Sand fly endosymbionts in Kenya: Rickettsia and Wolbachia associations with Leishmania and detection of Rickettsia africae
Steve Kiplagat, Damaris Matoke-Muhia, Barrack O. Owino, David P. Tchouassi, Daniel K. Masiga, Gregory D. D. Hurst, Jandouwe Villinger

TL;DR
Kenyan sand flies host various microbes, including Rickettsia and Wolbachia, which are often found alongside Leishmania parasites.
Contribution
First detection of Rickettsia africae in sand flies and evidence of nonrandom associations between endosymbionts and Leishmania.
Findings
Rickettsia africae was detected in multiple sand fly species in Kenya.
Rickettsia and Wolbachia endosymbionts were positively associated with Leishmania DNA.
Gut bacteria in sand flies may influence vector competence for Leishmania.
Abstract
Sand flies are small hematophagous insects known as leishmaniasis vectors. Similar to most arthropods, they harbor nonobligate endosymbionts that may influence host adaptation and pathogen transmission, but these symbiont communities remain poorly characterized in Leishmania-endemic African sand flies. We screened 1700 wild-caught phlebotomine sand flies (1266 females, 434 males) from Kenya’s Baringo, Nakuru, and Kajiado counties, and 253 colony Phlebotomus duboscqi, for Rickettsia, Wolbachia, Spiroplasma, Cardinium, Arsenophonus, Microsporidia, and Leishmania by high-resolution melting analysis and sequencing of PCR products. In wild sand flies (Phlebotomus and Sergentomyia spp.), Wolbachia spp. were most common (8.5%, 145/1700), with particularly high prevalences in Ph. mireillae (92.3%, 12/13), Ph. guggisbergi (73.2%, 82/112), and Ph. saevus (48.6%, 18/37), followed by Spiroplasma…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
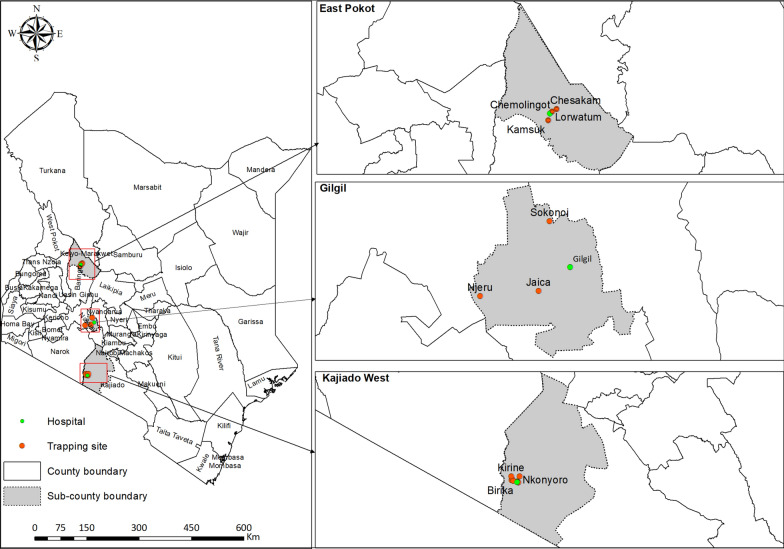 Figure 1
Figure 1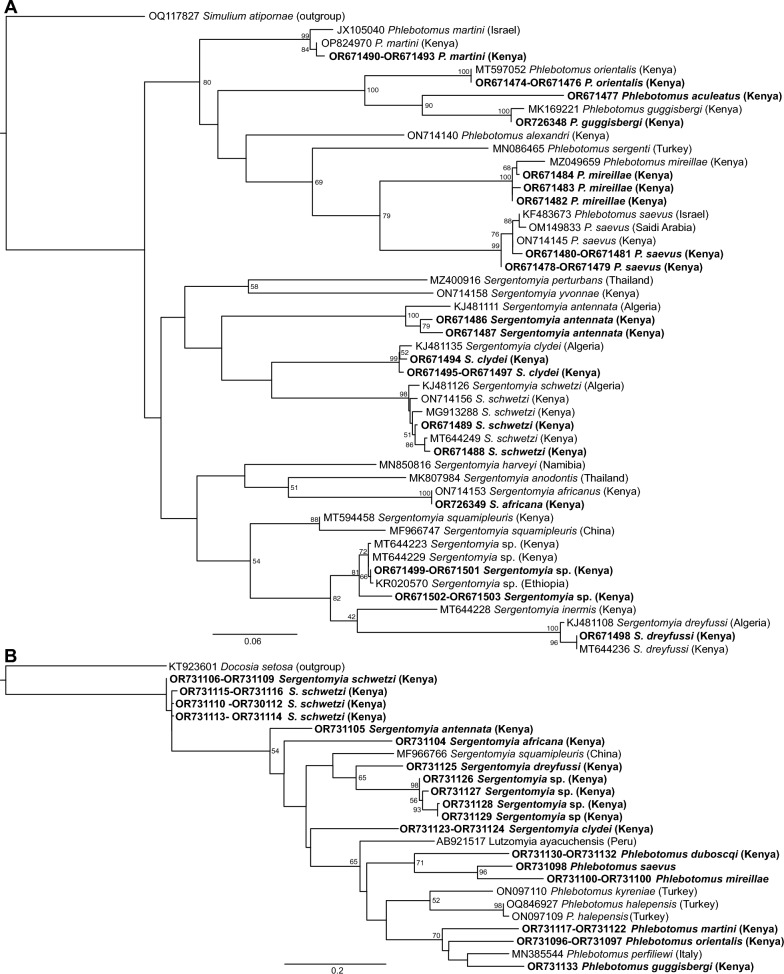 Figure 2
Figure 2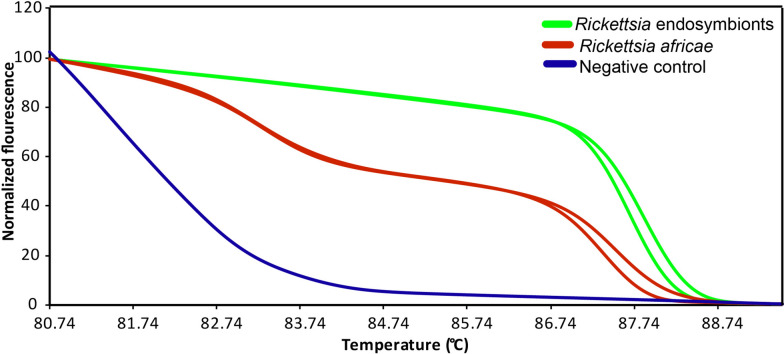 Figure 3
Figure 3 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6- —https://doi.org/10.13039/501100000268Biotechnology and Biological Sciences Research Council
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsResearch on Leishmaniasis Studies · Trypanosoma species research and implications · Insect symbiosis and bacterial influences
Background
Sand flies (Diptera: Psychodidae) are insects of medical importance because of their role in the transmission of various agents of human and veterinary disease [1], including phleboviruses, Bartonella bacilliformis, and most importantly, Leishmania (Kinetoplastida: Trypanosomatidae) parasites, which infect and cause human disabling disease leishmaniasis [1, 2]. Emergence of leishmaniasis outside known endemic areas [3], including complex transmission dynamics recently documented in Kenya [4–7], along with lack of vaccines, emergence of drug resistance, and paucity of new drugs against leishmaniasis [8] has created an imperative for novel disease mitigation approaches, including vector control strategies [9, 10].
Since the discovery of insect gut bacteria with promising impacts in arthropods against vector-borne disease [11–13], microbial endosymbionts of sand flies have gained attention owing to their potential impact on sand fly fitness and growth of Leishmania parasites in host sand flies [14]. They commonly include a range of bacteria and fungi such as Rickettsia (Rickettsiales: Rickettsiaceae), Wolbachia (Rickettsiales: Rickettsiaceae), Cardinium (Bacteroidales: Bacteroidaceae), Spiroplasma (Entomoplasmatales: Spiroplasmatacae), Arsenophonus (Enterobacterales; Morganellaceae), and microsporidia [15]. These microbes are of interest as they can play significant biological roles in controlling disease transmission by either modulating host insect behavior, physiology, or conferring resistance to pathogens and parasites development in the host [16]. Here, we distinguish between vertically transmitted (“heritable”) endosymbionts, such as Rickettsia, Wolbachia, Spiroplasma, Cardinium, and environmentally acquired or transient gut-associated bacteria, which may also modulate sand fly biology but are less likely to be inherited.
In arthropods, endosymbionts of the alphaproteobacterial order Rickettsiales include five major genera of importance as agents of disease or as symbionts of arthropods that spread disease: Anaplasma, Ehrlichia, Orientia, Rickettsia, and Wolbachia. Members of these taxa infect diverse eukaryotic hosts, including humans, livestock, insects, and protists [17]. They are gram-negative bacteria, some of which have a spectrum of endosymbiotic relationships from parasitism to obligatory mutualism in a wide range of arthropods [18]. Wolbachia, for example, has evolved strategies to manipulate host reproduction, including parthenogenesis, feminization, male killing, and cytoplasmic incompatibility to facilitate their own propagation, proliferation, transmission, and others provide an additional fitness benefit for the host to protect against pathogens [18, 19]. Successes in the experimental transinfection of selected strains of Wolbachia into mosquito species such asAedes aegypti have been deployed to prevent transmission of dengue viruses, preventing dengue and Zika virus transmission by either inhibiting binding of the viruses to mosquito cells, directly blocking the virus, or by decreasing the lifespan of the vector [20, 21].
Similar to Wolbachia, Rickettsia have been found to influence host fitness by manipulating reproduction to enhance their transmission and cause thelytokous parthenogenesis (where mothers produce daughters from unfertilized eggs) in parasitoid wasp and Pnigalio soemius (Hymenoptera: Eulophidae) [22] and male-killing in the two spot ladybird Adalia bipunctata [23]. Both Rickettsia and Wolbachia are primarily transmitted vertically to host progeny, across the generations [22, 24], and along with Wolbachia, these two symbionts have the potential to enable biological control strategies against vectors of diseases and their pathogens [24].
Recent research has highlighted the role of bacterial symbionts in vectors of medical importance. A notable discovery is the microsporidian species, microsporidia MB, which impairs the development and transmission of Plasmodium in Anopheles arabiensis mosquitoes [25]. Remarkably, this impairment occurs without significantly affecting the host’s fertility, fecundity, development, or longevity, making this taxon an appealing candidate for pathogen blocking [26]. Consequently, there is need for more research to identify further endosymbionts relevant for innovative biocontrol strategies to curb pathogen transmission [27], especially in sand flies where most of the impactful bacterial endosymbionts remain largely unexplored [28].
To understand the effects and role of endosymbionts on the growth and development of Leishmania and the ecological evolution of their hosts, we screened sand flies collected in three study areas of Kenya’s Rift Valley region for diverse endosymbionts, determined their occurrence, prevalence, and diversities, and investigated potential associations of specific endosymbionts with naturally occurring Leishmania parasites. To our knowledge, this is the first study to jointly characterize multiple heritable endosymbionts, gut bacteria, Leishmania parasites, and the tick-borne R. africae, a zoonotic tick-borne pathogen responsible for African tick bite fever (ATBF), in sand flies from Kenya, and to quantify statistical associations between these microbes.
Methods
Study areas and sand fly collections
We collected sand flies from three study areas in the upper (East Pokot and Gilgil sub-counties) and lower (Kajiado West sub-county) parts of Kenya’s Rift Valley. We selected the study areas and sites (Additional file Supplementary1: Table S1) on the basis of active endemic areas of both cutaneous (CL) and visceral (VL) forms of leishmaniasis in Kenya. The trapping sites included Chesakam, Lorwatum, and Kamsuk in Chemolingot in East Pokot sub-county (Baringo County, 0.01419^o^E, 36.01557^o^N), Thugunui, Njeru, and Jaica in Gilgil sub-county (Nakuru County −0.582909^o^E, 36.083596^o^N), and Nguruman; Birika, Nkonyoro, Olosinyai, Olomanyatta, Empaleki, Enchanipus, Kirine, and Shompole in Kajiado West sub-county (Kajiado County −1.875549^o^S, 36.157326^o^N) (Fig. 1 and Additional file Supplementary1: Table S1). Generally, with exception of Gilgil sub-county where rock crevices were covered by green shrubs, acacia trees were predominant vegetation in all the areas along with shrubs and dry grass. These habitat types are known to influence sand fly population structure in Kenyan leishmaniasis foci [7]. We trapped sand flies predominantly from termite mounds in East Pokot and Kajiado West, while in Gilgil we set sand fly traps in rock crevices, caves, and animal burrows inhabited by rock hyraxes, bats, and rodents, respectively.Fig. 1. Sampling sites of Kenyan sand flies. East Pokot (Top) and Gilgil (Middle) in Baringo and Nakuru counties lie in the upper Rift Valley, whereas Kajiado West Sub-County (bottom) in Kajiado County lies in the lower Rift Valley. Temporary field laboratories were established at Chemolingot Dispensary, Gilgil Sub-County Hospital, and icipe’s field station at Nguruman (Green)
Sand fly collection in East Pokot also involved trapping sand flies in caves and rock crevices in hillocks of volcanic origin. We also sampled sand flies in peridomestic environments, such as nearby animal sheds and the periphery of homesteads, using standard Center for Disease Control (CDC) miniature light traps between 6PM and 7AM the following day. The samples were transported to a temporary field laboratory where we processed sand flies for dissections, morphological identification and preservation in Liquid Nitrogen awaiting transportation to Emerging Infectious Diseases (EID) laboratory at icipe’s Duduville campus in Nairobi, Kenya, where they were stored at −80 °C freezers awaiting DNA extraction for subsequent molecular screening and identification.
Sand fly dissections and morphological identification
After sand fly processing that involved sorting and washing in 2% teepol detergent with subsequent washing in distilled water twice, we dissected out the head and the genitalia in each sand fly for morphological identification while the thorax and the remaining part of the abdomen was preserved in liquid nitrogen for later molecular analysis. We mounted the sand fly parts dissected out onto a labeled microscope slide using gum chloral-hydrate (locally prepared Hoyer’s mounting medium) on dissecting microscope and kept in a room temperature surface to dry. Morphologically, we identified each sand fly using combination of phlebotomine dichotomous keys [29, 30] using Olympus CX31 compound microscope.
Rearing of Ph. duboscqi sand flies
All the Phlebotomus duboscqi specimens used in this study were bred at icipe’s sand fly rearing unit. Sand flies were maintained on apple fruits until they were 2–3 weeks old under laboratory condition of temperature between 26–28 °C and 75–80% humidity.
Extraction of genomic DNA from sand flies
In the laboratory, we homogenized each preserved sand fly body (thorax with part of abdomen) in 180 µl of buffer ATL (Qiagen GmbH, Hilden, Germany). We extracted the genomic DNA from each homogenate using DNeasy Blood and Tissue Kit (Qiagen GmbH, Hilden, Germany) following the manufacturers’ instructions. We eluted obtained DNA in 30 µl of buffer ET and stored at −20 °C for later use.
Molecular identification of sand flies
We verified morphological sand fly species identifications by amplifying sand fly cytochrome oxidase 1 gene (CO1) (Table 1) using COI primers and cytochrome b gene (cyt b) using cyt b primers (Table 1), which yield DNA amplicons sizes of about 700 bp and 386 bp, respectively. The two sand fly biomarkers (COI and cyt b regions) were amplified in 20-µl PCR reactions that included 4 µl of X5 Blend (Biodyne, Solis) ready-to-load master mix, 1 µl of each specific primers, and 12 µl of nuclease free water (Sigma, St. Louis, USA). We used 2 µl (2.5–7.0 ng/ µm) of each template DNA. Table 1. List and description of primers used in this studyTarget organismPrimer namePrimer sequenceAnnealing (°C)& (No. of cycles)Cycling conditionsAmplicon size (bp)ReferenceWol bachiawsp81FTGGTCCAATAAGTGATGAAGAAAC55 (35)Denaturation: 95 °C for 30 s; Annealing: 45 s; Extension: 72 °C for 60 s550[32, 33]wsp691RAAAAATTAAACGCTACTCCAwspecFCATACCTATTCGAAGGGATAG62 (35)Denaturation: 95 °C for 30 s; Annealing: 30 s; Extension: 72 °C for 40 s440[34]wspecRAGCTTCGAGTGAAACCAATTCRickettsiaRick 16S FGAACGCTATCGGTATGCTTAACACA51 (35)Denaturation: 95 °C for 30 s; Annealing: 45 s; Extension: 72 °C for 30 s365/500[35]Rick 16S RCATCACTCACTCGGTATTGCTGGArOmpB 120–2788AAACAATAATCAAGGTCATGT53 (35)Denaturation: 95 °C for 30 s; Annealing: 30 s; Extension: 72 °C for 40 s856[31]rOmpB 120–3599TACTTCCGGTTACAGCAAAGTMicrosporidiaMsp SSU RNA FCACCAGGTTGATTCTGCC62 (35)Denaturation: 95 °C for 45 s; Annealing: 60 s; Extension: 72 °C for 90 s600[25]Msp SSU RNA RTTATGATCCTGCTAATGGTTCMsp SSU RNA F1492RCACCAGGTTGATTCTGCCGGTTACCTTGTTACGACTT60 (35)Denaturation: 95 °C for 30 s; Annealing: 60 s; Extension: 72 °C for 60 s1200[36]CardiniumCar-sp-FCGGCTTATTAAGTCAGTTGTGAAATCCTAG57 (35)Denaturation: 95 °C for 30 s, Annealing: 40 sExtension: 72 °C for 60 s544[37]Car-sp-RTCCTTCCTCCCGCTTACACGSpiroplasmaSpoulFGCTTAACTCCAGTTCGCC55 (35)Denaturation: 95 °C for 30 s; Annealing: 60 s; Extension: 72 °C for 60 s440[38]SpoulRCCTGTCTCAATGTTAACCTCArsenophonusArsFGGGTTGTAAAGTACTTTCAGTCGT54 (35)Denaturation: 95 °C for 30 s; Annealing: 30 s; Extension: 72 °C for 45 s1200[39]ArsR2GTAGCCCTRCTCGTAAGGGCCArs-23S-1CGTTTGATGAATTCATAGTCAAA60 (35)Denaturation: 95 °C for 30 s; Annealing: 45 s; Extension: 72 °C for 60 s600[40]Ars-23S-2GGTCCTCCAGTTAGTGTTACCCAACInsect COILepF1ATTCAACCAATCATAAAGATATTGG45 and 51 (5 and 35)Denaturation: 95 °C for 40 s; Annealing: 45 °C for 40 s and 51 °C for 40 s; Extension: 72 °C for 60 s704[41]LepR1TAAACTTCTGGATGTCCAAAAAATCAInsect cyt bCytb-J-1–933TCTTTTTGAGGAGCWACWGTWATTAC45 (35)Denaturation: 95 °C for 60 s; Annealing: 60 s; Extension: 72 °C for 120 s386[42]Cytb-N-11367AATTGAACGTAAAATWGTRTAAGCAALeishmaniaL5.8STGATACCACTTATCGCACTT58 (35)Denaturation: 95 °C for 20 s; Annealing: 30 s; Extension: 72 °C for 30 s365[5, 43]LITSRCTGGATCATTTTCCGATG
PCR conditions for cyt b amplification included an initial denaturation at 95 °C for 15 min, followed by 35 cycles of denaturation at 95 °C for 1 min, followed by annealing at 45 °C for cyt b for 1 min, extension at 72 °C for 2 min, and a final extension at 72 °C for 7 min. The COI PCR conditions included initial denaturation at 95 °C for 5 min, followed by five cycles with denaturation at 95 °C for 40 s, annealing at 45 °C for 40 s and extension at 72 °C for 1 min, and 35 cycles with denaturation at 95 °C for 40 s, annealing at 51 °C for 40 s, and extension at 72 °C for 1 min, and a final extension at 72 °C for 7 min. We used ProFlex PCR System (ThermoFischer Scientific Inc., USA), or the Kyratec Supercycler (POCD Scientific, Australia) in our subsequent PCR analysis.
PCR analysis and identification of specific endosymbionts in sand flies
To identify sand flies with specific endosymbionts, Wolbachia, Cardinium, Spiroplasma, Arsenophonus, and microsporidia, we screened whole sand fly genomic DNA using specific primers under their specific PCR conditions (Table 1) using Proflex or Kyratech thermocyclers. In total, we screened genomic DNA from all 1700 wild-caught sand flies (1266 females and 434 males) and 253 colony Ph. duboscqi for these endosymbionts and Rickettsia.
To improve the sensitivity for Rickettsia detection, we first used high-resolution melting (HRM) analysis of rickettsial 16S rRNA gene PCR products using Magnetic Induction Cycler (MIC) (Bio Molecular Systems, Queensland, Australia) machines [31]. Rickettsia with unique rickettsial 16S rRNA HRM profiles were further characterized by sequencing of the citrate rickettsial outer membrane protein B (ompB) gene PCR products [31].
We used 2 µl (2.5–7.0 ng/ µm) of each DNA as template in a master mix containing 2 µl of 5X Hot FIREPol Blend or HRM (Biodyne, Solis) ready-to-load master mixes, 1 µl of each specific primers, and nuclease free water (Sigma, St. Louis, USA). We used nuclease free water (Sigma, St. Louis, USA) also as negative control in each of our subsequent PCRs. PCRs were performed under each specific PCR condition, depending on target endosymbiont primers, that included initial denaturation at 95 °C for 2–15 min, followed by 35–40 cycles that included denaturation at 95 °C for 20–60 s, annealing temperatures (45–62 °C depending on each primer; Table 1), extension at 72 °C for 30–120 s and final extension at 72 °C for 5–7 min.
Detection and identification of Leishmania parasites in sand flies
We screened for Leishmania parasites in sand flies by amplifying internal transcribed spacer 1 gene (ITS1) using L5.8S and LITSR primers (Table 1). We screened 1266 wild-caught females (all morphologically identified females from the 1700 field-collected sand flies) for Leishmania DNA; males were not tested for Leishmania. We used 2 µl sample DNA of 2.5–7.0 ng/µm as template in a master mix with 2 µl of 2X Dream Taq Green PCR Master Mix, 0.5 µl of each primer and nuclease-free water (Sigma, St. Louis, USA). We used L. donovani DNA (NLB65), L. major DNA (LD63), and L. tropica DNA as positive controls and nuclease free water (Sigma, St. Louis, USA) as negative control in our subsequent PCRs. We performed PCRs under cycling conditions that involved initial denaturation at 98 °C for 2 min followed by 35 cycles of denaturation, annealing, and extension as indicated in Table 1, and a final extension at 72 °C for 7 min. We used Proflex or Kyratech thermocyclers in our PCR analysis.
We resolved all obtained amplicons in 1.5% agarose gels stained with × 1 ethidium bromide and visualized in Kodak Gel Logic 200 Imaging System (SPW Industrial, Laguna Hills, CA, USA). Amplicons of expected DNA band sizes were purified using the QIAquick PCR purification kit (QIAGEN, CA. USA) according to manufacturers’ protocol and submitted for Sanger sequencing using forward and reverse primers.
Sequencing analyses
We analyzed the obtained nucleotide sequence chromatograms using the MAFFT plugin in Geneious Prime software v. 2023.1.2. Each consensus sequence was queried for related reference sequences in the GenBank database using the Basic Local Alignment Search Tool (www.ncbi.nlm.nih.gov/BLAST/). A similarity of over 99% in each queried sequence against the subject sequences at the GenBank database was considered to be the most likely organism. Maximum-likelihood phylogenies of the studied sand fly species based on the organism target gene sequences, aligned with reference sequences retrieved from GenBank, were constructed using PhyML v. 3.0, with automatic model selection on the basis of the Akaike information criterion [44]. We estimated Tree topologies over 1000 bootstrap replicates with the nearest neighbor interchange improvements [45]. We visualized our Phylogenetic trees using FigTree v. 1.4.4 [46].
Data analysis
To understand the modulatory effects of endosymbionts to Leishmania parasite development in sand flies, we used a Fisher’s exact test to test for associations between Leishmania and endosymbionts in both Phlebotomus and Sergentomyia sand flies and also in each sand fly species considering P-values < 0.05 as statistically significant. All our data were entered in excel sheet v2006 and analyzed in R software v4.2.2. Prevalence estimates were calculated as the proportion of positive specimens out of the total screened, and 95% confidence intervals (95% CIs) were obtained using the Clopper–Pearson exact binomial method.
An overview of the laboratory screening workflow (from collection and morphological identification through DNA extraction, PCR-HRM screening and sequencing) is provided in Additional file 2: Supplementary Fig. S1.
Results
Morphological identification of sand flies
We collected 1700 sand flies (1266 females and 434 males) comprising 290 (17.1%) Phlebotomus spp. and 1410 (82.9%) Sergentomyia spp. from the three study areas (Table 2). We morphologically identified seven Phlebotomus and nine Sergentomyia sand fly species. Phlebotomus sand flies included Ph. aculeatus (0.1%, n = 1), Ph. duboscqi (0.2%, n = 3), Ph. guggisbergi (6.6%, n = 112), Ph. martini (6.2%, n = 106), Ph. mireillae (0.8%, n = 13), Ph. orientalis (1.1%, n = 18), and Ph. saevus (2.2%, n = 37). Phlebotomus martini was collected in Kajiado West and East Pokot sub-counties, while both Ph. saevus and Ph. guggisbergi were collected in areas of Kajiado West and Gilgil sub-counties. Phlebotomus orientalis was found only in Kajiado West. Phlebotomus aculeatus and Ph. mireillae were collected only in Gilgil, while Phlebotomus duboscqi sand flies were only found in areas of East Pokot in Baringo County. Sand flies of the genus Sergentomyia were commonly found in all the areas and included Sergentomyia sp*.* (21.8%, n = 371), Sergentomyia schwetzi (15.5%, n = 263), Sergentomyia africana (2.5%, n = 43), Sergentomyia antennata (4.4%, n = 74), Sergentomyia adleri (7.7%, n = 130), Sergentomyia bedfordi (4.7%, n = 79), Sergentomyia dreyfussi (0.1%, n = 1), and Sergentomyia ingrami (0.5%, n = 9). In addition, 253 (198 females and 55 males) colony Ph. duboscqi sand flies were also screened for heritable endosymbionts. Table 2. Sand fly composition and diversity in Kenya’s Baringo, Nakuru, and Kajiado CountiesSand fly speciesEast Pokot, Baringo CountyGilgil, Nakuru CountyKajiado West, Kajiado CountyTotalChesakamKamsukLorwatumNjeruJaicaThugunuiBirikaEmpalekiEnchanipusKirineOlomanyattaOloisinyaiNkonyoroShompolePh. aculeatus000100000000001Ph. duboscqi201000000000003Ph. guggisbergi00012306900000010112Ph. martini22700009725222435106Ph. mireillae00013000000000013Ph. orientalis0000000022590018Ph. saevus00000000000037037S. adleri000000071612010274130S. africana812300010410110343S. antennata200000050247425774S. bedfordi444000105143273579S. clydei31020000139663818330239440S. ingrami000000000000099S. schwetzi118805600000090000263Sergentomyia sp.1011100006642438184267371S. dreyfussi100000000000001Total:196991022630698611661272792641241491700%:11.55.86.01.51.84.10.53.69.87.516.415.57.38.8100
Molecular identification and phylogenetic analysis of sand flies
Phylogenetically, the two cryptic sand flies of the Paraphlebotomus subgenus identified, Ph. saevus (GenBank accessions: COI OR671478 to OR671481) and Ph. mireillae (GenBank accessions: COI OR671482-OR671484; cyt b OR731100 to OR731103), shared 99% similarity with sequences of Ph. saevus from Israel (GenBank: KF483673) and Ph. mireillae previously sequenced from Gilgil, Kenya (GenBank: MZ049659), respectively (Fig. 2). As expected, sequences of Ph. martini (GenBank accessions: OR671490 to OR671493; cyt b OR731117 to OR731122) from this study shared 98.5% similarity with those of Ph. martini (GenBank: JX105040) and Ph. celiae from Israel (GenBank: JX105041), while Ph. guggisbergi sequence (GenBank accessions: COI OR726348; cyt b OR731133) shared 99% similarity with another Ph. guggisbergi sequence from Kenya (GenBank: MK169221). Sequences of Ph. orientalis (GenBank accessions: COI OR671474 to OR671476; cyt b OR731096 and OR731097) shared 99% identity Ph. orientalis from Ethiopia (KC204967) and Kenya (MT597052).Fig. 2. Maximum-likelihood trees of sand fly A COI and B cyt b gene sequences. Sequences from this study are in bold. Bootstrap support values (percentages) are shown at major nodes (1000 replicates). Scale bar represents substitutions per site
The Ph. aculeatus (GenBank accession: COI OR671477) was more phylogenetically related to sequences of Ph. guggisbergi than to those of Ph. orientalis (Fig. 2A). Sequences of Sergentomyia sp. (GenBank accessions: COI OR671499-OR671503; cyt b OR731126 to OR731129) shared 99.5–100% identity with Sergentomyia sp. sequences from Kenya (MT644223 and MT644229) and Ethiopia (KR020570), while S. dreyfussi (GenBank accessions: COI OR671498; cyt b OR731125) shared 100% identity with a previously sequenced S. dreyfussi from Kenya (MT644236).
Molecular analysis of bacterial symbionts and pathogens
Using HRM analysis and representative 16S rRNA amplicon sequencing, we identified Wolbachia endosymbionts in Phlebotomus (GenBank accessions OR704218-OR704288, OR704290) and Sergentomyia (GenBank accessions OR704289, OR704291, OR704292) sand flies*, Rickettsia* spp. endosymbionts (GenBank accessions OR704173-OR704178), Spiroplasma sp. (GenBank accessions OR704211-OR704217), Serratia sp*.* (GenBank accessions OR704204-OR704210), Cardinium sp. (GenBank accession OR704293), and Tubulinosema sp. (GenBank accession OR717175). We also identified Rickettsia africae (GenBank accessions: 16S rRNA OR704165-OR704172). The distinct HRM profiles of the R. africae and Rickettsia endosymbionts are shown in Fig. 3. The R. africae 16S rRNA sequences shared 100% similarity with R. africae sequences accessed from GenBank (Fig. 4A). Representative R. africae ompB gene sequences from this study (GenBank accessions OR731090, OR731091) also shared 100% similarity with R. africae sequences obtained from GenBank (Fig. 4B). The Rickettsia spp. endosymbiont sequences clustered phylogenetically with Rickettsia endosymbionts of other insect species (Fig. 4A).Fig. 3. Normalized high-resolution melting (HRM) profiles of R. africae and Rickettsia endosymbiont 16S rRNA gene ampliconsFig. 4Maximum-likelihood trees of A Rickettsia 16S rRNA, B Rickettsia ompB, and C Wolbachia 16S rRNA sequences. Sequences from this study are shown in bold. Bootstrap support values (percentages) from 1000 replicates appear at major nodes. Scale bar indicates substitutions per site
The Wolbachia 16S rRNA sequences amplified from Phlebotomus sand flies shared 100% sequence identity with Wolbachia endosymbionts of Phlebotomus papatasi (GenBank accession MZ577348) and other insects, whereas those sequenced from Sergentomyia sand flies shared 100% sequence identity with a Wolbachia endosymbiont of Sergentomyia minuta (GenBank accession DQ402520) and other insects (Fig. 4C). In the 16S rRNA phylogeny, Wolbachia sequences from Phlebotomus clustered with the wPap strain from Ph. papatasi (supergroup A), whereas those from Sergentomyia grouped with Wolbachia from Se. minuta (supergroup B), indicating that Kenyan sand flies host at least these two major Wolbachia supergroups, although 16S alone cannot fully resolve strain diversity.
The Spiroplasma sp. sequences shared 99% identity with Spiroplasma citri (KR818831) from Malaysia. We obtained only one sequence of Cardinium sp. endosymbiont that shared > 97% identity with a Cardinium endosymbiont of Microzetorchestes emeryi (LC090049) from Spain. We also found a microsporidian, Tubulinosema sp. (GenBank accession OR717175), for the first time in sand flies, which shared 100% sequence similarity with sequences of Tubulinosema acridophagus from a human patient with microsporidiosis from Belgium (JQ247017) [47] and Tubulinosema acridophagus from a mosquito colony collapse in Argentina (AF024658).
Along with endosymbionts, we amplified gut bacteria DNA of ten genera: Acetobacter (n = 3), Botryotrichum (n = 1), Enterobacter (n = 1), Pantoea (n = 2), Halomonas (n = 1), Klebsiella (n = 3), Olivibacter (n = 1), Stenotrophomonas (n = 2), Rhizobiales (n = 1), and Asaia sp. (n = 1) bacteria (Table 3), using the Arsenophonus primers. As summarized in Additional file 1: Supplementary Table S2 (including all microbes found in this study), sequences of gut bacteria shared similarities to their corresponding GenBank sequences. Sequences of Asaia sp. shared 99% similarity with sequences of Asaia sp. (MN094403, MN094402, and MK598732) from China. Table 3. Incidences and prevalences of gut microbes in wild-caught sand fliesSand fly spp.HalomonasEnterobacterKlebsiellaOlivibacterPantoeaBotryotrichumAcetobacterStenotropho-monasRhizobialesAsaiaPh. aculeatus0000000000Ph. duboscqi0000000000Ph. guggisbergi000000001 (0.9%)1 (0.9%)Ph. martini0000000000Ph. mireillae0000000000Ph. orientalis0000000000Ph. saevus0000000000S. adleri0000001 (0.8%)000S. africana00001 (2.3%)00000S. antennata0000000000S. bedfordi0000000000S. clydei01 (0.2%)1 (0.2%)1 (0.2%)1 (0.2%)02 (0.5%)2 (0.5%)00S. ingrami0000000000S. schwetzi2 (0.8%)02 (0.8%)0000000S. squamipleuris1 (0.3%)01 (0.3%)001 (0.3%)0000S. dreyfussi001 (100.0%)0000000Total3 (0.2%)1 (0.1%)5 (0.3%)1 (0.1%)2 (0.1%)1 (0.1%)3 (0.2%)2 (0.1%)1 (0.1%)1 (0.1%)
The gut bacteria identified from the colony sand flies, also from Arsenophonus primers included Klebsiella sp. (GenBank accessions OR704201-OR704203) that shared 99% 16S rRNA sequence identity with Klebsiella pneumoniae (CP104678), Ochrobactrum sp. (GenBank accession OR704188) that shared 99% similarity with Ochrobactrum intermedium from Japan (MT649859) and South Arabia (KY194745), Pantoea sp. (GenBank accessions OR704190–OR704191) that shared 99% similarity to sequences of Pantoea septica (GenBank: KF913782, KF913784 to KF913786), Raoultella sp. (GenBank accession OR704192) that shared 99% similarity with Raoultella ornithinolytica (GenBank: MT071372) from China, and Tatumella sp. (GenBank accession; OR704196–OR704199) that shared 99% with Tatumella ptyseos (GenBank: MN367128) from the USA. Among the colony Ph. duboscqi specimens, we detected Serratia sp. symbiont sequences using the Arsenophonus primers (GenBank accessions OR704204–OR704210) with 99% identity to a Serratia marcescens from Ethiopia (GenBank: MN006026).
Prevalence of Rickettsia africae and key endosymbionts in sand flies
We detected DNA of the pathogen R. africae in 0.7% (11/1700; 95% CI 0.3–1.2%),of wild-caught sand flies, predominantly in Ph. martini (4.7%, 5/106) and Ph. guggisbergi (1.8%, 2/112), as well as in S. schwetzi, S. clydei, and Sergentomyia sp. (Table 4). Wolbachia spp. were the most prevalent endosymbionts (8.5%, 145/1700; 95% CI 7.2–9.9%), followed by Spiroplasma sp. (1.4%, 23/1700; 95% CI 0.9–2.0%), Rickettsia endosymbionts (0.7%, 12/1700; 95% CI 0.4–1.2%), Cardinium sp. (0.4%, 6/1700; 95% CI 0.1–0.8%), and Tubulinosema sp. (0.1%, 1/1700; 95% CI 0.0–0.3%). Notably, among wild-caught males we detected R. africae DNA in one Ph. martini male and Rickettsia endosymbiont DNA in two males (S. clydei and S. schwetzi), whereas no males were positive for Wolbachia, Spiroplasma, Cardinium, or Tubulinosema. Table 4. Incidences and prevalences of gut R. africae and endosymbionts in wild-caught sand fliesPathogenEndosymbiont generaSand fly spp.R. africaeWolbachiaRickettsiaSpiroplasmaCardiniumTubulinosemaPh. aculeatus000000Ph. duboscqi000000Ph. guggisbergi2 (1.8%)82 (73.2%)4 (3.6%)000Ph. martini5 (4.7%)2 (1.9%)0000Ph. mireillae012 (92.3%)0000Ph. orientalis000000Ph. saevus018 (48.6%)4 (10.8%)000S. adleri01 (0.8%)0000S. africana03 (7.0%)1 (2.3%)000S. antennata05 (6.8%)05 (6.8%)00S. bedfordi06 (7.6%)1 (1.3%)000S. clydei2 (0.5%)3 (0.7%)1 (0.2%)2 (0.5%)2 (0.5%)1 (0.2%)S. ingrami000000S. schwetzi1 (0.4%)6 (2.3%)1 (0.4%)14 (5.3%)1 (0.4%)0S. squamipleuris1 (0.3%)6 (1.6%)02 (0.5%)2 (0.5%)0S. dreyfussi01 (100.0%)001 (100.0%)0Total11 (0.6%)145 (8.5%)12 (0.7%)23 (1.4%)6 (0.4%)1 (0.1%)
At the species level, Wolbachia prevalences were particularly high in some Phlebotomus species, including Ph. saevus (48.6%), Ph. guggisbergi (73.2%), and Ph. mireillae (92.3%) (Table 4), contrasting with the generally low prevalences of other heritable endosymbionts and suggesting that Wolbachia is established in specific sand fly lineages in our study area.
Molecular identification of Leishmania parasites
We identified Leishmania donovani (GenBank accessions OR717168–OR717172), Leishmania tropica (GenBank accessions OR717161–OR717167), Leishmania adleri (GenBank accession OR717160), and Leishmania major (GenBank accession OR717173) from sand flies analyzed in this study (Fig. 5). The L. donovani (n = 5) sequences shared 100% similarity with L. donovani from Sri Lanka (GenBank: KT273404), Senegal (GenBank: MN244152), India (GenBank: MN031878), and Argentina (GenBank: MN496379). The L. tropica sequences from sand flies collected from Nguruman shared 100% identity with other L. tropica sequences (GenBank: MG515728, AJ000301), while L. adleri shared 99% identity with a L. adleri sequence from Russia (GenBank: AJ300480).Fig. 5. Maximum-likelihood tree of Leishmania ITS1 sequences. Sequences obtained in this study are in bold. Bootstrap support values (percentages) based on 1000 replicates are shown at major nodes. The branch length scale bar represents substitutions per site
Leishmania DNA in sand flies
We detected Leishmania DNA in 4.0% (50/1266) of the wild-caught female sand flies screened for Leishmania (Table 5). In sand flies from Nguruman, we detected L. donovani (5.6%, 47/844), Leishmania adleri (0.2%, 2/844) and* Leishmania tropica* (0.1%, 1/844). Generally, L. donovani was the most prevalent in three Phlebotomus sand fly species, Ph. martini (8.6%, 5/58), Ph. orientalis (13.3%, 2/15) and Ph. saevus (12.0%, 3/25), as well as in Sergentomyia species, S. clydei (6.1%, 17/278) and Sergentomyia sp. (3.9%, 13/338). In Gilgil, we found L. tropica (10.0%, 11/110) and Leishmania major (0.9%, 1/110), the latter only in Ph. guggisbergi. Table 5. Detection rates of sand fly species in sampling areas and Leishmania DNA in sand fly speciesParasitesPh. guggisbergiPh. martiniPh. saevusPh. orientalisS. adleriS. africanusS. bedfordiSclydei**S. squamipleurisSampling areasn = 110n = 58n = 25n = 15n = 84n = 32n = 70n = 278n = 338NgurumanL. donovani010.0% (5/50)12.0% (3/25)13.3% (2/15)4.8% (4/84)6.3% (2/32)1.4% (1/70)6.1% (17/278)3.9% (13/338)NgurumanL. tropica004.0% (1/25)000000NgurumanL. adleri00000000.4% (1/278)0.3% (1/338)GilgilL. tropica10.0% (11/110)–0––––––GilgilL. major0.9% (1/110)–0––––––Parasites prevalenceL. donovani08.6%12.0%13.3%4.8%6.3%1.4%6.1%3.9%L. tropica10.0%04.0%000000L. adleri00000000.4%0.3%L. major0.9%00000000
Associations between endosymbionts and Leishmania presence
We detected Leishmania DNA in significantly higher proportions of wild-caught sand flies with Rickettsia DNA (including both the Rickettsia endosymbionts and Rickettsia africae) (OR = 10.17; 95% CI [3.16, 28.39]; P = 0.0001). When considering only R. africae, this association was not significant (OR = 2.67; 95% CI [0.061, 19.28]; P = 0.34). However, there was still a positive association between Rickettsia endosymbionts and Leishmania (OR = 20.31, 95% CI [4.93, 77.03], P < 0.0001). The positive association between Rickettsia endosymbionts and Leishmania was also significant among Phlebotomus sand flies (OR = 13.54; 95% CI [2.33, 78.88]; P = 0.0017). Within individual Phlebotomus species, point estimates were elevated but confidence intervals were wide and included 1.0 due to small sample sizes: Ph. saevus (OR = 13.28; 95% CI [0.66, 295.46]; P = 0.05), Ph. guggisbergi (OR = 9.41; 95% CI [0.62, 143.19]; p = 0.056). No association was detected among Sergentomyia (OR = 11.93; 95% CI [0.22, 151.92]; P = 0.11). Similarly, we found a significant association between Wolbachia endosymbionts and Leishmania (OR = 2.46, 95% CI [1.17, 4.79]; P = 0.011) in wild-caught sand flies, but there was no association within either Phlebotomus (OR = 1.76; 95% CI [0.68, 4.59]; P = 0.27) or Sergentomyia (P = 1) genera.
Discussion
Our results show that Kenyan sand flies harbor diverse microbiota, including heritable endosymbionts (Rickettsia, Wolbachia, Spiroplasma, Cardinium), gut-associated bacteria (Serratia, Ochrobactrum, Asaia), and a microsporidian (Tubulinosema sp.; Family: Tubulinosematidae). This diversity mirrors findings in other sand fly populations [48, 49] and suggests a complex microbial community potentially influencing vector competence. Furthermore, we found that sand flies with Rickettsia endosymbionts were more likely to also have Leishmania DNA, particularly in Phlebotomus sand flies of subgenus Larroussius (Ph. guggisbergi) and Paraphlebotomus (Ph. saevus). Rather than implying that Rickettsia directly enhances Leishmania growth and survival, we interpret this pattern as consistent with an association between Rickettsia infection and conditions that favor Leishmania establishment, akin to the positive association between Sodalis endosymbionts and trypanosomes in tsetse flies [50].
Alternatively, environmental/physiological factors that favor Rickettsia endosymbiont persistence in sand flies may also favor Leishmania survival. Such potential symbiont–parasite interactions warrant deeper investigation, and our cross-sectional data do not allow inference of causality. The significant associations between endosymbionts and Leishmania observed in our study align with emerging evidence that sand fly microbiota composition is associated with Leishmania susceptibility [51], and our finding of Rickettsia and Wolbachia co-occurring with Leishmania is consistent with recent reports of Wolbachia and Rickettsia coinfections in Sergentomyia species [52], suggesting complex tripartite interactions that merit investigation. Together, these findings situate our data within a growing body of evidence that sand-fly microbial communities influence parasite dynamics across genera and continents, while the underlying mechanisms remain to be elucidated.
Recent metagenomic work supports both positive and negative interactions between Rickettsia and Leishmania. For instance, Tom et al. (2025) found abundant Rickettsia in nonvector Sergentomyia babu but minimal levels in vector-competent Ph. argentipes [53], though whether this between-species difference reflects a functional relationship remains to be tested experimentally. Conversely, our positive associations in Kenyan sand flies may indicate strain- or species–specific facilitative effects, underscoring the complexity of Rickettsia–Leishmania relationships across sand-fly taxa. Furthermore, Itokawa et al. (2025) reported coinfection of Sergentomyia squamirostris with Wolbachia and Torix-group Rickettsia (“Candidatus Tisiphia”), providing the first complete genome of this clade from sand flies [52]. Such coinfections demonstrate that Rickettsia lineages are widespread and may interact with other symbionts within the same host.
This study provides the first record of R. africae in two vectors of leishmaniasis, Ph. martini and Ph. guggisbergi, and three biting-nuisance sand flies [54] of genus Sergentomyia, S. clydei, S. schwetzi, and Sergentomyia sp., expanding its known ecological range beyond ticks [55–57]. Notably, S. clydei has also been found to harbor Leishmania DNA and feed on humans in northern Kenya [4], suggesting potential involvement in Leishmania transmission cycles.
Detection of R. africae in both males and nonblood-fed females across multiple sand fly species is consistent with infection rather than recent blood-meal carry-over and raises the possibility of vertical transmission, similar to other Rickettsia–insect associations [58–60]. However, we cannot rule out repeated horizontal acquisition in sand flies. These occurrences of R. africae in sand flies could be attributed to the fact that wildlife are in free range conservancies in these areas of East Pokot, Kajiado West and Gilgil sub-counties, with likely active transmission of R. africae. Given the overlap of R. africae and Leishmania foci in our study areas, clinicians should consider both infections in differential diagnoses of febrile illness, as ATBF is frequently under-diagnosed in rural areas where clinical presentations overlap with other febrile illnesses [55, 61]. Targeted sand-fly control for leishmaniasis could therefore offer dual benefits by also reducing potential R. africae transmission.
In particular, detection of R. africae DNA in male Ph. martini, a nonblood-feeding sex, reinforces the interpretation that at least some infections are not due to recent vertebrate blood meals. However, the low prevalence of R. africae in males and the absence of Wolbachia and other symbionts in males do not fit the classical pattern of a common vertically transmitted endosymbiont, and the transmission route of R. africae in sand flies therefore remains unresolved. Taken together, the occurrence of R. africae across six sand fly species from three counties indicates that this pathogen is widespread in Kenyan sand flies and warrants further investigation of the role sand flies may play in the ecology and possible transmission of ATBF, including formal vector competence studies.
Rickettsiosis is neglected and cases could be going unreported in most African countries due to lack of knowledge, suspicion, and effective diagnosis in affected poor rural areas [55, 61]. Although our associations of R. africae to Leishmania were not significant, R. africae could have been acquired by sand flies zoonotically. Anopheles mosquitoes [59] and tsetse flies [60] have similarly also been shown to harbor pathogenic Rickettsia. More research is needed to determine the role of sand flies in ATBF transmission.
Although sand flies are known to host diverse microbial pathogens [48, 49], their broader microbiome remains poorly characterized [62], as does their potential role in modulating human pathogens, especially Leishmania, during its development in the sand fly. We highlight the presence of diverse endosymbionts in primary leishmaniasis vectors in Kenya, including Rickettsia, Wolbachia, Spiroplasma, Cardinium, and for the first time, a microsporidian species (Tubulinosema genus). Further studies are required to ascertain the nature of the symbiotic relationship of these endosymbionts with sand flies, which could potentially be targeted for development of biocontrol strategies against transmission of Leishmania parasites to humans and animals.
The known arthropod-associated Wolbachia endosymbionts [63] seem to preferably infect Ph. guggisbergi, Ph. saevus, and Ph. mireillae, which are found at high altitude*.* This pattern underscores the complex environmental, geographic, and ecological factors that shape endosymbiont distribution in sand-fly populations. Moreover, similar to our finding with Rickettsia endosymbionts, Wolbachia also showed a significant positive association to Leishmania in sand flies when considering all genera, but this association was not significant within genera. A large-scale survey found Wolbachia in more than 45% of Phlebotomus and Sergentomyia populations in Spain and Morocco, with both A and B supergroup strains [63]. The consistent occurrence of Wolbachia in Leishmania infantum vectors across two continents supports our conclusion that these symbionts are globally widespread in sand flies and could modulate vector competence in ways yet to be resolved.
Low-prevalence taxa such as Spiroplasma, Cardinium, and the microsporidian Tubulinosema were also detected. These microbes, known in other insects to influence reproduction or immunity [64–69], were rare in our samples and may represent either transient gut residents or coextracted taxa derived from ingested material or environmental contamination [69–73]. Their potential roles in sand fly physiology warrant confirmation through deeper metagenomic sequencing. Clarifying whether these microbes are stable symbionts or transient environmental associates will refine our understanding of sand-fly microbial ecology.
We did not detect the endosymbiont Arsenophonus, but other gut bacteria of genera Serratia, Tatumella, Raoultella, Klebsiella, Ochrobactrum, Botryotrichum, Enterobacter, and Olivibacter did amplify using the Arsenophonus primers. In colony Ph. duboscqi sand flies, we found Serratia, a colony-destroying bacterium, along with gut bacteria including Tatumella, Raoultella, as well as Klebsiella and Ochrobactrum that opportunistically infected immunocompromised persons [74, 75]. Diverse gut bacteria, including Botryotrichum, Enterobacter, and Olivibacter, which are mostly known to be opportunistic pathogens [76, 77], also occur in wild sand flies. Because we relied on partial 16S rRNA sequences, which often cannot reliably discriminate among closely related species, we report most gut-associated taxa at the genus level, even where BLAST hits suggested candidate species, to avoid overinterpretation of taxonomic assignments.
Notably, Klebsiella pneumoniae is a well-known opportunistic pathogen in humans, frequently causing nosocomial infections, urinary tract infections, and infections of the respiratory tract predominantly [78]. Also included are Ochrobactrum, which has been found to cause infections including endocarditis and septicemia in immunocompetent hosts [77] and Tatumella ptyseos, an opportunistic pathogen causing human sepsis [79], are opportunistic pathogens and their role in sand fly biology is not yet known. Although we could not confirm whether these gut bacteria are symbiotic to sand flies, detection of Asaia, a genus of symbiont found to be associated with the malaria vector An. stephensi [80], and bacteria that reduce Leishmania establishment through colonization resistance, such as Ochrobactrum intermedium [81], in a sand fly vector of CL, Ph. guggisbergi, could provide a promising strategy against CL transmission. Recent work positions Ochrobactrum sp. as a promising paratransgenic symbiont due to its transstadial persistence and transformability [82, 83], and findings by Marialva et al. (2025) suggest that microbiota differences between susceptible and nonsusceptible sand fly populations could be exploited to enhance natural refractoriness to Leishmania [51]. Together, these insights reinforce the potential of microbiome-based vector-control strategies.
Our study has several limitations that should be considered when interpreting the endosymbiont and pathogen prevalences. First, although sand flies were externally washed prior to dissection, DNA was extracted from intact thorax and abdomen without separating the gut, meaning that some low-prevalence bacteria may represent transient gut residents rather than stable, heritable symbionts. In addition, head and genitalia were processed separately for morphological identification rather than molecular screening, so symbionts concentrated in reproductive tissues may have been under-detected [84]. Second, although detection of R. africae in males and nonblood-fed females is consistent with true infection, the low prevalence in males and the absence of Wolbachia and other symbionts in males do not conform to classical patterns expected for common vertically transmitted endosymbionts. This may reflect low statistical power due to the smaller male sample size (434 males versus 1266 females), sex-biased symbiont localization, or genuinely different transmission dynamics for R. africae compared to typical heritable symbionts. Finally, sample sizes for some sand fly species were modest, leading to wide confidence intervals for certain prevalence estimates. These findings therefore warrant confirmation in larger, tissue-targeted surveys that include reproductive tissues.
In our study areas of Gilgil and Nguruman, re-emerging foci of leishmaniasis, there is active transmission of Leishmania. We detected L. donovani DNA in Ph. martini, Ph. orientalis and several Sergentomyia species in Nguruman and a high prevalence of L. tropica in Ph. guggisbergi in Gilgil sub-county. This could be attributed to the fact that Ph. guggisbergi dwell mainly in rock crevices and caves inhabited by rock hyraxes that are known reservoirs of L. tropica [85]. We also found L. tropica for the first time in the newly collected Ph. saevus in Nguruman. Although Ph. orientalis, a species associated with cracked vertisols in dry seasons [6, 86], was rare during the rainy-season sampling, its detection with L. donovani DNA indicates its potential importance in L. donovani transmission in Nguruman. Although we found no Leishmania DNA in sand flies from East Pokot, we found Ph. martini, probably the main vector of VL in the region, also detected with R. africae and Rickettsia endosymbiont DNA.
Conclusions
In this study, we identified diverse microbes associated with sand flies, some of which are closely related to microbes that have been used to control vector-borne diseases in other arthropods (e.g., Wolbachia), providing a step toward identifying and developing novel strategies to reduce transmission of leishmaniasis, a disease whose spread is being reshaped by climate and environmental change [3] and by drug resistance and limited treatment options [8, 9]. Therefore, we recommend: (i) targeted investigations to test the vector competence of sand flies for ATBF transmission, given the detection of R. africae across multiple sand fly species and in male specimens; (ii) establishment of integrated vector surveillance programs that screen for both Leishmania parasites and Rickettsia pathogens in endemic areas, particularly in the Rift Valley region where both leishmaniasis and rickettsial diseases co-occur; (iii) physician awareness of potential sand fly-borne rickettsiosis in patients presenting with febrile illness in leishmaniasis-endemic areas; and (iv) further investigations into the potential modulatory effects of Rickettsia and Wolbachia endosymbionts on sand fly biology and Leishmania parasite development, which could inform novel biocontrol strategies against leishmaniasis transmission.
Supplementary Information
Additional file 1.Table S1. GPS coordinates for sand fly trapping sites, altitudes and ecological descriptions. Table S2. List of pathogens, endosymbionts, and gut bacteria 16S gene sequences obtained from this studyAdditional file 2.Figure S1. Laboratory screening workflow
The reference list from the paper itself. Each links out to its DOI / PubMed record.