Genetic evidence for a periplasmic protein as a third component for a subset of NtrYX family two-component systems
Alexa R. Wolber, Liliana S. McKay, Richard M. Johnson, Zain T. Hameed, Katlyn B. Mote, Steven M. Julio, Peggy A. Cotter

TL;DR
This study shows that a periplasmic protein, PlrP, acts as a third component in a bacterial signaling system, helping pathogens like Bordetella survive in the lungs.
Contribution
The paper identifies PlrP as a novel third component in a subset of NtrYX family two-component systems.
Findings
PlrP prevents PlrS from acting as a strong phosphatase, maintaining PlrR phosphorylation levels.
PlrP is essential for bacterial survival in the lower respiratory tract.
PlrP homologs are found with NtrYX-family TCSs in other bacteria, suggesting a conserved function.
Abstract
PlrSR, a member of the NtrYX family of two-component regulatory systems (TCSs), is required for the classical bordetellae, including the causative agent of whooping cough, Bordetella pertussis, to persist in the lower respiratory tract. The plrSR genes are in the middle of a six-gene cluster whose regulation and roles during infection were unknown. rsmB and plrP are often found 5′ to plrSR homologs in β- and γ-proteobacteria, while trkAH is often found 3′ to plrSR homologs in ⍺-proteobacteria. We investigated these genes to determine if they have a functional link to plrSR. We found that this gene cluster does not function as an operon. Rather, it contains two internal promoters: a weaker promoter in the 3′ end of rsmB and a stronger promoter in the 3′ end of plrS. Additionally, our results indicate that PlrP functions as a third component of the PlrSR TCS. Genetic manipulations of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
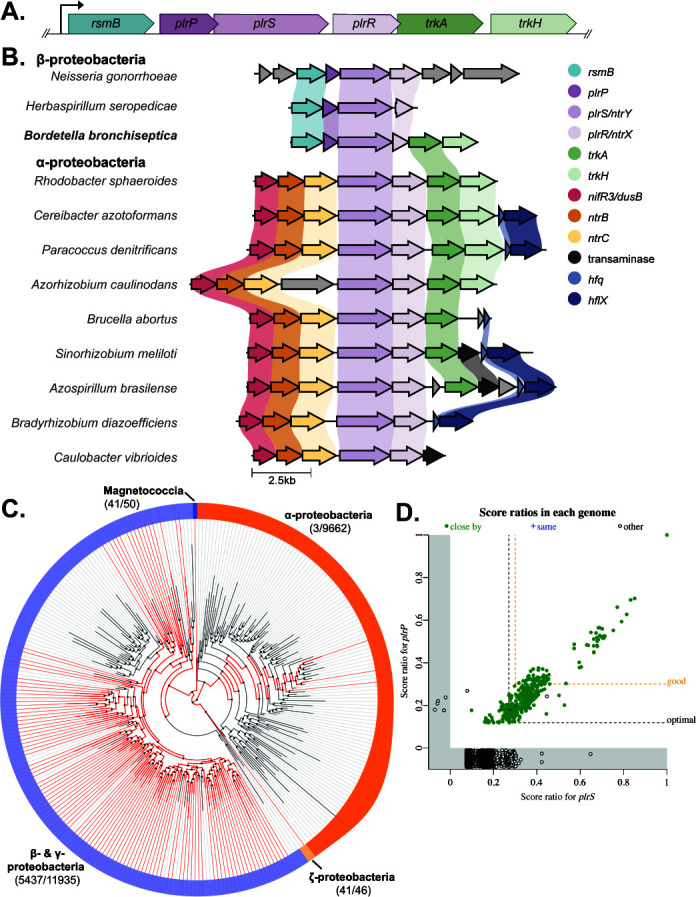 Fig 1
Fig 1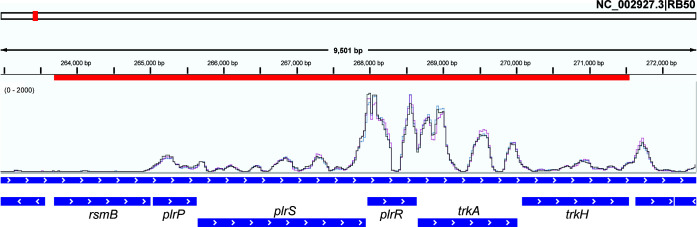 Fig 2
Fig 2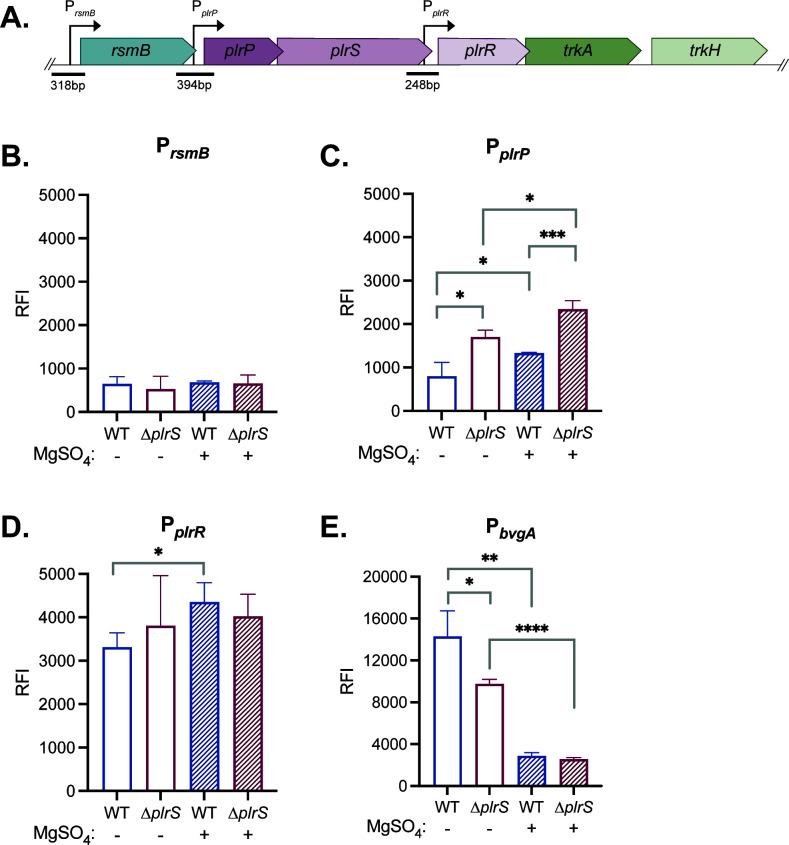 Fig 3
Fig 3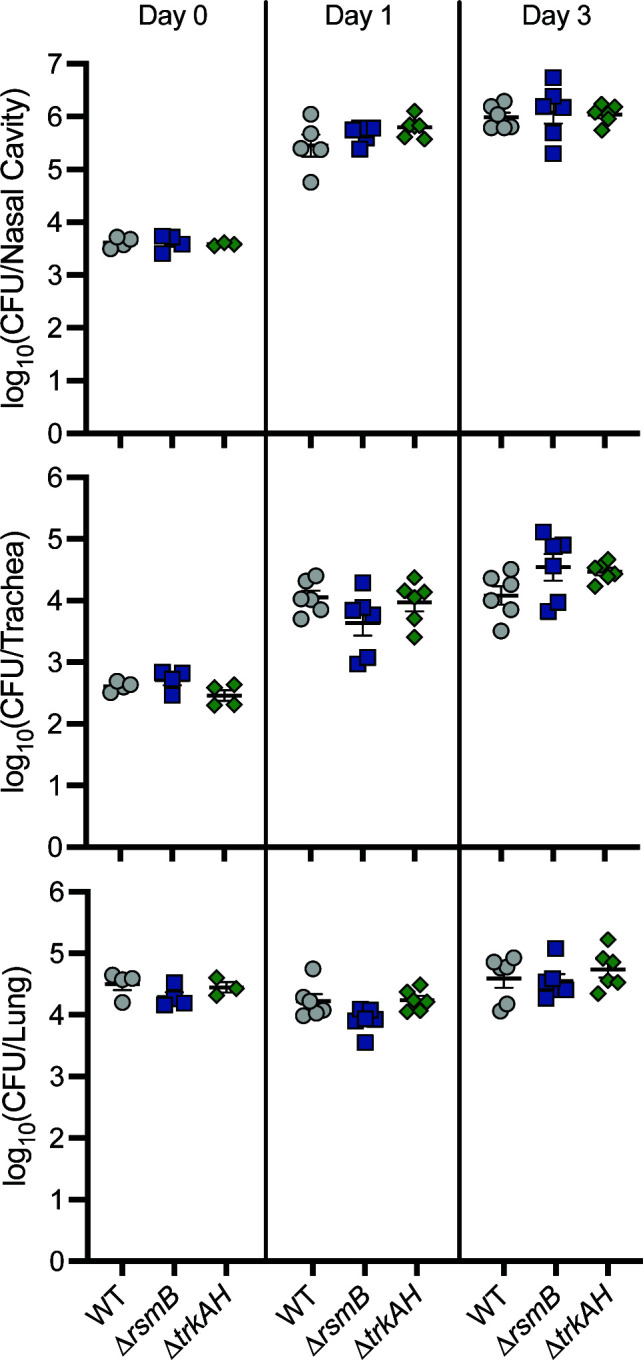 Fig 4
Fig 4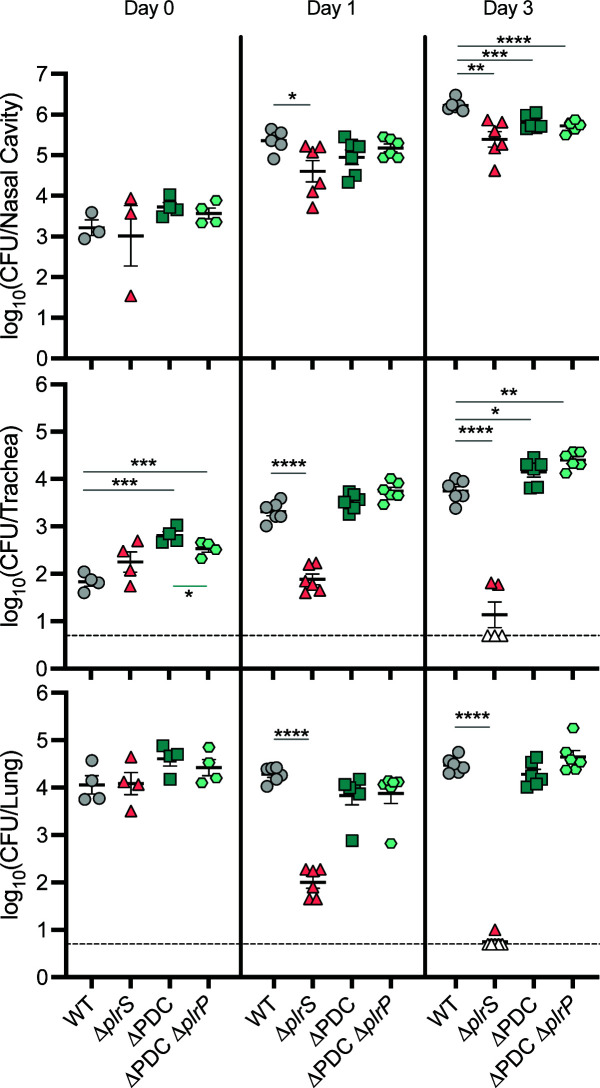 Fig 5
Fig 5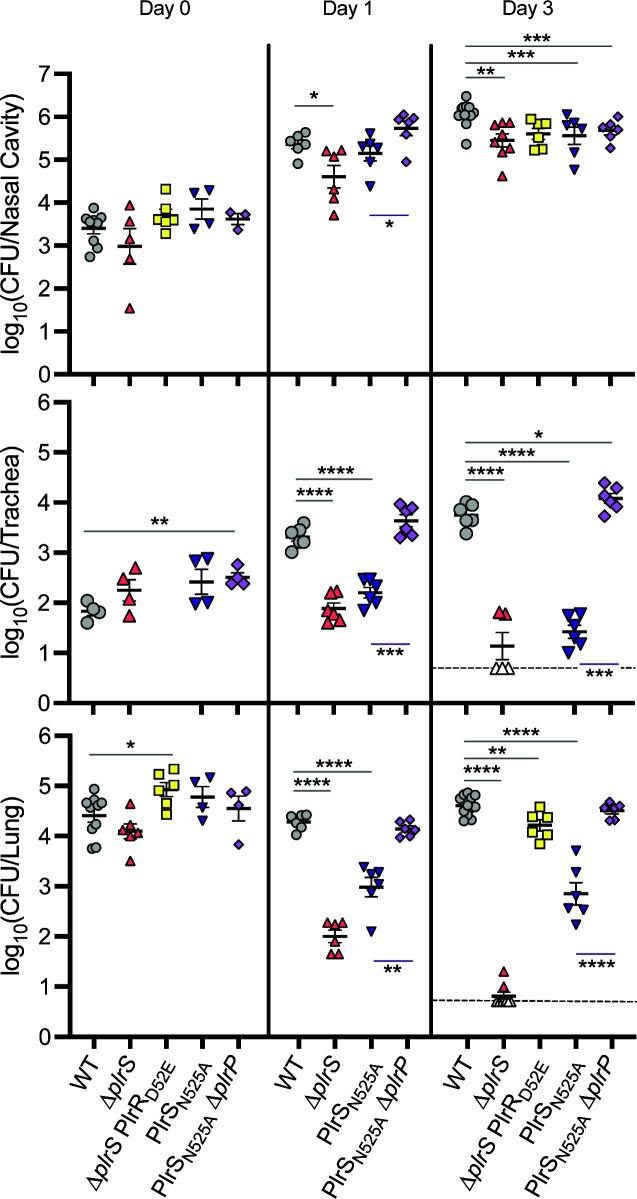 Fig 6
Fig 6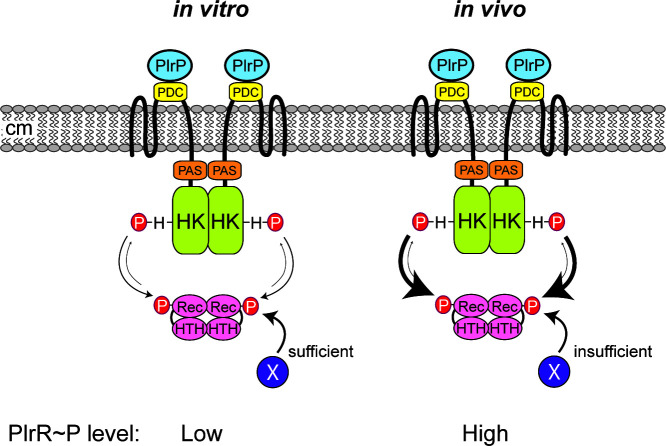 Fig 7
Fig 7| Species | Accession or assembly number | Gene IDs |
|---|---|---|
|
|
| AZC_3081–3088 |
|
|
| OH82_RS30760–RS30810 |
|
|
| BB0262–0267 |
|
|
| AAV28_RS19055–RS19085 |
|
|
| BruAb1_1120–1125 |
|
|
| CC1739–CC1744 |
|
|
| LV780_RS06670–RS06710 |
|
|
| ACP92_RS00340–RS00355 |
|
|
| HT085_RS10140–RS10180 |
|
|
| Pden_4123–4131 |
|
|
| RSP_2836–2844 |
|
|
| V7S97_RS16985–RS17025 |
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Bacterial Infections and Vaccines · Metalloenzymes and iron-sulfur proteins
INTRODUCTION
Bordetella species, including the classical bordetellae, are β-proteobacteria that survive in a broad range of environmental and eukaryotic niches. Bordetella bronchiseptica, the causative agent of kennel cough in dogs, infects most mammals, causing a chronic, often asymptomatic disease. It can also survive long-term in extra-host environments, such as filtered pond water (1). By contrast, Bordetella pertussis and Bordetella parapertussishu, which evolved from a B. bronchiseptica-like ancestor, are respiratory pathogens that survive only in the human host (causing whooping cough) and briefly during transmission. Despite high vaccination rates, whooping cough cases are on the rise globally, and better vaccination strategies are needed (2, 3).
All known protein virulence factor-encoding genes in Bordetella spp., including those that encode the components of current acellular vaccines, are regulated by the two-component regulatory system (TCS) BvgAS, which has been regarded as the “master virulence regulator” (4–8). BvgAS controls at least three distinct phenotypic modes. Bvg^+^ mode, produced when BvgAS activity is high and virulence-associated genes (vags), such as those encoding toxins and adhesins, are maximally expressed, is both necessary and sufficient for bacterial persistence in vivo (8–12). Bvg^–^ mode, produced when BvgAS is inactive and virulence-repressed genes (vrgs), required for flagella synthesis and motility and other phenotypes, are maximally expressed, does not appear to occur in vivo but is required for B. bronchiseptica survival in nutrient-limited environments (9, 13, 14). The Bvg^i^ mode is produced when BvgAS is partially active and is hypothesized to occur during transmission between mammalian hosts (15, 16).
In 2011, another TCS, PlrSR, was discovered to be required for B. bronchiseptica persistence in the lower respiratory tract (LRT) of rats and mice (17, 18). In addition, BvgAS activity was found to be dependent on PlrSR in the LRT (18), suggesting a functional link between the two TCSs. However, even when BvgAS is constitutively active, a mutant that produces non-functional PlrS (∆plrS) fails to survive in the LRT, indicating that the essentiality of PlrSR in the LRT is independent of its effects on BvgAS (18). Therefore, PlrSR must regulate one or more BvgAS-independent genes that is/are required for persistence in the LRT.
PlrS is a histidine sensor kinase (HK) protein with a predicted periplasmic PhoQ-DcuS-CitA (PDC) sensory domain, followed by cytoplasmic HAMP, PAS, HK, and HATPase domains. PlrS has a motif (HEIKN) at its primary site of phosphorylation (H521) that is characteristic of sensor kinases that can act as both a kinase and a phosphatase toward their cognate response regulator proteins (19, 20). PlrR is a typical response regulator protein with an N-terminal receiver domain and a C-terminal DNA-binding domain. A B. bronchiseptica mutant encoding PlrS with glutamine substituted for histidine at position 521 (H521Q) is as defective as a PlrS mutant missing aa 5-198 (∆plrS) for persistence in the LRT (17), indicating that phosphorylated PlrS, and thus, presumably, phosphorylated PlrR, is required in vivo. While the H521Q mutant is defective for kinase activity in vitro (21), its phosphatase activity is unknown. The asparagine residue at position H+4 (N525) is required for phosphatase activity, and biochemical analyses indicate that PlrS with alanine substituted for N525 (N525A) may also be partially defective for kinase activity (21).
PlrSR is a member of the NtrYX family of TCSs that are widely distributed in proteobacteria and involved in regulating nitrogen metabolism, cell envelope processes, respiratory enzymes, and iron homeostasis (22–29). ntrYX homologs appear to have evolved from ntrBC, which encodes a TCS important in controlling nitrogen metabolism in proteobacteria (30, 31). NtrYX systems evolved further such that homologs in ⍺-proteobacteria differ from those in β-proteobacteria. Specifically, NtrX response regulator proteins of β-proteobacteria lack the AAA+ domain that is present in NtrX proteins of ⍺-proteobacteria (32). Moreover, the ntrYX genes in β-proteobacteria are 3′ to two highly conserved genes: one predicted to encode an RNA methyltransferase (rsmB), and one predicted to encode a periplasmic proline-rich domain of unknown function protein (which we are calling plrP) (32). The role of this highly conserved genetic organization is currently unknown. Our analyses revealed genetic evidence that plrP is involved in PlrSR signaling, and we hypothesize it may be a conserved component of NtrYX TCSs in other β-proteobacteria.
RESULTS
plrSR are located within a six-gene cluster with operon-like structure
The plrSR genes are in the middle of a cluster of genes oriented in the same transcriptional direction (Fig. 1A; BB0262-0267). 5′ to plrS are two genes: rsmB, predicted to encode an rRNA methyltransferase, and BB0263, here named plrP, predicted to encode a protein containing a domain of unknown function. The rsmB homolog from Escherichia coli encodes a product that methylates C697 of the 16S rRNA (m^5^C967), a residue involved in interactions with tRNA (33–35). By stabilizing the interaction between the pre-initiation complex and the initiating tRNA bearing methionine, m^5^C967 has been shown to impact the efficiency of translation initiation and thus the overall bacterial proteome (36). plrP is predicted to encode a 203 amino acid protein containing DUF4390. SignalP analysis identifies a signal sequence (amino acids 1–35) for exportation to the periplasm with 96% confidence (37).
Conservation of genes surrounding ntrY and ntrX homologs in proteobacteria. (A) Schematic of the plrSR gene cluster in Bordetella spp. containing locus BB0262-BB0267. plrP and plrS overlap by four base pairs, as do plrR and trkA. (B) Schematic showing the organization of the gene region surrounding ntrYX homologs, centered on the ntrY homolog. Genes that are predicted to encode the same type of protein are similarly colored. Shaded lines connecting genes indicate >30% sequence identity. (C) Distribution of genes encoding DUF4390-containing proteins within proteobacteria. Red lines represent orders that contain genomes with genes encoding DUF4390-containing proteins. Gray lines represent orders that do not. (D) Gene presence/absence plot of plrS homologs vs plrP homologs. Score ratios closer to 1 indicate a greater degree of homology to the queried genes. A score ratio below 0 indicates no homolog was detected in that genome. Solid green dots indicate the plrS and plrP homologs are found within 5 kb of each other in the genome. Orange dashed line indicates the arbitrary threshold for good homology (30% of the maximum bit score).
The two genes 3′ to plrSR (trkA and trkH) are predicted to encode a potassium transporter. Studies with TrkA and TrkH from Vibrio parahymolyticus indicate that TrkH forms the membrane channel through which potassium ions can be transported, while TrkA remains cytosolic, regulating ion flux through TrkH by changing conformation in response to intracellular ADP/ATP levels (38).
The fact that all six genes in the plrSR-containing cluster are oriented in the same transcriptional direction with minimal or no intergenic sequence between predicted start and stop codons suggests they form an operon. The gene 5′ to rsmB is oriented in the opposite transcriptional direction with a 107 base pair (bp) intergenic region. While the gene 3′ to trkH is in the same transcriptional direction as the gene cluster, 79 bp separate the two genes. There are only 18 bp between the predicted stop codon of rsmB and the predicted start codon of plrP, and 9 bp between the predicted stop codon of plrS and the predicted start codon of plrR. The 3′ end of plrP overlaps with the 5′ end of plrS by 4 bp, and the 3′ end of plrR overlaps with the 5′ end of trkA by 4 bp. The largest intergenic region, 48 bp, is between the predicted stop codon of trkA and the predicted start codon of trkH. Based on these features, we hypothesized that there is a promoter 5′ to rsmB and that the six genes are expressed as an operon.
rsmB, plrP, and plrSR are colocalized in β- and γ-proteobacteria
A previous study examined the genes surrounding ntrYX and found a distinction between α- and β-proteobacteria (32). In ⍺-proteobacteria, ntrYX was 3′ to ntrBC, genes encoding another closely-related TCS. In β-proteobacteria, ntrYX was 3′ to an rsmB homolog and a gene encoding a proline-rich DUF4390-containing protein like PlrP. To further assess this evolutionary distinction, we compared the gene neighborhood structure of NtrYX-encoding systems within 12 bacterial species, including B. bronchiseptica, where the TCS has been studied*,* using the gene cluster comparison program CAGECAT clicker (39) (Fig. 1B). As reported, the three β-proteobacteria, including B. bronchiseptica, had rsmB and plrP homologs 5′ to their plrSR homologs. The nine ⍺-proteobacteria had ntrBC, as well as a nifR3 or dusB homolog, 5′ to their plrSR homolog. In both the α- and β-proteobacteria, the genes 3′ to their plrSR homologs varied. However, trkA and trkH, which are 3′ to plrSR in B. bronchiseptica, were also located 3′ to plrSR homologs in seven and four ⍺-proteobacteria species, respectively, but not in other β-proteobacteria.
To assess the relationship between the six genes within the plrSR gene cluster in a wider selection of genomes*,* we examined the rate of co-occurrence of these genes using the fast.genomics database (40). First, we examined the neighborhood architecture surrounding the top 200 closest homologs to plrS. Out of the 198 with complete sequencing of the region, 195 (98%) had homologs of rsmB and plrP 5′ to the plrSR homologs. By contrast, only 53 (27%) had trkA and 46 (23%) had trkH 3′ to the plrSR homologs, further indicating a weaker conserved relationship between plrSR and trkAH than between plrSR, rsmB, and plrP.
Given the conservation of the genes 5′ to plrSR, we further examined the distribution of rsmB and plrP within proteobacteria using AnnoTree, a tool for visualizing the distribution of genes across large phylogenetic trees (41). Genes encoding proteins containing DUF4390 were found within the genomes of β- and γ-proteobacteria, as well as Magnetococcia and ζ-proteobacteria, but not within the genomes of ⍺-proteobacteria (Fig. 1C). Of the β- and γ-proteobacteria included in AnnoTree, a minority (37%) encoded a plrP homolog. rsmB, however, was widely distributed; 94.3% of all proteobacteria examined encoded at least one copy (Fig. S1A).
We then focused our analysis on the co-occurrence of rsmB, plrP, and plrSR across the entire fast.genomics database. rsmB homologs were widely distributed even outside of proteobacteria and were found within 76% of the genomes queried, versus the 47% of genomes containing a plrS homolog (Fig. S1B). While many genomes contained either rsmB or plrS homologs, the likelihood of encoding both genes increased as the degree of homology increased. Using a cut-off of 30% of the maximum bit score as an indicator of good homology, 336 plrS and 490 rsmB homologs were identified. A total of 313 of these genomes contained good homologs of both genes, and 311 (99.4%) of these homologs were located within 5 kb of each other, indicating a conserved functional relationship.
Within the genomes queried, only 446 (6.1%) contained plrP homologs. However, 441 of those 446 genomes (98.9%) also contained intact plrS homologs (Fig. 1D). The other five genomes with plrP homologs contained incomplete sequences or pseudogenes with homology to plrS. Of the 441 genomes with annotated homologs of both plrP and plrS, 437 (99.1%) contained both genes within 5 kb of each other. Again, using a cut-off of 30% of the maximum bit score as an indicator of good homology, 336 plrS and 94 plrP homologs were identified. Of the 94 genomes containing good plrP homologs, all 94 (100%) also contained a good plrS homolog, and in every case, these homologs were within 5 kb of each other. Manual examination of the 336 plrS homologs with good homology determined that 333 of these homologs (99.1%) had a gene encoding a DUF4390-containing protein 3′ to it, even if these genes were not determined to have homology to plrP. While the vast majority (94%) of these good PlrS homologs were found in the genomes of β-proteobacteria, specifically Burkholderiales, this cut-off also included γ-proteobacteria, which had the same neighborhood architecture. This colocalization of plrS and plrP homologs strongly implies that plrP or the protein it encodes interacts in some capacity with plrS or PlrS and that this interaction is important to the function of the TCS in β-proteobacteria.
RNAseq data suggest internal promoters 5′ to plrP and plrR
Attempts to delete plrR or sequences near the 3′ end of plrS have been unsuccessful, suggesting that plrR is essential in vitro and that its expression is driven by a promoter within the 3′ end of plrS. To investigate this hypothesis, we analyzed RNAseq data across the rsmB, plrP, plrS, plrR, trkA, and trkH regions from a previous study (42). Among all the conditions assessed (ambient air [~21% O_2_], 5% O_2_, 2% O_2_, and 20% O_2_ 5% CO_2_), few or no transcripts were detected for rsmB (Fig. 2; Fig. S2). A moderate number of transcripts was detected at the 5′ end of plrP, reaching a peak ~200 bp within the gene and extending through plrS (Fig. 2). Many more transcripts were detected at the 5′ end of plrR, continuing, and decreasing, through trkA and trkH (Fig. 2). These data suggest that, under the conditions tested, rsmB is not expressed, that there is a weakly active promoter within the 3′ end of rsmB, and that there is a strongly active promoter within the 3′ end of plrS.
RNAseq analysis of the plrSR gene cluster suggests two internal promoters. Graph of RNA transcripts measured using RNAseq from B. bronchiseptica samples that were grown in ambient air conditions (42). Data were visualized using the Integrative Genomic Viewer (43).
PplrP and PplrR are active in vitro
To determine if sequences within the 3′ end of rsmB and the 3′ end of plrS, as well as those 5′ to rsmB (Fig. 3), contain promoter activity, we cloned DNA fragments from each region 5′ to the promoterless gfp gene in plasmid pMABgfp, delivered the putative promoter-gfp cassettes to the chromosomal attTn7 site in wild-type and ∆plrS bacteria, and measured fluorescence after growing the strains in Stainer-Scholte (SS) medium (standard liquid growth medium for Bordetella spp.) with and without the addition of 40 mM MgSO_4_ (40 mM MgSO_4_ inactivates BvgS [44]). As a control to show that 40 mM MgSO_4_ modulated BvgAS activity in our experiments, we included a strain containing a PbvgA-gfp fusion. bvgAS is positively autoregulated, and in addition to BvgAS being inactive in SS medium containing 40 mM MgSO_4_, BvgAS activity is somewhat diminished when PlrS is inactive in vitro (18). Consistent with these previously published reports, the wild-type strain containing the PbvgA-gfp fusion was highly fluorescent in SS medium and minimally fluorescent in SS medium containing 40 mM MgSO_4_, and the ∆plrS strain containing the PbvgA-gfp fusion was highly fluorescent in SS medium but less so than the wild-type strain, and minimally fluorescent in SS medium containing 40 mM MgSO_4_ (Fig. 3E).
*PplrP and PplrR are active under standard in vitro growth conditions. (A) Schematic of the plrSR gene cluster in Bordetella spp. containing locus BB0262–BB0267. Putative promoter regions are annotated with an arrow indicating the direction of transcription. Fragments cloned to the promoterless gfp gene are annotated with a line and the corresponding size in base pairs. Strains containing PrsmB-gfp (B), PplrP-gfp (C), PplrR-gfp (D), and PbvgA-gfp (E) cassettes at the chromosomal attTn7 site in wild-type and ∆plrS bacteria were grown in SS medium with and without the addition of 40 mM MgSO4. GFP fluorescence (excitation: 485 nm, emission: 535 nm) and OD600 were measured after 16–18 h of growth, and relative fluorescence intensity (RFI) was calculated by dividing GFP fluorescence by OD600 values. Statistical significance, as determined using unpaired Student’s t-test, is indicated as *, P < 0.05; **, P < 0.01; ***, P < 0.001; ***, P < 0.0001.
Fluorescence of strains containing the PrsmB-gfp fusion was minimal under all conditions (Fig. 3B), consistent with the 300 bp 5′ to rsmB not containing a promoter that is active in SS medium, with or without the addition of 40 mM MgSO_4_.
Fluorescence of the wild-type strain containing the PplrP-gfp fusion was low in SS medium and about twofold higher in SS medium containing 40 mM MgSO_4_ (Fig. 3C), suggesting a weak promoter that is more active in the Bvg^–^ mode than the Bvg^+^ mode. The ∆plrS strain was moderately fluorescent in SS medium and slightly more fluorescent in SS medium containing 40 mM MgSO_4_ (Fig. 3C). Under both conditions, fluorescence of the ∆plrS strain was higher than that of the wild-type strain (Fig. 3C). These data indicate that the 400 bp region 5′ to plrP contains a promoter that is weakly active under the conditions tested and that is somewhat negatively regulated by both PlrS and BvgAS. By contrast, the wild-type and ∆plrS strains containing the PplrR-gfp fusion were highly fluorescent under all conditions tested (Fig. 3D). These data indicate the presence of a promoter within the 3′ end of plrS.
Overall, these data indicate that plrP and plrS are likely transcribed from a promoter within the 3′ end of rsmB that is weakly active under the conditions tested, and that plrR, trkA, and trkH are likely transcribed from a promoter within the 3′ end of plrS that is moderately active under the conditions tested. However, the presence of other unidentified promoters within this region cannot be ruled out.
rsmB, trkA, and trkH are not required for B. bronchiseptica persistence during murine infection
To determine if rsmB, trkA, and trkH contribute to infection, we constructed strains with in-frame deletion mutations in each gene and compared them to wild-type B. bronchiseptica for their ability to persist in the murine respiratory tract. Mice were inoculated intranasally with 7.5 × 10^4^ CFU, and bacterial burdens in the nasal cavity, trachea, and right lung lobes were determined at days 0, 1, and 3 post-inoculation. Unlike the ∆plrS mutant, which is severely defective for persistence relative to the wild-type strain in the LRT (trachea and lung), but not the nasal cavity (17, 18), the ∆rsmB and ∆trkAH mutants were recovered at levels similar to those of the wild-type strain in the nasal cavity, trachea, and lung at every time point (Fig. 4). These data indicate that neither rsmB nor trkAH is required for B. bronchiseptica persistence during infection in this murine model.
rsmB, trkA, and trkH are not required for B. bronchiseptica persistence in the murine respiratory tract. Bacterial burden over time within the nasal cavity (top), trachea (middle), and right lung lobes (bottom) of mice infected with 7.5 × 104 CFU of wild-type (WT), ΔrsmB, or ΔtrkAH bacteria. n = 4 for day 0, and n = 6 for days 1 and 3, with each symbol representing a single mouse.
plrP is essential in vitro when plrS is unmutated
To determine if plrP is required for persistence in the LRT, we attempted to construct a strain containing an in-frame deletion in plrP. We used an allelic exchange plasmid designed to delete codons 127–600, leaving 12 bp at the 3′ end of plrP, which overlaps by 4 bp with plrS (Fig. 1A). We were able to obtain co-integrants with this plasmid, but all colonies (>64 screened) obtained after growth with no antibiotic selection and then plating on agar containing 20% sucrose (i.e., bacteria in which the plasmid has recombined out of the chromosome) contained wild-type plrP, suggesting that plrP is essential in vitro. Using the same allelic exchange plasmid, we were able to delete plrP in the PlrS_H521Q_ strain, in which PlrS is unable to be phosphorylated (21), as well as the PlrS_N525A_ strain, in which PlrS is defective for phosphatase activity (21). Using this allelic exchange plasmid, we could not attempt to delete plrP from the ∆plrS strain because the ∆plrS strain lacks appropriate homologous sequences. We were also able to delete plrP in a strain in which plrR was deleted from the native site and plrR in which the codon for D52 (the primary site of phosphorylation in PlrR) was replaced with a codon for glutamic acid (PlrR_D52E_), a substitution that is predicted to function as a phosphomimetic, was expressed at the attTn7 site. Together, these observations suggest that plrP is essential in vitro in a manner that is dependent on PlrS functionality, and they support the hypothesis that PlrR must be phosphorylated, at least at a low level, in vitro, and that PlrP prevents PlrS from fully dephosphorylating PlrR~P in vitro (i.e., that, during growth in SS, PlrP keeps PlrS in “kinase mode”).
plrP essentiality requires the PlrS PDC domain
Because PlrP is predicted to contain a signal sequence that directs it to the periplasm, we hypothesized that PlrP affects PlrS activity by interacting with the PlrS PDC domain. We constructed a strain in which codons 463–720 of plrS are deleted, resulting in the deletion of just the PDC domain. This strain has no obvious growth or colony phenotype in vitro, similar to the ∆plrS strain and the PlrS_H521Q_ strain. Unlike the case with wild-type bacteria, we were able to delete plrP in the ∆PDC strain, indicating that PlrP essentiality in vitro requires the PDC domain of PlrS, supporting the hypothesis that PlrP affects PlrS activity via the PlrS PDC domain.
The PlrS PDC domain is not required for B. bronchiseptica persistence in the lower respiratory tract, and if plrP plays a role in vivo, its role requires the PlrS PDC domain
We compared the ∆PDC and ∆PDC ∆plrP strains for their ability to persist in the murine LRT. The mutants were recovered from all sites in the respiratory tract at levels similar to those of wild-type bacteria (Fig. 5), indicating that the PlrS PDC domain is not required in vivo, and that if PlrP plays a role during infection, that role requires the PlrS PDC domain.
*B. bronchiseptica persistence in the murine respiratory tract does not require the PlrS PDC domain. Bacterial burden over time within the nasal cavity (top), trachea (middle), and right lung lobes (bottom) of mice infected with 7.5 × 104 CFU of wild-type (WT), ΔplrS, ∆PDC, or ∆PDC∆plrP bacteria. n = 4 for day 0, and n = 6 for days 1 and 3, with each symbol representing a single mouse. The dashed line represents the limit of detection. Empty symbols represent symbols below the limit of detection. Statistical significance, as determined using unpaired Student’s t-test, is indicated as *, P < 0.05; **, P < 0.01; ***, P < 0.001; ***, P < 0.0001. The absence of asterisks indicates P > 0.05 and no statistically significant difference.
Evidence that PlrR must be phosphorylated at a high level in vivo
Although the ∆plrS and PlrS_H521Q_ strains grow in vitro, they are cleared rapidly from the LRT (17), suggesting that PlrRP is required for bacterial survival in the LRT. The ∆plrS strain producing PlrR with the D52E phosphomimetic substitution (∆plrS PlrR_D52E_) is viable in vitro and, unlike the ∆plrS strain, the ∆plrS PlrR_D52E_ strain adheres to L2 cells at levels similar to wild-type bacteria when incubated in the presence of 5% CO_2_, indicating that PlrS activity is not required for PlrR activity in vitro if PlrR contains the D52E phosphomimetic substitution (18). These data suggest that PlrR is phosphorylated to some extent in vitro, and this level of phosphorylation is required for the increased adherence that occurs in the presence of CO_2_ (18). In the murine model, the ∆plrS PlrR_D52E_ strain was slightly defective for persistence in the LRT relative to wild-type bacteria (Fig. 6), suggesting that PlrR_D52E_ is not as active as PlrRP and that high levels of PlrR~P are required in vivo.
*PlrRD52E is not as active as PlrR~P and PlrP affects PlrS activity in the murine respiratory tract. Bacterial burden over time within the nasal cavity (top), trachea (middle), and right lung lobes (bottom) of mice infected with 7.5 × 104 CFU of wild-type (WT), ∆plrS, PlrSN525A, PlrSN525A∆plrP, or ∆plrS PlrRD52E bacteria. These data are compiled from two independent experiments, each performed in biological duplicate. For experiment 1, which includes WT, ∆plrS, PlrSN525A, and PlrSN525A∆plrP strains, n = 4 for day 0, and n = 6 for days 1 and 3. For experiment 2, n = 6 for days 0 and 3 for the WT and ∆plrS PlrRD52E strains, and n = 2 for days 0 and 3 for the ∆plrS strain. Each symbol represents a single mouse. Dashed line represents the limit of detection. Empty symbols represent symbols below the limit of detection. Statistical significance, as determined individually for each data set using unpaired Student’s t-test, is indicated as *, P < 0.05; **, P < 0.01; ***, P < 0.001; ***, P < 0.0001. The absence of asterisks indicates P > 0.05 and no statistically significant difference.
Evidence that PlrP affects PlrS activity in vivo
Because we could not delete plrP in wild-type bacteria, we could not determine directly if plrP is required during respiratory infection. The PlrS_N525A_ strain is defective for persistence in the LRT, but not as defective as the ∆plrS strain ([21], Fig. 6). Because PlrS_N525A_ is defective for phosphatase activity, we previously concluded that both PlrS kinase and phosphatase activity must be required in the LRT. However, the PlrS_N525A_ ∆plrP double mutant was recovered from the LRT at levels similar to the wild-type strain at both day 1 and day 3 post-inoculation (Fig. 6). The most likely explanation for these data is that PlrS_N525A_ is defective for both kinase and phosphatase activity, and that the absence of PlrP causes PlrS_N525A_ to have increased kinase activity—further supporting the hypothesis that PlrR~P levels must be higher in the LRT than when the bacteria are growing in SS medium in vitro. Most importantly, these data provide evidence that PlrP affects PlrS activity in vivo.
DISCUSSION
Our data strongly suggest that PlrP is a third component of the PlrSR (NtrYX) subfamily of TCSs, and they support the model shown in Fig. 7 and further explored in Fig. S3 and S4. According to this model, when B. bronchiseptica is grown under standard laboratory conditions in vitro, PlrS functions as a weak kinase (or kinase and phosphatase activities are balanced) such that PlrRP levels are low. PlrP, potentially by interacting with the PlrS PDC domain, prevents PlrS from acting as a strong phosphatase against PlrRP. Our data indicate that PlrRP is essential in vitro, and that in the absence of PlrS kinase activity, PlrR can obtain a phosphoryl group from another molecule. According to our model, a high level of PlrRP, which is dependent on PlrS kinase activity, is required in the LRT during infection. PlrP likely also affects PlrS activity in vivo, although its exact role in vivo is unclear.
Model for PlrPSR activity in vitro and in vivo. According to this model, PlrS functions as a weak kinase when B. bronchiseptica is growing in SS medium in vitro, and PlrP functions to prevent PlrS from acting as a phosphatase. Another phosphodonor (X) can support PlrR phosphorylation if PlrS is mutated. When B. bronchiseptica is growing in the LRT (in vivo), PlrS has strong kinase activity toward PlrR, and PlrR~P levels are high. Although PlrP affects PlrS activity in the LRT, its role is unknown.
Attempts to delete plrR have been unsuccessful (17, 18), and it was unknown whether PlrR (i.e., non-phosphorylated PlrR) or PlrRP is essential in vitro. Because strains in which PlrS cannot autophosphorylate, such as the ∆plrS and PlrS_H521Q_ strains, are viable, either PlrR is essential, or PlrR must be able to obtain a phosphoryl group from another molecule in vitro. Our new data showing that plrP can be deleted in the PlrS_N525A_ strain, which is defective for phosphatase activity, but not in the wild-type strain, suggests that PlrP prevents strong phosphatase activity by PlrS, which is lethal in vitro, presumably due to the dephosphorylation of PlrR. Therefore, the PlrS_H521Q_ strain must also be defective for phosphatase activity. The fact that plrP can also be deleted in the PlrR_D52E_ strain, in which the mimicked phosphorylation state of PlrR is independent of PlrS, indicates that the essentiality of plrP is dependent on the phosphorylation state (or mimicked phosphorylation state) of PlrR. Together, these data indicate that PlrRP is essential in vitro, and that PlrR must be able to obtain a phosphoryl group from another molecule in vitro.
How does PlrP prevent strong phosphatase activity by PlrS? Because PlrP is predicted to contain a signal sequence for export to the periplasm, we hypothesized that PlrP affects PlrS activity via its periplasmic PDC domain. Since DUF4390 composes the majority of PlrP (161 out of the 168 amino acids of the mature peptide), it is likely that this domain is involved in the interaction. The fact that plrP can be deleted in the ∆PDC strain suggests that the essentiality of PlrP depends on the PlrS PDC domain, and that the PDC domain controls PlrS phosphatase activity in vitro. While our genetic analyses suggest that PlrP interacts with the PlrS PDC domain in the periplasm to prevent phosphatase activity, further biochemical analyses are required to determine colocalization and direct versus indirect interactions. These analyses would also elucidate the function of DUF4390, a widely distributed but as-yet undefined domain.
Based on previous studies, replacement of the Asp that is the primary site of phosphorylation in a response regulator protein with Glu, a phosphomimetic, results in a response regulator that is at least partially active (45). We showed previously that the in vitro adherence defect of a ∆plrS mutant is complemented by PlrR_D52E_, suggesting that PlrR_D52E_ is active in vitro (18). Although the ∆plrS PlrR_D52E_ strain was not nearly as defective as the ∆plrS strain in the LRT, it did not persist quite as well as the wild-type strain, suggesting that PlrR_D52E_ is not as active as PlrRP and that high levels of PlrPP are required for persistence in the LRT. Because the PlrS_H521Q_ mutant is viable in vitro but not in vivo, the predicted alternate phosphodonor for PlrR must either not be present when the bacteria are in the LRT, or, more likely, it is unable to produce sufficient levels of PlrRP for in vivo survival. Together, these data suggest that while relatively low levels of PlrRP are sufficient in vitro, high levels of PlrR~P are required in vivo.
Determining the exact role of PlrP in vivo is a challenge, since we cannot delete plrP in the wild-type strain in vitro. Our results suggest that neither the PlrS PDC domain nor PlrP in the ∆PDC mutant is required for B. bronchiseptica persistence in the LRT (Fig. 6), but in the PlrS_N525A_ mutant, PlrP must affect PlrS activity in vivo (Fig. 6). While the N525A substitution results in PlrS that cannot dephosphorylate PlrR (as predicted), PlrS_N525A_ is also slightly defective for kinase activity (21). Therefore, the persistence defect of the PlrS_N525A_ mutant in vivo could be due to reduced kinase activity. In which case, PlrS_N525A_ in the absence of PlrP has kinase activity sufficient for B. bronchiseptica persistence in the LRT. These results are puzzling yet informative, as we continue to demonstrate that PlrSR activity significantly differs between B. bronchiseptica in vitro and in vivo growth conditions.
plrP is not unique to B. bronchiseptica. Not only are homologs of plrP found in the genomes of many β- and γ-proteobacteria, but they are also almost exclusively found 5′ to plrSR homologs. The conserved structure of this gene cluster indicates a conserved functional link between these genes. Therefore, it is likely that other PlrP homologs modulate the kinase and/or phosphatase activity of their cognate PlrS homologs.
PlrP is not the first protein proposed to contribute to the function of an NtrYX-family TCS. In the ⍺-proteobacterium Caulobacter vibrioides, NtrZ, a putative periplasmic protein, was found to regulate the levels of phosphorylated NtrX through NtrY, presumably by reducing NtrY phosphatase activity (29). Unlike plrP, ntrZ is not encoded adjacent to ntrYX and is not predicted to encode a DUF4390-containing protein. Additionally, NtrZ has only 15.1% sequence identity with PlrP. However, COBALT comparison of PlrP and NtrZ indicated conservation between the two proteins that includes all the amino acids encoded by NtrZ (46). This result opens the possibility that utilization of a periplasmic regulatory protein is common in NtrYX-family TCSs, even outside of proteobacteria.
Our results indicate that plrP is essential in wild-type B. bronchiseptica in vitro but possibly not in vivo. Given the essentiality of PlrSR during infection and the proposed interaction between PlrP and PlrSR, this finding was unexpected. However, NtrYX-family TCSs are not exclusively linked to pathogenesis. In many bacteria, NtrYX-family TCSs regulate critical cellular functions, including nitrogen fixation and response to oxygen tension (22, 23, 47). PlrP is likely important for responding to a signal that B. bronchiseptica does not encounter within a mammalian host but may encounter when living in the environment. Similarly, rsmB, which was very frequently found 5′ of plrP and plrSR, could contribute to survival outside a host. Alternatively, since rsmB expression remained below the detection limit under all the conditions we tested*, rsmB* may have been conserved due to the promoter in its 3′ end.
We began this analysis of the gene cluster surrounding plrSR in part to parse what contributed to the difficulty of performing genetic manipulations within this region. Through RNAseq analysis and promoter-gfp fusion assays, we have determined that, at least under laboratory conditions, this cluster does not function as an operon. Instead, there are two promoters for this region, beginning 5′ of plrP (PplrP) and 5′ of plrR (PplrR). The localization of these two promoters allows for differential regulation of plrS and plrR. In fact, under laboratory conditions, PplrR was a stronger promoter than PplrP (Fig. 3). The decoupling of plrS and plrR expression indicates that different stoichiometries of PlrS and PlrR are required under different conditions. Further studies are required to determine co-transcription of the genes within these regions. With these promoters identified, it will be possible to generate mutations that maintain regulation through this region, allowing deeper analysis of this unusual but clinically important two-component system.
MATERIALS AND METHODS
Bacterial culturing
B. bronchiseptica strains were grown on Bordet-Gengou (BG) agar plates (BD Biosciences) supplemented with 6% defibrinated sheep blood (Hemostat) at 37°C for 2–3 days. B. bronchiseptica strains were grown in Stainer-Scholte (SS) broth supplemented with SS supplement ([48], updated in reference 49) at 37°C on a rotating wheel to increase aeration overnight or until the desired density was reached. E. coli strains were grown in lysogeny broth (LB) at 37°C on a rotating wheel overnight or on LB agar plates at 37°C for 1–2 days. As needed, media were supplemented with streptomycin (Sm, 20 μg/mL), gentamicin (Gm, 30 μg/mL), kanamycin (Km, 50 μg/mL), diaminopimelic acid (DAP, 300 μg/mL), ampicillin (Ap, 100 μg/mL), or sucrose (15% wt/vol). All cultures were started from individual colonies from a clonal population when possible.
Construction of plasmids and strains
The strains and the plasmids that were used in this study can be found in Table S1. In-frame deletions were constructed via allelic exchange using derivatives of the pEG7S plasmid (50). Transcriptional reporter strains were constructed via transposase-mediated insertion at the attTn7 site using derivatives of the pUCgfpMAB vector (51). Plasmids were constructed and propagated within the DH5a E. coli strain. The RHO3 E. coli strain was used for introducing plasmids into B. bronchiseptica through conjugation. Plasmids were confirmed using sequencing, and all mutations introduced in B. bronchiseptica strains were confirmed by PCR.
Bioinformatic analysis of the plrSR gene cluster
The gene neighborhoods of the genes encoding NrYX-family TCSs outlined in Table 1 were aligned using CAGECAT clinker (39). An unbiased examination of the gene cluster was performed using fast.genomics (40). This database uses B. pertussis strain 18323 as the representative Bordetella strain. We used the “gene neighborhood tool” to identify the closest homologs to plrS (BN118_RS17975; 200 top hits, 9 kb neighborhood). This data set was manually examined for neighborhood structure similarities, with samples being removed if they lacked complete sequencing across the 9 kb region. We used the “compare gene presence/absence” tool to examine the co-occurrence of plrS, plrP (BN118_RS17980), and rsmB (BN118_RS17975). For plrs/plrP, the table of the best homologs of both genes was used for further analysis. The gene neighborhoods of the good plrS homologs were manually examined for the presence of an adjacent gene encoding a DUF4390-containing protein. AnnoTree (AnnoTree v2.0.0; GTDB Bacteria Release R214) was used to visualize the distribution of plrP (Pfam ID: PF14334) and rsmB (Pfam ID: PF01189) across proteobacteria (41).
RNA sequencing analysis
The RNA sequencing data used in this study have been previously published (42); the raw sequencing files can be retrieved from the GEO repository (GSE268598). Read counts were determined using the igvtools Count command on BAM alignment files within the Integrative Genomic Viewer (43). Counts were mapped to the RB50 genome (RefSeq NC_002927.3).
In vitro promoter activity analysis
The transcriptional reporter strains were grown for 16–18 h in standard SS medium or SS medium containing 40 mM MgSO_4_ at 37°C. A volume of 200 μL of each culture was transferred to a black, clear-bottom 96-well plate (ThermoScientific catalog no. 165305), and the OD_600_ and GFP fluorescence (excitation: 485 nm, emission: 535 nm) were measured on a BioTek Synergy H1 Hybrid Reader plate reader. The relative fluorescence intensity (RFI) was calculated by normalizing GFP fluorescence to OD_600_ and subtracting the empty vector control values. The experiments were performed in biological triplicate.
Bacterial infection of the mouse respiratory tract
Six-week-old female BALB/c mice from Charles River Laboratories (catalog no. BALB/cAnNCrl) were inoculated intranasally with 7.5 × 10^4^ CFU B. bronchiseptica in 50 μL of DPBS. At 0, 1, or 3 days post-infection, the right lung lobes, the trachea, and the nasal cavity tissues were harvested from each mouse. The tissues were homogenized in DPBS using a mini-beadbeater with 0.1 mm zirconia beads (Biospec catalog no. 11079110zx). The number of CFU was determined by plating dilutions of tissue homogenates on BG Sm blood agar and enumerating the number of colonies per tissue after at least 48 h of growth at 37°C. Each experiment was performed in biological duplicate.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Porter JF, Parton R, Wardlaw AC. 1991. Growth and survival of Bordetella bronchiseptica in natural waters and in buffered saline without added nutrients. Appl Environ Microbiol 57:1202–1206. doi:10.1128/aem.57.4.1202-1206.19912059041 PMC 182868 · doi ↗ · pubmed ↗
- 2CDC. 2025. Pertussis surveillance and trends. Whooping cough (Pertussis). https://www.cdc.gov/pertussis/php/surveillance/index.html. Accessed 27 January 2026
- 3Mattoo S, Cherry JD. 2005. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin Microbiol Rev 18:326–382. doi:10.1128/CMR.18.2.326-382.200515831828 PMC 1082800 · doi ↗ · pubmed ↗
- 4Weiss AA, Hewlett EL, Myers GA, Falkow S. 1983. Tn 5-induced mutations affecting virulence factors of Bordetella pertussis. Infect Immun 42:33–41. doi:10.1128/iai.42.1.33-41.19836311749 PMC 264520 · doi ↗ · pubmed ↗
- 5Cummings CA, Bootsma HJ, Relman DA, Miller JF. 2006. Species- and strain-specific control of a complex, flexible regulon by Bordetella Bvg AS. J Bacteriol 188:1775–1785. doi:10.1128/JB.188.5.1775-1785.200616484188 PMC 1426559 · doi ↗ · pubmed ↗
- 6Moon K, Bonocora RP, Kim DD, Chen Q, Wade JT, Stibitz S, Hinton DM. 2017. The Bvg AS regulon of Bordetella pertussis. m Bio 8:e 01526-17. doi:10.1128/mbio.01526-1729018122 PMC 5635692 · doi ↗ · pubmed ↗
- 7Decker KB, James TD, Stibitz S, Hinton DM. 2012. The Bordetella pertussis model of exquisite gene control by the global transcription factor Bvg A. Microbiology (Reading) 158:1665–1676. doi:10.1099/mic.0.058941-022628479 PMC 3542142 · doi ↗ · pubmed ↗
- 8Bock A, Gross R. 2001. The Bvg AS two-component system of Bordetella spp.: a versatile modulator of virulence gene expression. Int J Med Microbiol 291:119–130. doi:10.1078/1438-4221-0010911437335 · doi ↗ · pubmed ↗