΔ0857 in propionate-containing media. As expected, BCGΔ0857 failed to grow in propionate + Tween-80 media. In contrast, the wild-type strain exhibited higher growth (Fig. 7B). Overall, our results indicate that a balance exists between lipid synthesis and central carbon metabolism for growth, which is regulated by the catalytic activity and membrane localization of Rv0805.
*The addition of Tween-80 exacerbates the growth defect in propionate. (A) Probable effect of Tween-80 on the growth of BCGΔ0857 in propionate. Tween-80 is catabolized as oleic acid, which is further converted into acetyl-CoA. Supplementation of Tween-80 in propionate-grown BCGΔ0857 may lead to even higher synthesis of methyl-branched as well as other cell wall lipids in BCGΔ0857. This may replace more methylmalonyl-CoA from the propionyl-CoA node and reduce carbon flux towards CCM, adversely affecting the growth of BCGΔ0857 on propionate. (B) Growth curves in propionate media supplemented with 0.05% Tween-80 (n = 4), using propionate minimal media as control. Results show mean ± SEM from biological replicates, and statistical significance was determined by two-way ANOVA with Tukey’s multiple comparison test. ***P < 0.0001.
DISCUSSION
The ability of mycobacteria to thrive within the lipid-rich but nutrient-limited environment of the host is underpinned by precise coordination between carbon metabolism and cell envelope composition. In this study, we identify the cell envelope-associated metallophosphoesterase BCG_0857/Rv0805 as a factor involved in this coordination during growth on propionate, an intermediate of host cholesterol catabolism. Loss of BCG_0857 not only perturbs lipid homeostasis but also causes a metabolic bottleneck at the malate/aspartate node, highlighting how structural changes at the cell surface reverberate into central carbon metabolism. These findings define a mechanistic link between envelope architecture and carbon flux control in mycobacteria.
The phenotype of the bcg_0857 deletion mutant parallels features observed in M. tuberculosis Δicl1/2, one of the most attenuated strains of Mtb and a classic model for impaired propionate utilization (13, 18, 38, 39). Like Δicl1/2, BCGΔ0857 exhibits a pronounced growth defect in propionate and a depletion of critical CCM metabolites, accompanied by impaired membrane potential. However, unlike Δicl1/2, where isocitrate lyase activity is absent and toxic MCC intermediates accumulate downstream of propionyl-CoA, BCGΔ0857 retains ICL activity and does not accumulate these intermediates. Instead, the absence of BCG_0857 leads to an excessive diversion of propionyl-CoA into methylmalonyl-CoA and onward into the synthesis of methyl-branched polyketide lipids, resulting in elevated PDIM and PGL. This pattern is consistent with prior observations that mycobacteria couple cholesterol metabolism through excess propionyl-CoA pools to increased PDIM/PGL production and the formation of longer methyl-branched acyl chains (18, 20). In BCGΔ0857, this diversion of propionyl-CoA toward methyl-branched lipid synthesis appears to occur at the expense of MCC flux, resulting in reduced ^13^C-carbon incorporation into 2-methylcitrate, depletion of malate and oxaloacetate, and lowered hexose-6-phosphate steady-state levels, ultimately impairing gluconeogenesis and biomass production.
Although a modest increase in ^13^C labeling of pyruvate and transcriptional induction of pckA, ppdk, pca, and icl2 was observed in BCGΔ0857, fractional ^13^C enrichment remained low in all strains (~1%–3%). Absolute pyruvate levels were unchanged, and carbon flux into downstream anaplerotic outputs, including phosphoenolpyruvate (PEP), was not restored in BCGΔ0857. These changes, therefore, reflect a compensatory remodeling of the PEP-pyruvate-oxaloacetate or anaplerotic node rather than productive rerouting of the propionate-derived carbon, reinforcing that impaired MCC flux is the primary driver of defective anaplerosis and gluconeogenesis in BCGΔ0857.
BCGΔ0857 also showed reduced prpD and increased icl2 expression (Fig. S4F). However, reduced ^13^C incorporation into MCC intermediates and depletion of malate and oxaloacetate indicate that this altered gene expression was insufficient to restore flux, and impaired upstream carbon partitioning was the primary defect.
Proper gluconeogenesis is essential for infection, intracellular survival, and the biosynthesis of vitamins, lipids, nucleotides, reducing equivalents, and structural components needed for cell division (40). In BCGΔ0857, reduced propionate assimilation not only limits these anabolic processes but also compromises membrane potential—an equally critical determinant of active replication and long-term viability (41). The excess accumulation of PDIM and PGL in BCGΔ0857 may also have consequences for host–pathogen interactions (42–44). PDIM has been shown to spread into host epithelial membranes and promote infectivity (45). Thus, elevated PDIM levels in BCGΔ0857 could translate into altered infectivity in vivo, highlighting another way in which Rv0805-mediated carbon partitioning may shape both bacterial physiology and host interactions. Phosphatidylmyo-inositol Mannosides (PIMs) are key regulators of mycobacterial envelope integrity and permeability. Disruption of PIM biosynthesis alters the uptake of lipophilic compounds, such as norfloxacin and chenodeoxycholate (46, 47). Thus, the increased Ac_2_PIM_2_ levels in BCGΔ0857 likely contribute to its altered permeability to lipophilic agents.
An example of another PDE influencing cell wall physiology is Rv1339, whose overexpression in M. smegmatis lowers cAMP, perturbs peptidoglycan precursor pools, and compromises envelope integrity (48). Although the mechanistic link to cAMP signaling remains correlative, this underscores how cyclic-nucleotide-modulating enzymes can broadly affect cell envelope homeostasis. Given the altered envelope composition of the BCGΔ0857 strain and the requirement for Rv0805 membrane localization for optimal growth, Rv0805 may likewise regulate envelope-associated processes beyond cAMP hydrolysis. Indeed, despite previous reports linking Rv0805 to altered cAMP turnover (30), we did not detect significant changes in either extracellular or intracellular cAMP levels between the wild type and BCGΔ0857 in propionate. These findings suggest that Rv0805 acts through an alternative or membrane-proximal substrate, integrating its catalytic and membrane localization-dependent functions to coordinate envelope lipid balance with metabolic regulation during propionate utilization.
Notably, BCGΔ0857 also displayed a growth defect on glycerol, suggesting Rv0805 regulates carbon partitioning beyond propionate metabolism. During growth on glycerol and Tween-80, mycobacteria can operate a reverse MCC to supply propionyl-CoA for virulence lipid synthesis (14). In the absence of BCG_0857, this reverse routing may be enhanced, diverting more glycerol-derived carbon into methyl-branched lipid synthesis at the expense of CCM and biomass production.
The growth defect of BCGΔ0857 in propionate was further exacerbated when supplemented with Tween-80 (Fig. 7A). Tween-80 supplementation increases acetyl-CoA availability through fatty acid metabolism, which in the context of BCGΔ0857 likely intensifies the diversion of propionyl-CoA and methylmalonyl-CoA toward lipid biosynthesis at the expense of central carbon flux, thereby halting growth (Fig. 7A). This contrasts with the Mtb Δicl1 mutant, where acetate supplementation alleviates propionate toxicity by promoting lipid synthesis and thereby reducing accumulation of toxic MCC intermediates (20). In BCGΔ0857, where ICL activity is retained, growth impairment arises from carbon diversion into lipids rather than MCC toxicity. Consistent with our findings, mutations in the pta-ackA operon, that limit acetyl-CoA production and deplete acetyl-CoA pools, alleviate the propionate growth defect of BCGΔRv0805 (30). Together, these findings underscore the tight control of propionyl-CoA partitioning and reveal Rv0805 as a key regulator that prevents excessive lipid commitment, which can compromise metabolic fitness.
Vitamin B_12_ supplementation rescues the propionate growth defect in both Δicl1/2 (4) and BCGΔ0857, but via distinct routes. In Δicl1/2, vitamin B_12_-driven activation of the MM pathway bypasses the MCC block, reducing accumulation of toxic 2-methylcitrate/2-methylisocitrate and replenishing CCM intermediates. In BCGΔ0857, where MCC flux is reduced rather than blocked, vitamin B_12_ enables methylmalonyl-CoA mutase (MCM) to convert methylmalonyl-CoA into succinyl-CoA, shunting carbon back into the CCM (Fig. 6A). This effect is reinforced by higher mutAB and sucCD (encoding MCM and SCS subunits) expression in BCGΔ0857, which likely supports the observed recovery of malate and other metabolites. While aspartate (proxy for oxaloacetate) levels remain low after vitamin B_12_ supplementation, restoring malate and downstream gluconeogenic intermediates is sufficient to reestablish growth. Oxaloacetate is rapidly consumed in gluconeogenesis, transamination, and condensation reactions (6), so its steady-state levels may remain low despite restored anaplerotic flux. Supporting this, elevated pca, pckA, and ppdk expression in BCGΔ0857 suggests increased turnover rather than accumulation (Fig. 5F). Thus, vitamin B_12_ restores effective carbon flux without fully normalizing oxaloacetate-derived pools.
The ability of Mtb rv0805 to restore growth and metabolic phenotypes in BCGΔ0857 demonstrates functional conservation of Rv0805 across MTBC species, supporting the use of BCG as a valid and informative model for dissecting its metabolic role. However, while this study suggests that Rv0805-mediated regulation of carbon partitioning may influence host-pathogen interactions, it is important to note that these experiments were conducted in BCG, which is attenuated due to loss of several virulence-associated regions, including ESX-1 (31–33). Therefore, direct extrapolation to Mtb pathogenesis should be made with caution, and future studies using Mtb infection models are required to determine how Rv0805-dependent control of propionyl-CoA flux and cell envelope composition impacts virulence, persistence, and immune modulation in vivo.
Our findings, therefore, suggest that BCG_0857/Rv0805 is involved in both cell envelope lipid homeostasis and carbon routing at the propionyl-CoA node. The parallels with the highly attenuated Δicl1/2 strain further support a role for Rv0805 in intracellular survival (30) and perhaps persistence, a hypothesis that warrants direct in vivo testing. Beyond its contribution to propionate utilization, BCG_0857 may act as a general integrator of envelope architecture and metabolic adaptability. The fact that vitamin B_12_ supplementation can bypass the BCGΔ0857 defect underscores the metabolic plasticity of mycobacteria. It raises the possibility of targeting propionyl-CoA partitioning or cofactor availability to disrupt this balance. Such interventions could represent novel strategies to weaken the metabolic and structural robustness that underlie mycobacterial pathogenesis.
MATERIALS AND METHODS
Mycobacterial strains and culture conditions
A list of all strains used in this study is provided in Table 1. BCG was cultured in OADC (oleic acid-albumin-dextrose-catalase) agar or OADC-glycerol media (Table 2) under static conditions at 37°C in a humidified incubator located in the bio-safety level 2 (BSL2) facility. Hygromycin B and kanamycin sulfate were included at a final concentration of 50 μg/mL and 10 μg/mL, respectively, wherever necessary.
Generation of BCGΔ0857 and complement strains
Upstream and downstream flanking regions of bcg_0857 were amplified using BCG genomic DNA with primers listed in Table 3 and cloned separately into pBKS, generating pBKS-bcg_0857 upstream and pBKS*-bcg_0857* downstream. The upstream and downstream flanking portions of bcg_0857 were then subcloned into p2NIL, generating p2NIL-bcgΔ0857. A marker cassette (~8 kb) was then cloned into this (by PacI digestion) to generate p2NIL-bcgΔ0857-Pac.
BCG was electroporated with p2NIL-bcgΔ0857-Pac and, after recovery, plated onto OADC agar containing cycloheximide (100 μg/mL), hygromycin, kanamycin, and X-gal (50 μg/mL). Following 4–5 weeks of incubation, blue colonies [putative “single-cross overs” (SCOs)] were confirmed by colony PCR using the primers bcg_0857fwd and bcg_0857downstreamrev (Table 3). SCOs were then streaked out onto OADC-glycerol agar (no antibiotics) to allow a second recombination event, generating either wild type or deleted bcg_0857 locus. Next, a scoopful of SCOs was plated onto OADC-glycerol agar supplemented with 2% sucrose. Only bacteria that have undergone a second crossover event would grow on sucrose-containing media. Colonies were screened for the deletion of the bcg_0857 gene by colony PCR using primers bcg_0857fwd and bcg_0857downstreamrev (Table 3). Complement strains were generated by introducing the rv0805 or rv0805_N97A_ gene, along with an upstream ~150 bp intergenic region (available in the laboratory), to serve as an endogenous promoter at the L5 att locus in the BCGΔ0857 genome (Fig. S1A). BCGΔ0857 was electroporated with pMV306-rv0805prom-rv0805/rv0805_N97A_/rv0805_Δ40_ and, after recovery, plated onto OADC agar containing kanamycin. Individual transformants were confirmed by PCR using BCG attB FWD and rv0805/bcg_0857 Nterm RT FWD, which yielded the expected 1.5 kbp PCR amplicon.
Growth analysis, spot assay, and susceptibility to propionate
For all experiments, BCG was grown in OADC-glycerol medium until the mid-exponential phase. For growth analysis, cells were inoculated in either glycerol or propionate minimal media (Table 2) in biological and experimental duplicates, to a starting optical density at 570 nm (O.D.) of 0.05, and incubated at 37 °C under static conditions. Culture was resuspended at specific time points, and bacterial growth was assessed by measuring optical density (O.D.) at 570 nm.
For spot assay, 200 µL O.D.570 nm = 1 were washed twice in assay buffer (phosphate-buffered saline [PBS] supplemented with 0.025% Tyloxapol), resuspended in the same buffer, and 5 µL spotted on cholesterol, OADC, or OADC agar supplemented with 10 µg/mL malachite green, 10 µg/mL crystal violet, or 0.005% SDS. The plates were incubated at 37°C for 16–25 days and imaged.
To monitor the effect of propionate toxicity, cells were inoculated in glycerol minimal media (Table 2) with increasing concentrations of sodium propionate. Cultures were seeded with a starting O.D. at 570 nm of 0.05 and incubated at 37°C under static conditions until the control culture (glycerol alone) reached the mid-exponential phase. Bacterial growth (O.D.570 nm) was measured, and relative growth compared to propionate-free media (glycerol alone) was determined.
To monitor susceptibility to ionophores and antibiotics, cells were inoculated in glycerol medium containing varying concentrations of a specific antibiotic and incubated at 37°C under static conditions until the control culture (glycerol alone) reached the mid-exponential phase. Bacterial growth (O.D.570 nm) was measured, and relative growth compared to ionophore or antibiotic-free media (glycerol alone) was estimated.
Preparation of mycobacterial cell lysate and Western blot analysis
For preparation of cell lysates, cells were harvested, washed with TBST buffer (10 mM Tris-Cl (pH 8.2), 0.9% NaCl, and 0.1% Tween-80), and lysed by bead beating against 0.5 mm diameter glass beads (BioSpec Products, USA) in buffer containing 50 mM Tris-Cl (pH 8.2), 100 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol (β-ME), 2 mM phenylmethyl sulfonyl fluoride (PMSF), and 1 mM benzamidine hydrochloride. The protein concentration of the lysates was estimated using the Bradford method.
Protein samples were electrophoresed on SDS (sodium dodecyl sulfate) PAGE polyacrylamide gel (12% acrylamide, 1.2% bis-acrylamide) and transferred to PVDF (polyvinylidene fluoride) membranes (Millipore, USA) in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol, pH 8.3) for 2 h at 200 mA. After transfer, the membrane was incubated in blocking agent for 1 h at 25°C, followed by incubation with primary antibody overnight at 4°C with gentle agitation. Rv0805-specific monoclonal antibody was used at a 1:50 dilution. The anti-CRP polyclonal antiserum was used at a dilution of 1:5,000. Next, membranes were washed thrice with TBST and incubated with anti-mouse IgG conjugated to horse-radish peroxidase (1:5,000) or anti-rabbit IgG conjugated to horse-radish peroxidase (1:25,000) for 1 h at 25°C. The membranes were washed three times with TBST, and the bound antibody was detected using Enhanced Chemiluminescence substrate (ECL Plus, GE Healthcare, UK).
Promoter activity and growth analysis
The bcg_0857 promoter (150 bp upstream region of bcg_0857) was cloned upstream of the luciferase reporter gene (luxAB) in the pMV1025 vector, and the BCG wild-type strain was electroporated with this construct. A control strain harboring pMV1025-no promoter-luxAB was also generated as a control to ascertain no expression of luxAB in the absence of any promoter (Table 1). BCG_WT_bcg_0857 prom_luxAB strain was grown in glycerol or propionate minimal media, and bcg_0857 promoter activity was monitored at specific time-points along with growth. For promoter activity analysis, 20 μL of the resuspended culture was diluted in 90 μL of TBST on a Luminance white plate, and 10 μL of 1% decanal (Sigma), prepared in 70% ethanol, was added. Luciferase activity was measured immediately using an Infinite M200 PRO (Tecan, Switzerland) luminometer and normalized to the growth rate.
Radioimmunoassay
Iodinated 2′-O-monosuccinyl adenosine-3′,5′-cyclic monophosphate tyrosyl methyl ester (2′-O-MS-cAMP-TME) (BRIT, India) was used for radioimmunoassay. Radioimmunoassay was carried out by setting up a competition between iodinated 2′-O-MS-cAMP-TME and unlabeled cAMP. The amount of cAMP-specific antiserum used was standardized so that 30%–50% of input iodinated 2′-O-MS-cAMP-TME (input ~25,000 cpm) bound to the antiserum in the absence of competing cAMP. The assay was set up in 50 mM sodium acetate buffer (pH 4.75) containing 5 mg/mL BSA in a total volume of 300 μL and allowed to equilibrate by overnight incubation at 4°C. Free cAMP was separated from antibody-bound cAMP by the addition of 1 mL activated charcoal (2 mg/mL) and BSA (1 mg/mL) in 50 mM potassium phosphate buffer, pH 6.3. Charcoal was then recovered by centrifugation at 4,000 × g for 20 min at 4°C. The supernatant was discarded, and the radioactivity associated with the charcoal pellets was measured using a γ-counter (PerkinElmer, USA). A standard curve, consisting of 12–15 known concentrations of cAMP, was used to calculate the concentrations of cAMP in the unknown samples.
For sample preparation, samples were diluted to appropriate amounts in 50 mM Na-acetate buffer (pH 4.75) and acetylated to increase the sensitivity of the radioimmunoassay. For acetylation, the samples were mixed with 0.02 volume of triethylamine by vortexing. This was followed by the rapid addition of 0.01 volume of acetic anhydride just below the surface of the sample solution and vortexing. The cAMP reference standard was acetylated in a similar manner. Appropriate amounts were used for radioimmunoassay.
For the preparation of intracellular cAMP measurement, approximately 200 µL of O.D. 1 equivalent culture was centrifuged and resuspended in a 200 µL volume of 0.1 N HCl. The sample was then boiled at 95°C for 10 min and stored at −70°C until measurement. For extracellular cAMP measurement, 0.1 volume (50 µL) of 1 N HCl was added to 450 µL of culture supernatant, boiled at 95°C for 10 min, and then stored at −70°C until measurement.
RNA isolation, cDNA preparation, and gene expression analysis by qRT-PCR
Mid-exponential-phase BCG cultures were centrifuged, resuspended in TRI Reagent (RNAiso Plus, TaKaRa, Japan), and lysed using 0.5-mm-diameter glass beads (BioSpec Products, USA). The lysate was then heated at 65°C for 5 min and centrifuged at 16,000 × g for 10 min at 4°C. The supernatant was collected and mixed with chloroform, followed by centrifugation at 16,000 × g for 15 min at 4°C. The upper aqueous phase was collected, and RNA was precipitated with isopropyl alcohol. The RNA pellet was washed with 75% ethanol, dissolved in RNase-free Milli-Q water, and treated with RNase-free DNase (Thermo Scientific). Two micrograms of RNA was used for reverse transcription using 200 units of reverse transcriptase (Thermo Scientific, USA). Sequences of primers used to study the transcript level of different genes are shown in Table 4. Real-time qRTPCR was performed using SYBR Premix Ex Taq (Tli RNase H Plus) on a CFX96 Touch real-time PCR detection system (Bio-Rad, USA). The housekeeping gene 16S rRNA was used as an internal control for normalization.
Metabolite extraction, liquid chromatography-mass spectrometry
Strains were cultured on OADC-glycerol agar. BCG was initially grown in 7H9 media supplemented with 0.2% glucose, 0.5% fraction V BSA (HiMedia, India), 0.085% NaCl, and 0.05% Tyloxapol until the late exponential phase and then inoculated onto 22 mm diameter, 0.22 μm pore size nitrocellulose filters under vacuum filtration. Laden filters were then placed on top of chemically equivalent agar media (described above) and allowed to grow at 37°C for 5 days. Filters were then transferred for 24 h into 7H10 plates supplemented with 0.5% fraction V BSA, 0.05% Tyloxapol, 0.085% NaCl, containing 0.2% propionate, 0.2% propionate supplemented with 10 μg/mL of vitamin B_12_, or 0.2% [U-^13^C]-propionate. Bacteria were metabolically quenched by plunging cell-laden filters into acetonitrile/methanol/H_2_O (2:2:1, by vol.), pre-cooled to −40°C, and metabolites were extracted by mechanical lysing of the entire bacteria-containing solution with 0.1 mm acid-washed Zirconia beads (Sigma). Lysates were clarified by centrifugation and then filtered through 0.22 μm Spin-X column filters (Costar). The bacterial biomass of individual samples was determined by measuring the residual protein content of the metabolite extracts using the BCA Protein Assay Kit (Thermo Fisher Scientific).
Aqueous normal-phase liquid chromatography of the metabolites was performed using an Agilent 1290 Infinity II LC system equipped with a binary pump, temperature-controlled auto-sampler (set at 4°C), and temperature-controlled column compartment (set at 25°C), containing a Cogent Diamond Hydride Type C silica column (150 mm × 2.1 mm; dead volume 315 µL). A flow rate of 0.4 mL/min was used. Elution of polar metabolites was carried out using solvent A, consisting of deionized water (Resistivity ~18 MΩ cm), 0.2% acetic acid, and solvent B, composed of 0.2% acetic acid in acetonitrile. The following gradient was used: 0 min 85% B; 0–2 min 85% B; 3–5 min to 80% B; 6–7 min 75% B; 8–9 min 70% B; 10–11 min 50% B; 11.1–14 min 20% B; 14.1–25 min hold 20% B followed by a 5 min re-equilibration period at 85% B at a flow rate of 0.4 mL/min. Accurate mass spectrometry was carried out using an Agilent Accurate Mass 6545 QTOF apparatus. Dynamic mass axis calibration was achieved by continuous infusion, post-chromatography, of a reference mass solution using an isocratic pump connected to an ESI ionization source, operated in the positive-ion mode. Nozzle voltage and fragmentor voltages were set at 2,000 V and 100 V, respectively. The nebulizer pressure was set at 50 psig, and the nitrogen drying gas flow rate was set at 5 L/min. The drying gas temperature was maintained at 300°C. The MS acquisition rate was 1.5 spectra/s, and m/z data ranging from 50 to 1,200 were stored. This instrument enabled accurate mass spectral measurements with an error of less than 5 parts-per-million (ppm), a mass resolution ranging from 10,000 to 25,000 over the m/z range of 121–955 atomic mass units, and a 100,000-fold dynamic range with picomolar sensitivity. The data were collected in the centroid 4 GHz (extended dynamic range) mode. Detected m/z ratios were deemed to be identified metabolites based on unique accurate mass-retention time identifiers for masses exhibiting the expected distribution of accompanying isotopomers. The typical variation in abundance for most metabolites stayed between 5% and 10% under these experimental conditions. All non-labeled metabolomics data were analyzed using Agilent MassHunter Qualitative Analysis B.07.00. Targeted labeled metabolic data were performed using targeted features extractions using Profinder 8.0 (Agilent Technologies, USA), exported, and analyzed via Agilent VistaFlux software for data visualization.
As 2-methylcitrate and 2-methylisocitrate are structural isomers with identical exact masses and similar chromatographic behavior, the LC-MS platform used in this study cannot distinguish them without targeted analytical standards or MS/MS fragmentation (49). Accordingly, the measured signal represents the collective pool of methylcitrate and methylisocitrate (methyl[iso]citrate), which we refer to as methylcitrate for simplicity of presentation.
Measurement of membrane potential
Propionate-grown and mid-exponential phase BCG cultures were centrifuged and immediately resuspended in 7H9 base media supplemented with 0.2% glycerol, 0.2% glucose, 0.085% NaCl, and 0.025% Tyloxapol. Two hundred and fifty microliters of resuspended cells was incubated with 15 µM of the fluorescent membrane-permeable dye 3,3′-diethyloxicarbocyanine chloride (DiOC_2_, Sigma) for 20 min at 25 °C in the dark. Cells were washed with 1 mL of sterile 1× PBS and resuspended in 250 μL of sterile 7H9 base medium. One hundred microliters in duplicates was aliquoted on a Costar black 96-well flat-bottom plate (Costar), and fluorescence was monitored by following excitation/emission at green 488/530 nm and red 488/610 nm using an Infinite M200 PRO (Tecan, Switzerland). The membrane potential was calculated as a ratio of red to green fluorescence. The positive control for membrane depolarization was achieved by incubating cells with 5 μM ionophore: carbonyl cyanide 3-chlorophenylhydrazone (CCCP, Sigma) along with the dye in the reaction.
Lipid extraction
[1,2-^14^C] sodium acetate (specific activity ~37.70 mCi/mmol; BRIT, India) was added to propionate-grown exponential-phase BCG cultures at a concentration of 1 µCi/mL and incubated for an additional 7 days. The cells were then centrifuged, and the cell pellets were dried at 65°C in glass tubes. To the dried biomass, 2 mL of methanol/0.3% NaCl (10:1, vol/vol) was added, and the mixture was vortexed and sonicated at 25°C for 45 min. Two milliliters of petroleum ether was added, and the tubes were vortexed and mixed on a rotator for 20 min. The suspension was centrifuged at 3,000 rpm for 5 min, and the upper layer was collected. The extraction was repeated, and the petroleum ether fractions were pooled. The tubes were dried at 65°C, and the dried residue represented the apolar lipids.
To extract the polar lipid fraction, the cell residue was treated with 2.3 mL of chloroform/methanol/0.3% NaCl (9:10:3, by volume) and mixed on a rotator for 60 min. The sample was centrifuged for 5 min at 3,000 rpm, and the supernatant was collected. The cell pellet was again treated with 750 µL chloroform/methanol/0.3% NaCl (5:10:4, by volume), mixed on a rotator for 30 min, and then centrifuged for 3 min at 3,000 rpm. The supernatants were pooled with the supernatant from the first extraction. This was repeated once more, and 1.3 mL of chloroform and 1.3 mL of 0.3% NaCl were added to the pooled extracts, allowing two phases to form. After mixing for 5 min, the sample was centrifuged for 5 min at 3,000 rpm, and the lower layer containing polar lipids was collected using a long Pasteur pipette, transferred to a separate tube and dried at 65°C. The dried residue represented the polar lipids.
Dried polar and apolar lipids were resuspended in 200 µL chloroform: methanol (2:1, vol/vol), and equal radioactivity (~25,000 dpm) from each sample was analyzed by one- or two-dimensional thin-layer chromatography (2D-TLC). Samples were applied to silica gel 60 F_254_ plates (Merck) and resolved using solvent system A, B, or D in the case of specific apolar lipids and solvent system E for polar lipids (Table 5). Radioactivity was monitored using a phosphorimager (Bass 1800; Fuji). The intensity of spots or bands was monitored by densitometric analysis using ImageJ.
Intrabacterial pH measurement
pUV15-pHGFP (Addgene; plasmid #70,045), an episomal plasmid containing a hygromycin-resistant cassette and encoding a pH-sensitive ratiometric GFP [pHGFP] under the mycobacterial promoter Psymc, was used to electroporate BCG strains. BCG harboring pUV15-pHGFP was grown in glycerol + hygromycin till they reached the mid-exponential phase. Cells were centrifuged and resuspended in phosphate-citrate buffer at pH 7.4 or pH 4.4, supplemented with 0.025% Tyloxapol, to an O.D. at 570 nm of 0.5. Cells were incubated in biological replicates in 24-well plates (JetBiofil, China) at 37°C for 15 min or 8 h. Following incubation, cells were concentrated fivefold by centrifugation to increase the GFP signal. One hundred microliters of aliquots was analyzed in Infinite M200 PRO reader (Tecan, Switzerland) (Excitation = 395 & 475 nm, emission = 510 nm). Interbacterial pH was determined by interpolating the ratios of (Ex_395 nm_, Em_510 nm_) and (Ex_475 nm_, Em_510 nm_) on a standard curve.
Standard curves were generated by placing 70 µg lysates (at a concentration of 14 µg/mL) prepared from glycerol-grown BCG electroporated with pUV15-pHGFP in 100 µL of a phosphate citrate buffer series at pH 5.6 to pH 8.0 in 0.4 pH increments. The 395/475 fluorescence ratios were fitted to pH with the sigmoidal-Hill equation using Prism software (GraphPad Prism 10.6.0).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goletti D, Meintjes G, Andrade BB, Zumla A, Shan Lee S. 2025. Insights from the 2024 WHO global tuberculosis report - more comprehensive action, innovation, and investments required for achieving WHO End TB goals. Int J Infect Dis 150:107325. doi:10.1016/j.ijid.2024.10732539631498 · doi ↗ · pubmed ↗
- 2Rustad TR, Sherrid AM, Minch KJ, Sherman DR. 2009. Hypoxia: a window into Mycobacterium tuberculosis latency. Cell Microbiol 11:1151–1159. doi:10.1111/j.1462-5822.2009.01325.x 19388905 · doi ↗ · pubmed ↗
- 3Russell DG. 2001. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol 2:569–577. doi:10.1038/3508503411483990 · doi ↗ · pubmed ↗
- 4Eoh H, Wang Z, Layre E, Rath P, Morris R, Branch Moody D, Rhee KY. 2017. Metabolic anticipation in Mycobacterium tuberculosis. Nat Microbiol 2:17084. doi:10.1038/nmicrobiol.2017.8428530656 PMC 5540153 · doi ↗ · pubmed ↗
- 5Ehrt S, Schnappinger D, Rhee KY. 2018. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat Rev Microbiol 16:496–507. doi:10.1038/s 41579-018-0013-429691481 PMC 6045436 · doi ↗ · pubmed ↗
- 6Beste DJV, Bonde B, Hawkins N, Ward JL, Beale MH, Noack S, Nöh K, Kruger NJ, Ratcliffe RG, Mc Fadden J. 2011. 13C metabolic flux analysis identifies an unusual route for pyruvate dissimilation in mycobacteria which requires isocitrate lyase and carbon dioxide fixation. P Lo S Pathog 7:e 1002091. doi:10.1371/journal.ppat.100209121814509 PMC 3141028 · doi ↗ · pubmed ↗
- 7de Carvalho LPS, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. 2010. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol 17:1122–1131. doi:10.1016/j.chembiol.2010.08.00921035735 · doi ↗ · pubmed ↗
- 8Beste DJV, Nöh K, Niedenführ S, Mendum TA, Hawkins ND, Ward JL, Beale MH, Wiechert W, Mc Fadden J. 2013. 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem Biol 20:1012–1021. doi:10.1016/j.chembiol.2013.06.01223911587 PMC 3752972 · doi ↗ · pubmed ↗