Defining the role of Pseudomonas aeruginosa PilY1 in signaling and virulence
Christopher L. Pritchett, F. H. Damron, M. Barbier

TL;DR
This study explores how the PilY1 protein in Pseudomonas aeruginosa prevents a signaling system from activating, which affects bacterial virulence and adaptation.
Contribution
The study identifies new genes regulated by the AlgZ/R system and reveals how PilY1 controls cAMP levels to impact virulence.
Findings
PilY1 prevents the AlgZ/R system from activating the major adenylate cyclase cyaB.
PilY1 also inhibits activation of a putative c-di-GMP phosphodiesterase, PA4781, by the AlgZ/R system.
Bacterial survival in the lung, liver, and blood does not depend on PilY1.
Abstract
Pseudomonas aeruginosa (Pa) is an important opportunistic pathogen that has many virulence factors expressed in a coordinated manner to cause infection. The PilY1 protein is a component of the type IV pili (T4P) but has additional signaling functions outside of the pilus. A major function of PilY1 is to prevent the AlgZ/R two-component system from functioning. However, the complete effects of PilY1 signaling through the control of the AlgZ/R or other regulatory systems are not well understood. In this study, we determine new genes controlled by the AlgZ/R system that are impacted by PilY1. We discovered that PilY1 impacts cAMP by preventing the AlgZ/R system from activating the major adenylate cyclase, cyaB. PilY1 also prevents the AlgZ/R system from activating itself and a putative c-di-GMP phosphodiesterase, PA4781. Furthermore, PilY1 functions in different P. aeruginosa strains,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
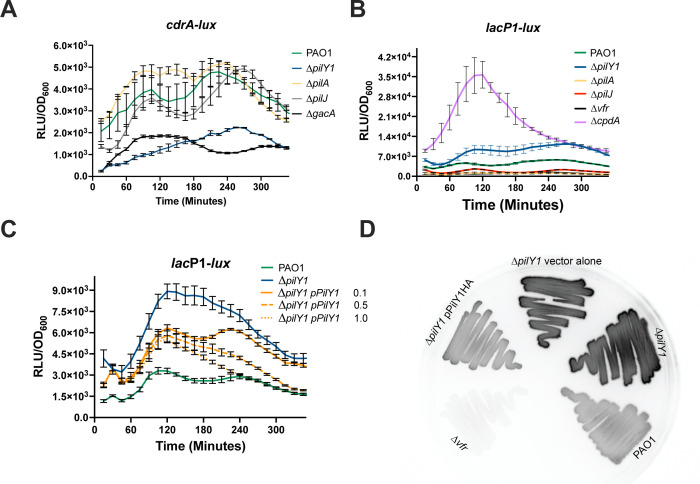 Fig 1
Fig 1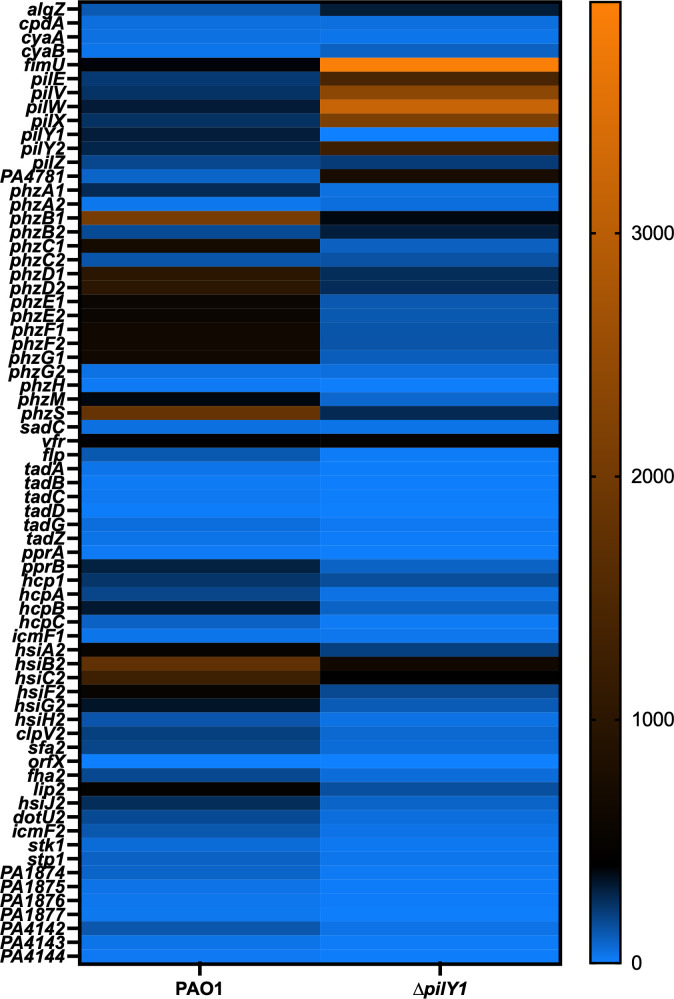 Fig 2
Fig 2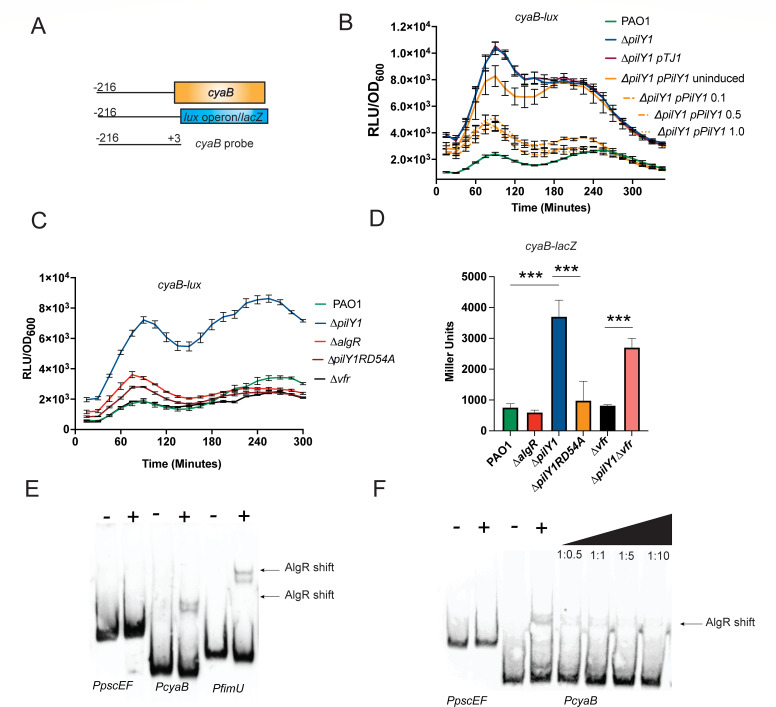 Fig 3
Fig 3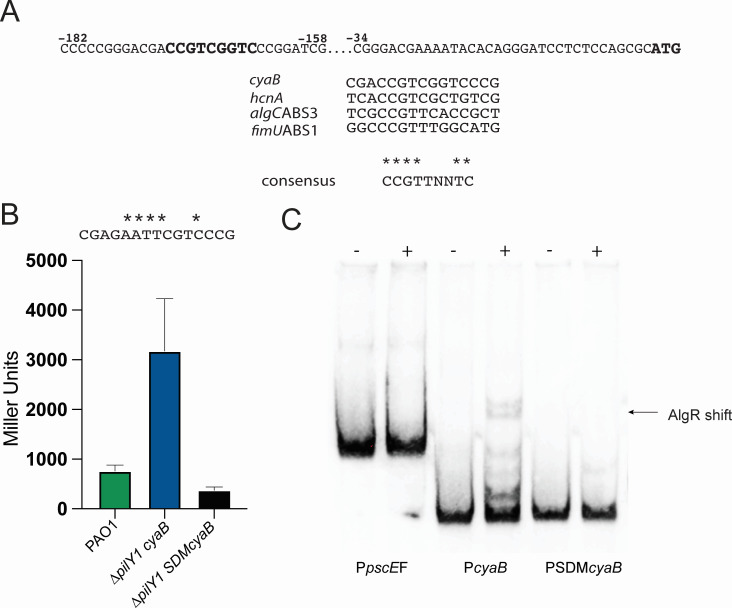 Fig 4
Fig 4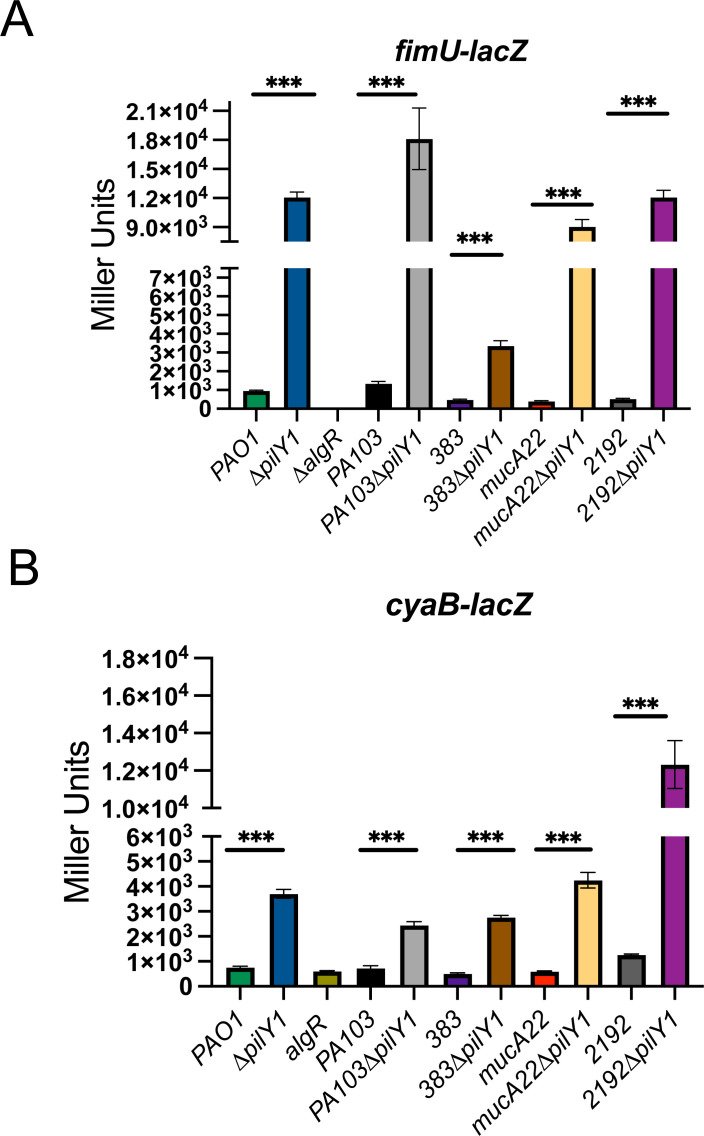 Fig 5
Fig 5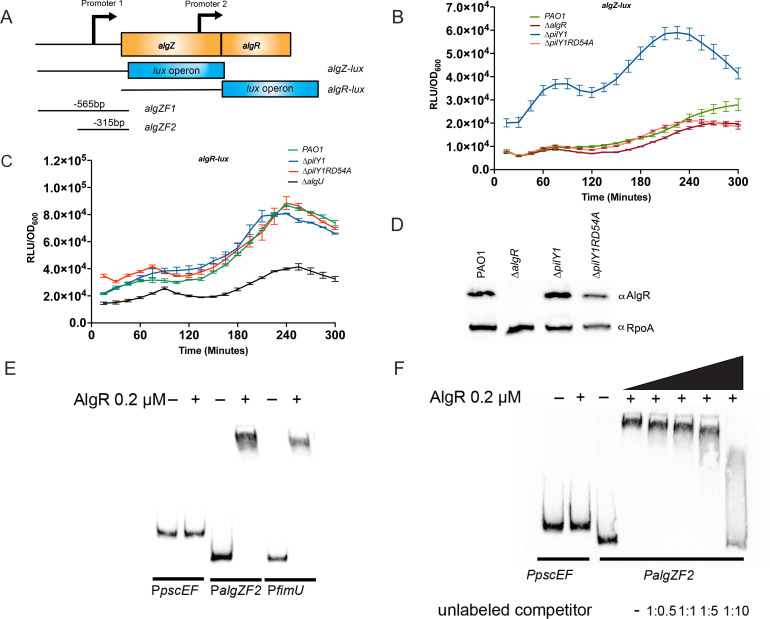 Fig 6
Fig 6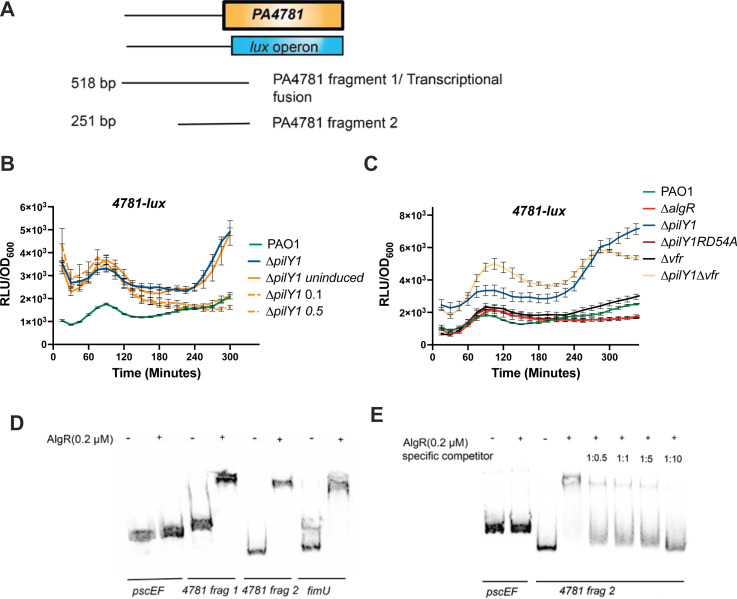 Fig 7
Fig 7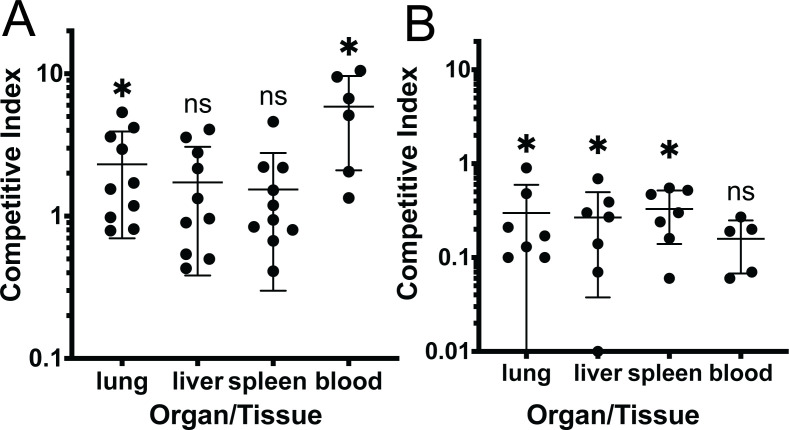 Fig 8
Fig 8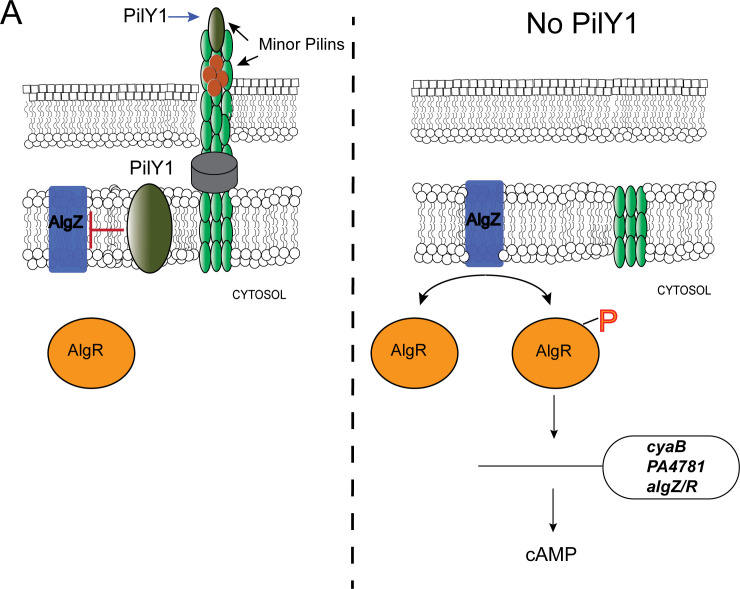 Fig 9
Fig 9- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Bacterial biofilms and quorum sensing · Antimicrobial Resistance in Staphylococcus
INTRODUCTION
Pseudomonas aeruginosa is a human opportunistic pathogen capable of causing fatal infections in immunocompromised individuals such as those undergoing chemotherapy, those suffering from severe burn wounds, chronic obstructive pulmonary disease (COPD), or cystic fibrosis (CF) (1–3). Intrinsic and acquired drug resistance has made P. aeruginosa a microbe to fear in the clinical setting (4, 5). In the disease process, the adherence to host tissues is a prerequisite for colonization, and the type IV pili (T4P) play a dominant role in adhesion (6–9). Upon inhalation or aspiration, P. aeruginosa uses the T4P to colonize the respiratory tract by allowing movement along epithelial cells and forming microcolonies to initiate infection (9). A component of the T4P, PilY1, also has an additional role outside of the T4P. Recent work has discovered that PilY1 is also important in signaling when the bacteria have encountered a surface (10–12). Understanding the molecular events that transpire during these initial events can inform the development of new treatment modalities to combat P. aeruginosa infection.
The T4P are made up of the major pilin subunit PilA and the minor pilins FimU-PilVWXE (13–16). PilY1 is the tip adhesin of the T4P and functions in twitching motility, adherence, and mechanosensing (7, 12, 17–19). The transcriptional regulators Vfr and the AlgZ/R two-component system control expression of the fimU operon, which is a polycistronic operon that encodes pilY1 (20–23). The AlgZ/R system is comprised of the membrane histidine kinase AlgZ and the response regulator AlgR which activate transcription of numerous genes (24). AlgR can function in both the unphosphorylated and phosphorylated form (25–28). Once made, PilY1 prevents activation of the AlgZ/R system resulting in a negative feedback loop (12, 29). The fimU operon is the only AlgZ/R target known to be affected by PilY1. As the AlgZ/R system controls numerous genes, we hypothesized that PilY1 affects additional genes through the AlgZ/R system other than the fimU operon (which includes pilY1), but this has not been investigated.
PilY1 has also been found in the inner membrane as well as outside the cell as part of the pilus (7, 30, 31) suggesting that PilY1 has functions outside the T4P. Indeed, another study found PilY1 was important in controlling secondary metabolites, cell density, and persistence in a non-motile strain (32). This study found that PilY1 was functional even in chronic-infecting strains of P. aeruginosa. Strains found in chronic infections, such as cystic fibrosis (CF), can acquire mutations in mucA causing an overproduction of the polysaccharide alginate (33–35). Therefore, PilY1 may be important in both acute and chronic infections. However, whether PilY1 signaling is similar in different strains has not been determined.
PilY1 signaling impacts second messengers with one known mechanism for regulating c-di-GMP. PilY1 senses surfaces upregulating c-di-GMP levels to increase biofilm formation (10, 12, 36). In contrast, PilY1 was shown to be important for upregulating cAMP upon surface binding (11, 12). However, no mechanism for how PilY1 controls cAMP is known. Other studies have demonstrated that cAMP and c-di-GMP are inversely correlated (37, 38), so how PilY1 affects both c-di-GMP and cAMP has not been clarified. If PilY1 were to increase cAMP levels, then this could explain decreased virulence of pilY1 mutants in various pathogenesis models (29, 32) due to decreased virulence gene expression. The transcriptional activator Vfr binds cAMP leading to increased production of several virulence factors, including the type III secretion system (11, 22). PilY1 involvement in cAMP has not been closely examined, and this would greatly benefit our understanding of the interactions of various regulatory systems.
While PilY1 is important for signaling, the full extent of the downstream impacts of PilY1 is not well understood. Using RNAseq and other genetic and biochemical methods, we identify other targets in the PilY1 signal cascade. We determined that many of the upregulated genes were due to the AlgZ/R two-component system adding to the AlgZ/R regulon. This work furthers our understanding of the PilY1 signaling cascade and the AlgZ/R system.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this study are listed in Table S1. P. aeruginosa was grown at 37°C in Luria Bertani (LB) broth supplemented with 50 mM MOPS or LB agar for routine growth. Transcriptional fusion assays were performed in LB Mops (50 mM), quorum sensing media (QSM, 22 mM KH_2_PO_4_, 22 mM Na_2_HPO_4_, 85 mM NaCl, 10% tryptone, 1 mM MgSO_4_, 0.1 mM CaCl_2_). Media used for P. aeruginosa was supplemented with tetracycline (180 μg/mL), gentamicin (250 μg/mL), carbenicillin (300 μg/mL), or trimethoprim (500 μg/mL) as needed. E. coli was cultivated at 37°C in LB and supplemented when necessary with ampicillin (100 μg/mL), kanamycin (35 μg/mL), or chloramphenicol 34 μg/mL. Yeast-tryptone media (YT, 1% tryptone, 0.5% yeast extract) was used for allelic exchange experiments and was supplemented with the appropriate antibiotics or with 10% sucrose. In the case of complementation, spectinomycin (25 μg/mL) was used in combination with trimethoprim (500 μg/mL).
Mutant strain construction
PCR-generated fragments were amplified using Q5 polymerase (New England Biolabs) and cloned directionally into either pEX18Tc or pEX18Gm (39) after performing cross-over PCR (40). Mutant constructs were introduced into PAO1 or other strains via tri-parental mating using the helper strain pRK2013 (41). Single crossover mutants were selected on the appropriate selective media, and merodiploids were grown without selection to select for a second crossover event. Mutants were plated on YT 10% sucrose and then patch-plated on selective media and PIA. Colonies not growing on selective media were screened via PCR for the appropriate mutation. Complementation was accomplished by PCR of the corresponding wild-type gene using Q5 (NEB) and cloning into the integrating vector pTJ1 (42). Sequencing was used to confirm all constructs and mutant strains. Strains are listed in Table S1, and primers used for making strains are listed in Table S2.
RNA seq
Total RNA was isolated from the selected strains using the RNA SNAP procedure (43) and DNase-treated. The RNA was purified using RNeasy kit (Qiagen), quantified using Nanodrop ND-1000 (Nanodrop), and assessed for RNA integrity using Agilent BioAnalyzer RNA Pico chip (Agilent). A total of 50 μg from each sample was sent for RNAseq analysis (Admera Health). All samples were submitted to rRNA depletion (RiboMinus Transcriptome Isolation Kit, ThermoFisher) and reassessed for RNA integrity. rRNA-depleted mRNA samples were then fragmented and prepared into libraries using Illumina TruSeq RNA library prep kit v2 (Illumina). The libraries were then sequenced on an Illumina HiSeq 2 × 150 bp reads with a total of 10^6^ reads per sample and 3 replicates per experimental condition. Sequencing data were deposited to the GEO website and are available under the reference number GSE278651. RNA-seq reads were analyzed using the software CLC Genomics Workbench (Qiagen). The P. aeruginosa PAO1 genome and annotations were downloaded from the Pseudomonas Genome Database, and reads were mapped using the following settings: mismatch cost = 2, insertion cost = 3, deletion cost = 3, length fraction = 0.8, similarity fraction = 0.8. RPKM values were generated using default parameters for CLC Genomics. Fold changes in gene expression and statistical analyses were performed using an Extraction of Differential Gene Expression (EDGE) test with the Bonferroni correction.
Transcriptional fusion analysis
Upstream DNA fragments containing promoter regions were generated by using primers listed in Table S2 in conjunction with Q5 polymerase (New England Biolabs, Ipswich, MA). PAO1 genomic DNA was used as template. PCR products were cloned into pMiniT (NEB) and then sub-cloned into miniCTXlacZ or miniCTXlux using the restriction enzymes HindIII*/Bam*HI, HindIII/EcoRI, or KpnI/BamHI (NEB). PCR products were purified, cut with restriction enzymes, and inserted into the EcoRI/BamHI sites of miniCTXlacZ using T4 DNA ligase (NEB). Strains were selected for tetracycline resistance and then conjugated with pFLP2 to remove vector sequences in the case of lacZ fusions (39). Strains were selected for carbenicillin resistance, grown overnight without selection, and plated on YT media with 10% sucrose to select for the loss of pFLP2. Individual colonies were patch-plated onto VBMM CB300 and PIA to ensure the loss of pFLP2. To confirm the presence of the fusion constructs, PCR was performed using the forward primer used to construct the fusion and the reverse primer, lacZRforTF (Table S2). β-Galactosidase activity was determined by incubating cell extracts with ONPG (4 mg/mL) as per Miller (44). A strain carrying the empty vector, miniCTXlacZ, was also conjugated into PAO1 and assayed, and this background (28 Miller Units) was subtracted from all transcriptional fusions. All mucoid strains were confirmed mucoid at the end of each experiment by plating on PIA plates to ensure all colonies were mucoid. All assays were performed in buffered LB (LB + 50 mM MOPS). At least three biological replicates were reproduced for all assays.
For lux fusions, promoter fragments were cloned into miniCTXlux and conjugated into P. aeruginosa strains (45). The vector sequences were not removed. White-walled plates in a Synergy HTX (Bio Tek) were used for assays. Strains were grown overnight and diluted 1:50 in LB MOPS (50 mM), and 200 μL was used to inoculate white-walled plates containing LB MOPS (50 mM). Strains were measured every 30 min for 5–10 h using a Synergy HTX or a Synergy LX plate reader. For complementation studies, arabinose (0.1%–1%) was added to media before diluting strains grown to logarithmic phase. Data were analyzed using GraphPad Prism. All experiments were repeated at least three times in triplicate for each strain. Data are reported as relative luminescence by dividing luminescence readings by the OD_600_ for luminescence assays and in Miller Units for β-galactosidase assays. Area under the curve analysis was used to compare strains using GraphPad Prism. Error bars indicate the standard error of the mean.
AlgR purification and antibody production
The algR gene was cloned into the expression vector pMALc6t expression vector (NEB). AlgR was expressed as a fusion with maltose-binding protein (MBP). The fusion protein was batch purified using agarose resin and cleaved using TEV protease, and the TEV and MBP were removed using Ni-affinity chromatography. AlgR was dialyzed using a slidalyzer (ThermoFisher) and Storage Buffer (20% glycerol, 20 mM Tris pH 7.5, 5 mM MgCl_2_, and 1 mM DTT) overnight at 22°C. The purity of AlgR was visually determined in a Coomassie stained 4%–12% gradient electrophoresis gel (SDS-PAGE), and AlgR was confirmed by western blots using AlgR-specific sera. Antibodies were produced to AlgR by ProSci (Poway, CA).
Electrophoretic mobility shift assays
Gel mobility shift assays are described previously with some modifications (46, 47). PCR fragments were generated using biotinylated primers (see Table S2) and gel purified using the Monarch Gel Extraction kit (NEB). Binding reactions were carried out using purified AlgR. The DNA (50–200 ng) probes were mixed with AlgR protein containing 20 mM Tris-HCl (pH 8.0), 0.5 mM dithiothreitol, 20 mM KCl, 0.5 mM MgCl_2_, 2 mM EDTA, and 5% glycerol. The nonspecific competitor poly (dI-dC) was added at 10 μg/mL for all gel shift reactions. After incubation for 20 min at room temperature (25°C), the samples were separated by electrophoresis on a 6% native polyacrylamide gel with 0.375 × TBE used as running buffer for approximately 1.5 h at 100 V. Purified AlgR was incubated at increasing concentrations to determine suitable concentrations to be used. The gel shifts were developed using the chemiluminescent detection kit (Life Technologies) and visualized using a Bio-Rad chemi-doc. Gel shifts were repeated at least three times, and a representative gel shift is shown.
Site-directed mutagenesis
Primers are listed in Table S1. The primers were phosphorylated and used in site-directed mutagenesis as per the manufacturer’s instruction using Q5 (NEB). Constructs were analyzed by restriction enzyme analysis and sequencing. Mutant strains were constructed using homologous recombination as described above and were checked using PCR and primer pairs in Table S1. Site-directed mutants had an EcoRI site engineered to allow easier detection of the promoter mutation. Additional PCR amplicons were sequenced to confirm the mutation in each strain using the same primers. Further confirmation of mutants was done using phenotypic or biochemical assays.
Western blot analysis
The bacteria were collected by centrifugation and resuspended in 50 mM Tris-HCl pH 7.5 and lysed using sonication or B-PER (Fisher Scientific). Total protein concentrations were quantified by the Bradford protein assay (Bio-Rad). Cell extracts (10 μg) were separated by SDS-PAGE on 10% polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (GE Osmonics). The membranes were probed using a 1:10,000 dilution of anti-AlgR rabbit polyclonal antibody followed by a 1:20,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit monoclonal antibody (48), and the signal was detected using chemiluminescence (Bio-Rad). The anti-AlgR polyclonal antibody was pre-absorbed with cell extract from the ∆algR strain to remove other non-specific antibodies.
Westerns were developed using ECL reagent (ThermoScientific) and imaged using a Chemi-Doc (Bio-Rad).
Phenotypic assays
Pyocyanin assays were performed as described in reference 49. Briefly, overnight cultures in QSM media were pelleted and the supernatant filtered through a 0.2 μm filter. Three milliliters of supernatant was extracted twice with 1 mL of chloroform and vortexed vigorously. The organic layer was removed, and 1 mL of 0.2N HCl was added. The OD_520_ was taken, and the samples were normalized to the OD_600_ of the culture. Blanking of the spectrophotometer was done using 0.2 N HCl. Biofilm assays were performed as described previously (50).
Competition experiments
All animal protocols were approved by the Institutional Animal Care and Use Committee at East Tennessee State University (P220301) following the guidelines of the Office of Laboratory Animal Welfare. Overnight cultures were diluted 1:100 in LB and grown for 3–5 h, pelleted, and washed two times with PBS. The OD_600_ of each strain was adjusted to an OD_600_= 0.2. Further dilution with PBS was done to achieve the final dose of 1 × 10^7^ CFUs. Mutant and wild-type strains were mixed 1:1, and the input was determined by plating on PIA and PIA gent. Groups of three to five 12–16 week CD-1 mice (Envigo) were infected by oropharyngeal aspiration after anesthetization with 2.5% isoflurane with P. aeruginosa at doses of 1 × 10^7^ CFUs in 40 μL. Sixteen hours after infection, mice were euthanized, and blood, lungs, livers, and spleens were collected. Organs were homogenized in 1 mL of PBS, and all tissues were serially diluted and 100 μl plated in duplicate on both PIA and PIA Gent. Competitive indices were determined by dividing the output ratio by the input ratio.
Statistical analysis
Statistics were performed using GraphPad/Prism 6.0 (GraphPad software, La Jolla, CA). Transcriptional fusions and pyocyanin levels were compared using a one-way analysis of variance (ANOVA) with a Tukey posttest. Image J software was used for western blot analysis.
A student’ t-test was used for beta-galactosidase assays for multiple strains comparing the mutant to the wild-type. All kinetic assays using lux reporter and EMSA’s were performed at least three times. Kinetic lux assays, beta-galactosidase, and pyocyanin assays were performed in duplicate or triplicate with three biological replicates.
RESULTS
PilY1 affects cAMP levels differently than other pilin components
One consequence of mechanosensing is the increase in cyclic di-GMP (10, 19). Notably, cAMP levels inhibit c-di-GMP levels in P. aeruginosa (37, 38, 51). This led us to speculate that PilY1 might impact cAMP levels in P. aeruginosa, and this could explain decreased c-di-GMP in the pilY1 mutant. Transcriptional lux reporters, encoding the luxCDABE genes responsible for luminescence, were constructed to monitor cAMP and c-di-GMP production over time. The advantage of using lux reporters is this allows real-time evaluation of expression in live cells without the need for lysing cells and adding exogenous substrates. We integrated the second messenger sensors into the P. aeruginosa chromosome to analyze the transcriptional fusions in single copy (45, 52). A cdrA transcriptional fusion was constructed and used as a sensor for c-di-GMP as this promoter has been shown to correlate with c-di-GMP levels (53). The strains were grown with shaking in triplicate in buffered LB (LB + 50 mM MOPS) and monitored for luminescence and optical density over time (300 min) at 37°C. The area under the curve was determined for these strains and demonstrated a significant decrease in relative luminescence for ∆pilY1 compared to the wild-type strain (P < 0.0001, Table S3). A previous study used a lux reporter and also showed a decrease in luminescence in ∆pilY1 (29); however, no statistical analysis was performed. A ∆gacA strain was used as a negative control (54), supporting the use of this construct to evaluate c-di-GMP levels. The cdrA reporter was also tested in other pilin mutants that are deficient in components that affect cAMP levels and T4P biogenesis (10). A ∆pilA and ∆pilJ had luminescence like the wild-type strain (Fig. 1A). Neither ∆pilA nor ∆pilJ were significantly different from PAO1 (Table S3). These data suggest that our cdrA reporter can also be used to assess c-di-GMP levels in P. aeruginosa.
PilY1 affects both cAMP and c-di-GMP in P. aeruginosa. Kinetic analysis of c-di-GMP and cAMP levels. (A) cdrA promoter activity representative of c-di-GMP levels. (B) lacP1-lux reporter activity representative of cAMP levels. (C) Complementation of ∆pilY1 reduces cAMP. (D) Luminescence of strains containing the lacP1-lux reporter on solid media after 16 h. Assays represent three biological replicates done in triplicate, and measurements were taken every 15 min for 5 h. Complementation studies were with L-arabinose (0%–1%). An uninduced control was indistinguishable from the ∆pilY1 strain and was left off the graph for simplicity.
The ∆pilY1 mutant was complemented by incorporating a C-terminal influenza hemagglutinin-tagged (HA-tag) version of the wild-type pilY1 under the control of an arabinose-inducible promoter using an integrating vector to provide single copy complementation (42). This construct was fully functional as determined by complementing twitching motility and biofilm formation (Fig. S1). When ∆pilY1 was complemented, there was a delay in the response, but increasing arabinose concentration restored cdrA reporter activity (Fig. S2). In fact, increasing arabinose to 0.5% and 1.0% resulted in increased cdrA reporter activity after 180 min suggesting that increasing the amount of PilY1 leads to increased cyclic di-GMP (Fig. S2). This further supports the importance of PilY1 in controlling c-di-GMP levels.
As cAMP and c-di-GMP levels are often inversely related (38), we also tested a cAMP biosensor to determine if cAMP levels were affected by the same mutations. The lacP1 promoter previously shown to correlate with cAMP levels (12, 37, 55, 56) was fused to the lux operon (Fig. 1B). A ∆vfr mutant was used as a negative control as this strain is not responsive to increased cAMP levels (55). We also tested the cAMP reporter in a ∆cpdA mutant as a positive control (57), which had the highest luminescence. A ∆pilY1 mutant had modestly increased cAMP levels compared to PAO1 using the lacP1-lux reporter in a liquid assay, and this was significant when comparing the area under the curve for these two strains (P < 0.0001, Table S3) (Fig. 1B). Previous work indicated that both PilA and PilJ were necessary for optimal cAMP levels after surface attachment (10). We would then expect that the cAMP reporter would have decreased activity in these two pilin mutant strains. A ∆pilA strain had decreased Lux activity compared to PAO1 and was like the ∆vfr strain. Similarly, a ∆pilJ strain had slightly decreased luminescence compared to PAO1. Both ∆pilA and ∆pilJ were significantly decreased from PAO1 when comparing the area under the curves (Table S3). Therefore, our results are consistent with published results for ∆pilA and ∆pilJ (11, 12, 55) and indicate our reporters function as expected. This further supports that PilY1 functions differently than other pilin components concerning both c-di-GMP and cAMP. Complementation of the ∆pilY1 strain containing the lacP1-lux reporter demonstrated decreased cAMP levels even without induction with arabinose (Fig. 1C). Increasing arabinose concentration lowered cAMP reporter activity indicating that PilY1 is responsible for lowering cAMP levels. Complementation using 0.5% or 1% arabinose reduced cAMP to wild-type levels in the complemented strain after 240 min. From these data, we conclude that PilY1 affects cAMP levels in P. aeruginosa.
A previous study found that cAMP levels are increased upon growth on surfaces (12). The strains containing the lacP1-lux reporter were grown on solid media and produced luminescence that was visually striking. The luminescence of the ∆pilY1 was much brighter than the wild-type and the ∆vfr mutant (Fig. 1D). Complementation with pilY1 restored the level of luminescence to the wild-type strain (Fig. 1D). These data suggested that PilY1 is important for decreasing cAMP levels and that PilY1 is different from other components such as PilA and PilJ that allow increased cAMP levels (11, 56, 58). This is also consistent with the idea that PilY1 signaling is not due to its presence in the pili as a ∆pilA mutant does not express functional pili.
Transcriptomic analysis reveals genes affected by pilY1 mutation
RNAseq was performed to provide an unbiased view of what other genes PilY1 affects that might explain cAMP regulation as well as determine other transcriptional changes. Comparing the ∆pilY1 mutant to the wild-type strain PAO1, RNAseq identified 168 genes that were upregulated in the ∆pilY1 strain and 118 that were downregulated (Fig. 2, Supplementary Excel file). PilY1 is known to control pyocyanin (32) and control expression of its own operon (29). As expected, many downregulated genes were involved in pyocyanin production (Fig. 2). There are two operons capable of producing pyocyanin (59). Twelve of the downregulated genes (Supplementary Excel file) were from both operons involved in pyocyanin biosynthesis. The finding of pyocyanin biosynthetic genes validates our use of RNAseq to identify new PilY1 targets.
RNAseq analysis defines the PilY1-controlled genes. Heat map comparing differentially regulated genes. The heat map shows the reads per kilobase per million reads (RPKM) for 150 differentially expressed genes between the wild-type PAO1 and the ∆pilY1 strains.
Other downregulated genes included several secretion systems. Two Type I secretion systems (PA1874–PA1877 and PA4142–4144) that are poorly understood were decreased in the pilY1 mutant. PA1874–1877 is an ABC transport system that has been shown to be an efflux pump when P. aeruginosa is growing as a biofilm (60). Even less is known about the putative secretion cluster PA4142–4144, but it is regulated via quorum sensing (61). However, both type I secretion systems have been increased in expression in burn wounds (62). In addition, the second type VI secretion system (H2-T6SS [63) was also decreased in expression when pilY1 was deleted.
Interestingly, PilY1 appears important in the expression of type IVb pili. P. aeruginosa is one of very few bacteria that expresses both type IVa and type IVb pili (64). Several mRNAs encoded by the tad locus and flp were severely decreased as well as the pprAB system that controls the type IVb structural genes. The type IVb pili are important for DNA transfer (65). Therefore, the RNAseq analysis suggests that there is a hierarchy in the expression of the type IVa and type IVb pili in P. aeruginosa with the type IVa pili controlling type IVb expression.
Upregulated genes include fimU, pilX, pilW, pilE, and pilY2 contained within the fimU operon that also contains pilY1 (Fig. 2, Supplementary Excel file). The RNAseq data indicated that PilY1 also impacts the expression of algZ, the protein that phosphorylates AlgR (24) and also regulates the fimU operon . Numerous unannotated genes were also increased in expression. Interestingly, the major adenylate cyclase, cyaB (22), was increased in the pilY1 mutant. Overall, our RNAseq results identified new genes affected by PilY1 and suggested a possible mechanism for PilY1 control of cAMP levels.
Another goal was to determine other possible explanations for PilY1 control of c-di-GMP or cAMP. There was no decreased expression of any known diguanylate cyclase, but we did find one putative phosphodiesterase that had increased expression: PA4781 (Fig. 2, Supplementary Excel file). An increase in PA4781 could explain the decreased c-di-GMP levels in addition to PilY1 affecting SadC (19, 66).
P. aeruginosa controls cAMP levels by two adenylate cyclases, CyaA and CyaB, and one known phosphodiesterase cpdA (22, 57). The RNAseq data did not indicate an increase in the main cAMP phosphodiesterase cpdA. We confirmed this result using a transcriptional reporter to the cpdA promoter and found that cpdA expression was slightly reduced (Fig. S3A). However, we did find an increase in the major adenylate cyclase cyaB in the RNAseq analysis. Therefore, the RNAseq results suggested that increased cAMP was due to increased cyaB expression and not increased expression of the phosphodiesterase that breaks down cAMP.
The major adenylate cyclase, cyaB, is increased in the ∆pilY1 strain
The RNAseq data showed that cyaB was increased in the ∆pilY1 mutant. This could explain the slight increase in cAMP detected in the ∆pilY1 strain (Fig. 1B). CyaB provides most of the cAMP in the P. aeruginosa cell (22). We constructed a cyaB-lux transcriptional fusion to monitor cyaB expression kinetically and detected increased reporter activity in the ∆pilY1 strain (Fig. 3A and B). A comparison of PAO1 and ∆pilY1 containing the cyaB-lux fusion was statistically significant (P < 0.0001, Table S3). This result confirms the RNAseq data and shows that cyaB expression is increased in the ∆pilY1 strain. We complemented the ∆pilY1 strain containing the cyaB-lux fusion (Fig. 3B). As little as 0.1% arabinose decreased expression of cyaB-lux in the complemented strain (Fig. 3B). From these data, we conclude that PilY1 does affect the expression of cyaB.
PilY1 controls cyaB using the AlgZ/R system. (A) Schematic showing the region of the cyaB promoter used for constructing transcriptional fusions and making the probe for EMSA analysis. (B) A cyaB-lux transcriptional fusion has increased activity in the ∆pilY1 strain, and overexpression of pilY1 leads to decreased cyaB expression. (C) The AlgZ/R system is important in regulating cyaB-lux. (D) Confirmation of AlgZ/R regulation of cyaB using a cyaB-lacZ transcriptional fusion. (E) EMSA analysis of the cyaB, pscEF, and fimU probes using purified AlgR. (F) Competition using unlabeled cyaB probe demonstrates the specificity of AlgR binding to cyaB promoter. Competitions were done using 0.5:1, 1:1, 1:5, and 1:10 ratios of unlabeled to labeled probe, respectively. A one-way ANOVA with Tukey’s post-test was used for statistical significance (**, P < 0.0001).*
To determine if PilY1 affects the expression of other genes encoding proteins that make or degrade cAMP, transcriptional lux reporters were constructed of cyaA and cpdA (Fig. S3). We also tested a ∆vfr mutant as Vfr and cAMP are intimately linked and Vfr has been demonstrated to directly activate cpdA expression (57). Compared to the wild-type strain, only cyaB differed significantly in the expression of the adenylate cyclase genes in the pilY1 mutant (Fig. 3C; Fig. S3). Interestingly, Vfr did not affect the expression of cyaB, but cyaA was slightly increased in a vfr mutant at later timepoints (Fig. S3B). There was a slight decrease in cpdA expression in the pilY1 mutant; however, when complemented, the activity of the reporter was not restored to wild-type levels (data not shown). There was also decreased expression of cpdA in the ∆vfr strain used as a control (57) (Fig. S3A). Altogether, these data suggest that Vfr only plays a significant role in regulating the phosphodiesterase cpdA and not the adenylate cyclases, cyaA and cyaB. Based on these data, PilY1 affects the expression of the major adenylate cyclase, cyaB. However, we cannot rule out that there is increased CyaB activity as well.
AlgZ/R is responsible for increased cAMP levels in a pilY1 mutant
A previous study demonstrated that two regulators, Vfr and the AlgZ/R two-component system, are increased in activity in a pilY1 mutant (12). We investigated the role of Vfr and the AlgZ/R system in cyaB regulation to determine which regulator is involved in regulating cyaB expression. As shown in Fig. 3C, a double mutant containing a deletion in pilY1 and encoding a phosphorylation-incompetent version of AlgR (∆pilY1RD54A) compared to the ∆pilY1 strain suggests that phosphorylated AlgR is required for the increased cyaB. Luminescence from ∆pilY1RD54A was significantly decreased from ∆pilY1 (P < 0.0001, Table S3) but was not significantly different from PAO1. There was no difference between PAO1 and the ∆vfr strain (Table S3). Even the wild-type strain had very little cyaB-lux reporter activity. These results suggest that phosphorylated AlgR, and not Vfr, was responsible for increased cyaB expression.
We constructed a second cyaB reporter using lacZ to further confirm the lux reporter data and further investigate if Vfr might play a role in cyaB expression. The ∆pilY1 strain had a >3-fold increase (P < 0.001) in lacZ expression compared to the wild-type confirming the lux fusion data and the RNAseq data (Fig. 3D). Once again, introducing the allele encoding a phosphorylation-incompetent version of AlgR into the ∆pilY1 strain (∆pilY1RD54A) abrogated the increased reporter activity (Fig. 3D). A ∆vfr strain did not differ from PAO1, the wild-type strain. When vfr was deleted in the ∆pilY1 mutant (∆pilY1∆vfr), there was still almost a threefold increase in lacZ activity indicating that Vfr does not increase cyaB expression and is consistent with the cyaB-lux data (P < 0.001) (Fig. 3D). Additionally, the ∆pilY1∆vfr strain did not differ significantly from the ∆pilY1 strain (Fig. 3D). These data indicate that AlgR, and not Vfr, is responsible for the increased cyaB expression in the ∆pilY1 mutant.
To further support AlgZ/R regulation of cyaB, we constructed a ∆algZ/R strain that was complemented with the algZ/R operon under arabinose control. While no significant difference was found between PAO1 and ∆algZ/R, overexpression of the algZ/R operon resulted in increased cyaB-lacZ reporter activity (Fig. S4). These data further support the role of AlgZ/R in control of cyaB expression.
A putative AlgR-binding site was found in the cyaB upstream sequence (Fig. 4A). To determine if AlgR directly controlled cyaB expression by binding this sequence, purified AlgR (0.1 μM) was tested with the cyaB promoter using gel shift analysis (Fig. 3E and F). As a negative control, the pscEF genes were used. As a positive control, the fimU promoter (which also regulates pilY1 expression) was also included. A shift comparable to the fimU positive control was evident (Fig. 3E). Competition experiments using the unlabeled cyaB promoter confirmed that the binding of AlgR to the cyaB promoter was specific (Fig. 3F). The gel shift studies indicated that AlgR directly binds the cyaB promoter.
Site-directed mutagenesis of the AlgR-binding site of the cyaB promoter abolishes increased expression. (A) Sequence of the cyaB promoter region. The putative AlgR-binding site is indicated in bold. Below is an alignment of the putative AlgR-binding site compared to other AlgR binding sites. The consensus sequence is shown below. (B) Transcriptional fusion analysis of the cyaB promoter after site-directed mutagenesis. Sequence changes made to the putative AlgR-binding site are indicated above the sequence by asterisks. The ∆pilY1 SDMcyaB (far right) contains the mutagenized cyaB-lacZ fusion. (C) EMSA demonstrating the importance of the single AlgR-binding site in the cyaB promoter. PSDMcyaB is the mutated cyaB promoter (far right) used in the EMSA. A one-way ANOVA with Tukey’s post-test was used for statistical significance (**, P < 0.0001).*
The putative AlgR-binding site was located 170-162 bp upstream (Fig. 4A). The putative binding site only deviated at one nucleotide from the consensus sequence “CCGTTNNTC” (24) (Fig. 4A). We used site-directed mutagenesis to mutate the AlgR-binding site in the cyaB-lacZ transcriptional fusion (Fig. 4B) to determine if AlgR might bind to this site. Mutation would abolish AlgR activation if AlgR bound this site and provides an in vivo relevance of this promoter region. Once again, cyaB expression was more than threefold higher in activity in the ∆pilY1 mutant when using the wild-type cyaB promoter. However, the site-directed fusion had a greater than 4.5-fold reduction from the ∆pilY1 parent strain. Interestingly, mutation of the AlgR binding site in the fusion also reduced the reporter activity to approximately half of the wild-type strain PAO1 but was not significant (Fig. 4B).
Gel shift analysis was used to further define the nucleotides necessary for AlgR binding. We compared the native cyaB promoter to the site-directed mutagenized promoter. The native promoter bound AlgR as previously shown (Fig. 4C). However, the mutagenized promoter was unable to bind to AlgR (Fig. 4C). These data clearly demonstrate that AlgR directly regulates the cyaB promoter and indicate that the putative AlgR-binding site is legitimate. Therefore, the AlgZ/R system is important in the regulation of cAMP levels in P. aeruginosa by controlling cyaB expression. Overall, our data indicate that PilY1 is necessary to prevent AlgZ/R activation of cyaB.
PilY1 functions in multiple strain types, including in mucoid strains
Given the diversity of P. aeruginosa strains, we wanted to assess the functionality of PilY1 in other backgrounds. We used another strain PA103 (67) as this strain is representative of strains causing high rates of pneumonia and sepsis (68–72). We also used clinical strains, strain 383, and its mucoid derivative 2192 (73), and PDO300, the mucA mutant of PAO1. A fimU-lacZ transcriptional fusion was used to assess the functionality of PilY1 in these strains as previous studies have shown that pilY1 mutation led to increased expression of its own operon (12, 29). A ∆algR strain was used as a negative control as the AlgZ/R system positively regulates the fimU operon (21). As expected, there was a >20-fold increase in fimU expression in ∆pilY1 vs PAO1 (Fig. 5A). Analysis of strain PA103 also resulted in increased fimU expression (P < 0.0001) when pilY1 was deleted in this background (Fig. 5A). There was a sevenfold difference between the 383*∆pilY1* strain and the parental strain 383 for fimU expression (P < 0.0001) (Fig. 5A). Overall, these data suggest that PilY1 plays an important role in inhibiting the AlgZ/R system in numerous strains.
PilY1 functions similarly in both laboratory and clinical of P. aeruginosa strains. (A) Beta-galactosidase activity of a fimU-lacZ reporter to indicate that PilY1 is functional in five strains of P. aeruginosa. (B) Beta-galactosidase activity of a cyaB-lacZ reporter in multiple P. aeruginosa strains to demonstrate PilY1 functionality. Transcriptional fusions were performed in triplicate at least three times. P <**, 0.0001 based on Student’s t-test comparing the parent and the mutant strain.
In cystic fibrosis patients, P. aeruginosa frequently loses motility and persists as a biofilm (74). Whether pili components, such as PilY1, are produced in mucoid strains has not been investigated. The rationale for these experiments was that PilY1 in a chronic P. aeruginosa isolate had significant phenotypic changes (32), suggesting that PilY1 can function independently of being incorporated into the pilus. The laboratory strain PDO300 (derived from PAO1) and a clinical isolate 2192 (75), which both contain mutations in mucA, were used. The fimU reporter had increased activity (>9-fold) in the pilY1 mutants in both PDO300 and 2192 (Fig. 5A). These results suggested that PilY1 does function in mucA mutants found in chronic infections.
We also wanted to determine if AlgZ/R regulation of cyaB was also conserved in P. aeruginosa strains. The cyaB-lacZ transcriptional fusion was tested in various strains to monitor cyaB expression. There was increased activity of the fusion whenever a strain had been deleted for pilY1 (Fig. 5B). Both PAO1*∆pilY1* and 383∆pilY1 had increased cyaB reporter activity compared to their respective parent strains (Fig. 5B). We also saw an increase in cyaB-lacZ activity in the mucoid ∆pilY1 strains in both genetic backgrounds (Fig. 5B). When pilY1 was deleted in PDO300 (PDO∆pilY1), there was a ~7-fold increase in cyaB reporter expression. In the case of 2192∆pilY1, there was an ~10-fold increase (Fig. 5B). This further confirms PilY1 is functional in mucA mutants and indicates that the signaling cascade is found in more than just the laboratory strain PAO1. These data also support that a major role of PilY1 is to repress the AlgZ/R system to prevent improper cAMP levels.
PilY1 affects algZ/R expression
A previous study indicated that the AlgZ/R system was hyperactive in a ∆pilY1 mutant, and this was responsible for attenuation in a C. elegans model (29). Additionally, our RNAseq data indicated increased algZ expression in the ∆pilY1 strain. This suggested to us that the algZ/R operon might be subject to autoregulation, a common characteristic of two-component systems (76), but has not been directly tested for the algZ/R system. The algZ/R operon is under complex genetic regulation where promoters lie upstream of algZ and within the algZ coding region (77) (Fig. 6A). At least one of the promoters upstream of algZ (Promoter 1) is regulated by Vfr (20, 77). The promoters within the algZ coding region controlling only algR expression are activated by the alternative sigma factor AlgU/T (77). To begin to investigate autoregulation of the algZ/R promoters, we constructed algZ and algR transcriptional fusions to separate the two promoter regions (Fig. 6A) and assayed the activity in the minor pilin mutant ∆pilY1. Confirming the RNAseq data, the pilY1 mutant had significantly increased activity of the algZ transcriptional fusion (P < 0.0001, Table S3), but there was no increased activity of the promoter located within the algZ coding region that controls only algR expression (Fig. 6B and C). If the minor pilin mutant has increased AlgZ/R activity, then this suggests that AlgR should exist in a phosphorylated state. When a double mutant consisting of the phosphodeficient allele of algR in the ∆pilY1 background (∆pilY1RD54A) was analyzed with the algZ transcriptional fusion, the reported luminescence was not significantly different from the wild-type (Fig. 6B; Table S3). The transcriptional fusion analyses indicate that AlgR activates the expression of the algZ/R operon, but only from the most distal promoter upstream of algZ. Interestingly, Vfr was previously shown to regulate this same promoter (20, 78).
PilY1 prevents autoregulation of the algZ/R operon. (A) Schematic depicting the transcriptional fusions and the EMSA probes used. (B) An algR-lux transcriptional fusion tested in the pilY1 mutant strains indicating no change in expression levels. (C) An algZ-lux transcriptional fusions tested in pilY1 mutant strains showing increased algZ promoter activity in the ∆pilY1 background. (D) Western blot analysis confirms increased AlgR in the ∆pilY1 strain and decreased AlgR in the ∆pilY1RD54A strain. Ten micrograms of protein was loaded in each lane. A ∆algR strain was used as a negative control. RpoA was used as a loading control. (E) EMSA showing AlgR binding to the algZ promoter region. Purified AlgR bound both promoter probes algZF1 and algZF2. Shown is the EMSA using the shorter probe algZF2. (F) Competition EMSA showing AlgR specificity for algZ fragment 2. Transcriptional fusions were performed in triplicate at least three times. Western blotting was performed four times. A representative western is shown. EMSAs were performed at least three times. A representative EMSA is shown.
Western analysis was used to confirm increased AlgR levels in the ∆pilY1 mutant (Fig. 6D). AlgR was increased in the ∆pilY1 strain compared to the wild-type strain PAO1 (Fig. 6D) indicating that algR is increased in ∆pilY1. However, when the ∆pilY1RD54A strain was tested, AlgR levels were decreased compared to the ∆pilY1 parental strain (Fig. 6D). A ∆algR strain was used as a negative control and RpoA was used as a loading control. These results suggested that increased expression of the algZ/R operon results in increased AlgR levels.
To determine if AlgR activation of the algZ promoter was direct, we performed gel shift analyses using biotinylated probe fragments representing the promoter region upstream of algZ. Purified AlgR (0.2 mM) was able to shift both a fragment containing 565 bp upstream sequence and a shorter fragment consisting of 315 bp of upstream sequence (data not shown and Fig. 6E). We used the 315 bp fragment in competition experiments and inhibited AlgR binding using increasing concentrations of the unlabeled probes for the algZF2 probe (Fig. 6F). These results indicated that AlgR specifically binds to the algZ promoter.
PilY1 affects PA4781 expression by preventing AlgR phosphorylation
PilY1 signals through the diguanylate cyclase SadC to increase c-di-GMP levels (19), and this explains why ∆pilY1 strains have decreased c-di-GMP. However, it is also possible that PilY1 affects c-di-GMP level by other means as well. The RNAseq results demonstrated increased PA4781 transcripts in the ∆pilY1 strain. PA4781 was previously suggested to degrade c-di-GMP (79). We hypothesized that PA4781 might contribute to the decreased c-di-GMP levels in the ∆pilY1 mutant by degrading c-di-GMP.
We first wanted to determine how PilY1 controls PA4781. We constructed a PA4781 transcriptional fusion (Fig. 7A and B). Confirming the RNAseq data, PA4781 expression was significantly increased in a ∆pilY1 (P < 0.0001, Table S3). Complementation of the ∆pilY1 mutant with a single copy of pilY1 and induction of 0.1% arabinose (or 0.5%) was able to fully restore levels to the wild-type after 120 min (Fig. 7B). Therefore, PilY1 affects the expression of PA4781.
PA4781 expression. (A) Schematic depicting the PA4781 transcriptional fusion and fragments of the PA4781 promoter region used for EMSA analysis. (B) Complementation of PilY1 reduces PA4781 expression. A PA4781 transcriptional fusion was tested in a complemented strain consisting of pilY1 under an arabinose-inducible promoter. Percentages to the right (0.1 and 0.5) indicate the percentage of arabinose used. (C) Testing the PA4781-lux fusion to demonstrate AlgR-dependence and Vfr independence. (D) EMSA demonstrating that AlgR binds to the PA4781 promoter region. (E) Competitive EMSA showing specificity of AlgR binding to the PA4781 promoter (4781 frag 2). Transcriptional fusions were performed in triplicate at least three times. EMSAs were performed at least three times. A representative EMSA is shown.
We also analyzed the PA4781-lux fusion in strains to understand how PA4781 is controlled in the context of PilY1. Because the AlgZ/R system and Vfr are important regulators in the ∆pilY1 background (12), we investigated these regulators. When the allele of algR that encodes a phosphorylation-incompetent form of AlgR in the pilY1 mutant was tested (∆pilY1RD54A), PA4781 expression was significantly decreased from ∆pilY1 (Table S3), suggesting that phosphorylated AlgR is required for the activation of the PA4781 promoter (Fig. 7C). A ∆algR mutant was also tested and had similar expression levels to the wild-type strain. We also tested this fusion in the ∆vfr and ∆pilY1∆vfr double mutant to determine if Vfr might also regulate PA4781 expression. Mutation of vfr did not play a role in regulating PA4781 compared to the wild-type (Fig. 7C). The ∆pilY1∆vfr mutant had similar PA4781-lux activity as the ∆pilY1 mutant and was not statistically different suggesting that increased PA4781 expression is not due to Vfr (Table S3). These data suggest that PilY1 prevents the AlgZ/R system from activating PA4781 expression and that phosphorylated AlgR is required for transcriptional activation.
To determine if AlgR directly regulates PA4781, we performed gel shift analyses. Biotinylated PCR products were used to determine if AlgR directly bound to the PA4781 promoter region. A putative AlgR-binding site was located 425 bp upstream of PA4781. Two probes were made and incubated with 0.2 μM of purified AlgR (Fig. 7A). A probe containing 581 bp of upstream sequence bound purified AlgR that contained a potential AlgR-binding site. A shorter probe encompassing 251 of upstream sequence also bound AlgR even though no AlgR consensus sequence was found in this region (Fig. 7D). Competition experiments with the smaller upstream fragment using unlabeled probe were able to reduce AlgR binding indicating a specific interaction of AlgR with the shorter promoter fragment (Fig. 7E). From these data, we conclude that AlgR directly controls the expression of PA4781. Altogether, PilY1 decreases PA4781 expression by preventing AlgR activation.
PilY1 does not contribute to fitness in an acute pneumonia model
Several studies have indicated that pilY1 mutants are attenuated (10, 29, 32, 80). However, only one of these studies used a mouse model. The strain used was an uncharacterized clinical isolate, and the bacteria were encased in alginate beads to model a chronic infection (32). We tested the ∆pilY1 strain (derived from the PAO1 background) in a competition experiment with the wild-type strain to determine the fitness of this strain in an acute pneumonia. Both PAO1 and the ∆pilY1 strains were labeled using either a gentamicin resistance cassette (PAO1) or a tetracycline resistance cassette (∆pilY1). The PAO1 and ∆pilY1 strains were mixed in a 1:1 ratio at a dose of 1 × 10*^7^* CFU/mouse (a lethal dose at 24 h). We expected the ∆pilY1 mutant to be less competitive to the wild-type based on previous studies. After 16 h of infection, the lungs, spleen, and liver were homogenized and plated on differential media to distinguish between the wild-type and mutant to calculate the competitive index (Fig. 8). Blood was also analyzed; however, there were no bacteria detected in the blood from 4 mice. Results showed that the ∆pilY1 mutant outcompeted the wild-type as indicated by CI values greater than 1.0 in the lung (CI = 1.63) and blood (CI = 5.9) (Fig. 8A). However, in the liver and spleen, the CI was close to 1 indicating that the ∆pilY1 mutant was able to compete as well as PAO1 in these tissues (CI = 1.15 and 1.1, respectively) (Fig. 8A). Overall, these results suggest that the ∆pilY1 mutant is more competitive in certain tissues and at least as competitive as the wild-type strain using the acute pneumonia model.
*Assessment of PilY1 in P. aeruginosa virulence in an acute pneumonia model. Mice were infected for 16 h via oropharyngeal aspiration. Each point represents a competitive index. n = 10 for PAO1/∆pilY1 and n = 7 for PAO1/∆pilA. (A) Competitive index of ∆pilY1 vs PAO1 during infection. Four mice had no detectable colonies from the blood. (B) Competitive index of ∆pilA vs PAO1 during infection. Two mice had no detectable bacteria in blood (∆pilA/PAO1). One mouse liver was not used due to technical error. Statistical analysis was using the Wilcoxon signed rank test. , P < 0.05.
The type IV pili mediate close interaction of bacteria with host cells in in vitro studies, but few studies have tested if type IV pili are important in pathogenesis in vivo. We tested a ∆pilA mutant in a competition experiment as described for ∆pilY1. In contrast to the ∆pilY1 data, a ∆pilA mutant was attenuated in the lung, liver, and spleen (Fig. 8B). The CI of ∆pilA was not significantly different in the blood, but this was likely because we were only able to enumerate CFUs from five mice. Altogether, these data indicate that a ∆pilA mutant is attenuated in the acute pneumonia model.
DISCUSSION
** **PilY1 is an adhesive component of the T4P and is important in mechanosensing (7, 10, 12, 29, 32, 81). However, there are still questions regarding the role of PilY1 in pathogenesis and the outcome of PilY1 signaling. Previous studies have established that PilY1 prevents activity of the AlgZ/R two-component system (12, 29). Therefore, a ∆pilY1 strain provides an opportunity to determine the role of phosphorylated AlgR in P. aeruginosa. Here, we identify new AlgZ/R-controlled genes using a ∆pilY1 strain. We found that the expression of the major adenylate cyclase, cyaB, and an unannotated putative cyclic di-GMP phosphodiesterase, PA4781, are increased in a ∆pilY1 mutant. We demonstrate that both cyaB and PA4781 are directly controlled by the AlgZ/R two-component system identifying new members of the AlgZ/R regulon. This signaling cascade was conserved in multiple P. aeruginosa strains. Additionally, we demonstrate that PilY1 plays a nuanced role in virulence in the acute pneumonia model. Overall, our study determined new members of the AlgZ/R regulon and demonstrated genes that are dysregulated in a ∆pilY1 strain.
Using transcriptional fusion analyses, gel shift studies, and site-directed mutagenesis, we firmly establish that the AlgZ/R system can activate cyaB expression. Control of cAMP levels is important to allow bacteria to move from a planktonic lifestyle to a biofilm lifestyle (37, 38). Other pili components control the activity of CyaB after expression (11, 12, 56), but this would occur after PilY1 has bound to a surface. It may be that there is expression of cyaB in the planktonic phase, but the CyaB enzymatic function is not active until the T4P extend and contract after binding a surface. This would help explain the surface-activated virulence seen previously (10).
The AlgZ/R system was also found to be subject to autoregulation. Previous studies have alluded to this autoregulation (82, 83), but no direct test of this has been done to date. The results presented here demonstrate direct regulation of the algZ/R operon by AlgR. Additionally, these data demonstrate that phosphorylated AlgR binds to the promoter upstream of algZ. Therefore, PilY1 control of the AlgZ/R system results in decreased expression of this system as well as decreased activity. Further work is necessary to determine how PilY1 prevents AlgR phosphorylation.
Our study also determined that the AlgZ/R system controls the expression of the putative phosphodiesterase, PA4781. The role of PA4781 in controlling c-di-GMP is still unclear (79, 84). While we cannot ascribe a role for PA4781, we have aided in understanding its transcriptional regulation. Understanding how PA4781 is involved in secondary messenger degradation may be facilitated by using a pilY1 mutant strain where its expression, and perhaps activity, is increased.
Our use of the ∆pilY1 strain provides a more physiological context to decipher AlgR function than other studies. None of the genes in our study were found in previous investigations studying AlgR using microarray or ChiP-seq analysis (82, 85). A possible explanation for why our study detected these changes in cyaB, algZ, and PA4781 is our use of the ∆pilY1 strain that allows increased phosphorylated AlgR (29). Previous studies may not have had phosphorylated AlgR whether using an algR overexpression strain (82) or purified AlgR (85). The stability of the acyl-phosphate bond in response regulators can be unstable and can have half-lives of seconds to hours (86, 87). The lack of phosphorylated AlgR in the previous studies could explain why these genes were not identified in those studies.
The role of PilY1 in virulence is still unclear. Previous studies have indicated that PilY1 is important in virulence (10, 29, 32). Only one of the previous studies used a mouse model of infection. This study used a chronic isolate and chronic infection model using alginate beads and only examined the lungs. However, the authors also found an increase in CFUs of the ∆pilY1 strain compared to the wild-type in a competition experiment (32) similar to our study. The competitive index (CI) is a sensitive measure of comparing virulence between mutant and wild-type strains (88). We found that the ∆pilY1 strain was at least as fit in the liver and spleen and more fit than the wild-type in the lungs and blood. While we did not evaluate the immunopathology during these infections, the fact that the host is incapable of eliminating the ∆pilY1 strain complicates assigning the ∆pilY1 strain as attenuated. It may be that the timepoint used in the acute pneumonia model of 16 h was not sufficient to see a difference between the two strains. For instance, if the ∆pilY1 strain caused more neutrophil infiltration, this could result in more pathology early on, but later timepoints might find that the mutant strain is eliminated at greater numbers than the wild-type.
A previous study indicated that a pilA mutant was attenuated in a survival study (9). Our work confirms this result using a slightly different model and a different strain. Using oropharyngeal aspiration instead of intranasal inoculation to introduce bacteria ensures more bacteria in the lungs (89, 90). This makes our model relevant to the study of ventilator-associated pneumonia. A recent transcriptomic analysis of a ∆pilA strain suggested that the main reason for attenuation is due to cAMP production (91). It is possible that the dysregulated cAMP levels in the ∆pilY1 are responsible for the increased virulence. Given the importance of careful regulation of second messengers, inappropriate levels at a given time may attenuate P. aeruginosa virulence. Additionally, our study has demonstrated that while some pili components are required for virulence, such as PilA, others, such as PilY1, can be unnecessary after the colonization phase.
Based on our data and the work of others, we propose that the major function of PilY1 is to regulate the activity of the AlgZ/R system (Fig. 9). In the case of pilY1 expression, phosphorylated AlgR and Vfr activate the fimU operon (which includes pilY1) (20, 21). The work presented here suggests that the AlgZ/R system can also activate cyaB expression allowing cAMP production that can then activate Vfr. Phosphorylated AlgR and Vfr can then activate the fimU operon (including pilY1). After the expression of the fimU operon, PilY1 turns off the AlgZ/R system. This would then allow Vfr to activate other targets such as the T3SS. Given that AlgR can inhibit T3SS expression (92) and that the T3SS is activated via surface contact (93, 94), PilY1 deactivation of AlgZ/R is required for Vfr to activate the T3SS. Could phosphorylated AlgR be used to prevent Vfr from activating the T3SS expression? This could explain the reduced virulence of algR overexpressing strains previously described (95). In addition, does PilY1 continue to turn off the AlgZ/R system when PilY1 is engaged with its cognate ligand on a host cell? Further studies using pilY1 variants instead of deletion mutants are required to answer these questions.
PilY1 exerts its effects by turning off the AlgZ/R system. (A) PilY1 turns “off” AlgZ/R to prevent increased cyaB, PA4781, and algZ/R expression. PilY1, through unknown mechanisms, prevents AlgZ phosphorylation of AlgR. Mutation of pilY1 results in increased AlgR phosphorylation that causes aberrant gene expression. This study uncovered new AlgZ/R targets and suggests that PilY1 signaling requires turning off expression of cyaB, PA4781, and the algZ/R operon to allow P. aeruginosa to modify its lifestyle upon surface contact.
PilY1 has garnered attention for its role in mechanosensing and virulence. In this report, we have identified an important role of PilY1 in controlling cAMP as well as c-di-GMP. This aids our understanding of how the T4P can engage in signaling but also suggests that PilY1 function is important in other locations as well. We also have aided in the understanding of how different regulatory pathways intersect. Vfr and AlgZ/R can work together to activate expression of the fimU operon. Once pilY1 is expressed, PilY1 turns off the AlgZ/R system and other pili components can activate CyaB allowing Vfr to activate expression of virulence genes. Further work is necessary to unravel the role of PilY1 in P. aeruginosa pathogenesis. Understanding signal transduction networks can identify new therapeutic targets and allow new ways to treat P. aeruginosa and other antibiotic-resistant bacteria.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gaynes R, Edwards JR, National Nosocomial Infections Surveillance System. 2005. Overview of nosocomial infections caused by gram-negative bacilli. Clin Infect Dis 41:848–854. doi:10.1086/43280316107985 · doi ↗ · pubmed ↗
- 2Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, Sievert DM. 2016. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the national healthcare safety network at the centers for disease control and prevention, 2011-2014. Infect Control Hosp Epidemiol 37:1288–1301. doi:10.1017/ice.2016.17427573805 PMC 6857725 · doi ↗ · pubmed ↗
- 3Crabtree TD, Gleason TG, Pruett TL, Sawyer RG. 1999. Trends in nosocomial pneumonia in surgical patients as we approach the 21st century: a prospective analysis. Am Surg 65:706–709;10432077 · pubmed ↗
- 4Hancock REW, Nikaido H. 1978. Outer membranes of gram-negative bacteria. XIX. Isolation from Pseudomonas aeruginosa PAO 1 and use in reconstitution and definition of the permeability barrier. J Bacteriol 136:381–390. doi:10.1128/jb.136.1.381-390.1978101518 PMC 218670 · doi ↗ · pubmed ↗
- 5Breidenstein EBM, de la Fuente-Núñez C, Hancock REW. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi:10.1016/j.tim.2011.04.00521664819 · doi ↗ · pubmed ↗
- 6Chi E, Mehl T, Nunn D, Lory S. 1991. Interaction of Pseudomonas aeruginosa with A 549 pneumocyte cells. Infect Immun 59:822–828. doi:10.1128/iai.59.3.822-828.19911671777 PMC 258333 · doi ↗ · pubmed ↗
- 7Heiniger RW, Winther-Larsen HC, Pickles RJ, Koomey M, Wolfgang MC. 2010. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell Microbiol 12:1158–1173. doi:10.1111/j.1462-5822.2010.01461.x 20331639 PMC 2906647 · doi ↗ · pubmed ↗
- 8Kang PJ, Hauser AR, Apodaca G, Fleiszig SM, Wiener-Kronish J, Mostov K, Engel JN. 1997. Identification of Pseudomonas aeruginosa genes required for epithelial cell injury. Mol Microbiol 24:1249–1262. doi:10.1046/j.1365-2958.1997.4311793.x 9218773 · doi ↗ · pubmed ↗