Antimicrobial Pharmacokinetic and Pharmacodynamic Considerations in Special Populations: A Call to Action
Marguerite L Monogue, James M Sanders, Nicholas J Mercuro, Crystal K Hodge, Esther Golnabi, Tyla Carettini, Christina F Yen, James B Cutrell

TL;DR
This paper emphasizes the need to improve antimicrobial dosing in special patient groups by addressing pharmacokinetic and pharmacodynamic variability.
Contribution
The paper calls for inclusive clinical trials and precision dosing to address underrepresented PK/PD data in special populations.
Findings
Special populations often face suboptimal antimicrobial dosing due to PK/PD variability.
Current data gaps hinder effective treatment in patients with conditions like obesity and renal dysfunction.
Improved modeling and real-world data collection are needed to enhance treatment outcomes.
Abstract
Pharmacokinetic (PK) and pharmacodynamic (PD) variability in special populations can impact antimicrobial efficacy and safety. This review highlights the importance of PK/PD optimization in patients known to have altered PK, including those with obesity, cystic fibrosis, renal dysfunction, critical illness, transplantation, pregnancy, and/or significant burns. Historically, PK/PD data are underrepresented in these populations, leading to suboptimal dosing recommendations and increased risks of therapeutic failure or toxicity. Herein, we discuss key physiological alterations affecting antimicrobial PK/PD, regulatory challenges, and currently available solutions. To bridge these knowledge gaps, we advocate for broader patient inclusion in clinical trials, improved PK modeling, real-world data collection, and increased investment in precision dosing strategies. Addressing these issues has…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
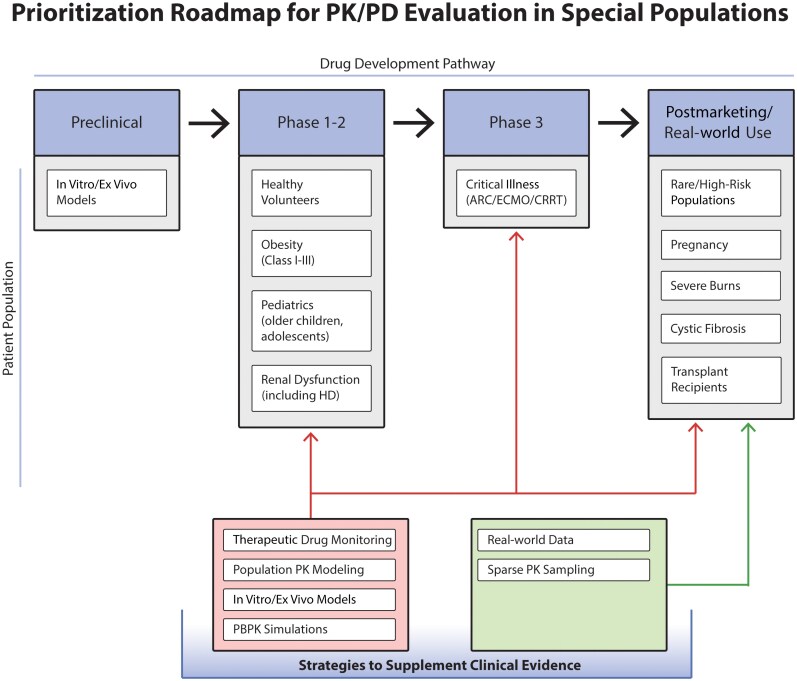 Figure 1
Figure 1| Agent | Approved Initial Dosage/Administration Strategy | Accepted or Suggested Updates | Indication | References |
|---|---|---|---|---|
| Ceftaroline | 600 mg q12h, 1-h infusion | 600 mg q8h, 1-h infusion | MRSA with MICs 2–4 µg/mL | [ |
| Ceftolozane-tazobactam | 1.5 g IV q8h, 1-h infusion | 3 g IV q8h, 3-h infusion | Pneumonia; MDROs | [ |
| Daptomycin | 4–6 mg/kg q24h | 8–12 mg/kg q24h |
| [ |
| Minocycline | 100 mg q12h | 200 mg q12h |
| [ |
| Tigecycline | 100 mg loading dose, 50 mg q12h | 200 mg loading dose, 100 mg q12h | MDROs; severe infections | [ |
| Patient Population | Notable Changes | Examples of Antimicrobials Impacted | Antimicrobial Property Impacting PK | Key PK Parameter Changes | Impact on PTA | References | |||
|---|---|---|---|---|---|---|---|---|---|
| Absorption | Distribution | Metabolism | Elimination | ||||||
| Renal dysfunction | AKI, CKD | Aminoglycosides, β-lactams | Hydrophilicity | … | … | … | ↓ | ↑ | [ |
| Critical illness | Fluid resuscitation | Aminoglycosides, β-lactams | Hydrophilicity, protein binding | … | ↑ | … | … | ↓ | [ |
| Hypoalbuminemia | Ceftriaxone, ertapenem | Protein binding | … | ↑ | … | ↑ | ↓ | [ | |
| ECMO | Amphotericin B, meropenem | Lipophilicity, protein binding, sequestration | … | ↑ | … | … | ↓ | [ | |
| ARC | Aminoglycosides, β-lactams | Hydrophilicity, protein binding | … | … | … | ↑ | ↓ | [ | |
| Obesity | Increased mass (adipose and lean tissue) | Daptomycin | Protein binding, hydrophilicity, renal elimination | … | ↑ or = | … | ↑ or = | ↓ or = | [ |
| Cystic fibrosis | CFTR channel modifications, gut physiology | Azithromycin, doxycycline, linezolid | Bioavailability | ↓ | … | … | … | ↓ | [ |
| Increased plasma volume, urine flow | Aminoglycosides, β-lactams | Hydrophilicity | … | ↑ | … | ↑ | ↓ | [ | |
| HSCT/SOT | Renal graft recovery | β-lactams | Hydrophilicity | … | … | … | ↓ or ↑ | ↓ or ↑ | [ |
| Altered gastric motility, delayed absorption | Letermovir, voriconazole, posaconazole | Bioavailability | ↓ | … | … | … | ↓ | [ | |
| Mucositis | Posaconazole | Bioavailability | ↓ | … | … | … | ↓ | [ | |
| Biliary complications and reduced total bilirubin | Tetracyclines, cefotaxime | Lipophilicity | ↓ | … | … | ↓ | ↓ | [ | |
| Drug–drug interactions, polymorphisms | Voriconazole | CYP450 | … | … | ↓ or ↑ | … | ↓ or ↑ | [ | |
| Pregnancy | Increased stomach pH, delayed gastric emptying | Ampicillin, artesunate | Acid labile, bioavailability | ↓ | … | … | … | ↓ | [ |
| Volume and fluid expansion | Amoxicillin, imipenem, trimethoprim | Hydrophilicity | … | ↑ | … | … | ↓ | [ | |
| ARC | β-lactams | Hydrophilicity, time-dependent PD | … | … | … | ↑ | ↓ | [ | |
| Burn injury | Disrupted skin integrity | Aminoglycosides | Topical administration | ↑ | … | … | … | ↑ | [ |
| Fluid resuscitation | β-lactams, daptomycin, aminoglycosides | Hydrophilicity | … | ↑ | … | … | ↓ | [ | |
| Hypercatabolic state (hepatic and renal) | Piperacillin-tazobactam, ceftazidime, cefepime | CYP450, protein transporters, time-dependent PD | … | … | ↑ | ↑ | ↓ | [ | |
| Patient Population | Agent | Standard Dosage/Administration Strategy | Recommended or Proposed Updates | Rationale and Considerations | References |
|---|---|---|---|---|---|
| Renal dysfunction | Daptomycin | Intermittent HD: standard dose q48h | Intermittent HD: standard dose on 48-h interdialytic days; increase dose by 50% on the 72-h interdialytic day | Due to daptomycin’s high renal excretion, elimination is substantially slower in patients with renal dysfunction, allowing for prolonged doses between HD sessions. | [ |
| Critical illness | Meropenem | 1–2 g q8h, 30-min infusion | 2 g q8h, 3-h infusion | Pathophysiological changes in critically ill patients can alter volume of distribution and total drug clearance, resulting in altered meropenem pharmacokinetics and variable plasma concentrations. | [ |
| Obesity | Acyclovir | Dose based on ABW | Dose based on adjBW | Use of ABW in obese patients resulted in adverse drug events (eg, renal failure) but use of IBW has led to inadequate exposures; the Vd (L/kg) does not increase in direct proportions to the increase in adipose tissue in obese patients. Limited data available. | [ |
| Cefazolin | 2 g for surgical prophylaxis | 3 g of cefazolin for surgical prophylaxis in persons >120 kg | Inadequate subcutaneous tissue concentrations and a “favorable” side effect profile. A systematic review suggested that outcomes data do not warrant the use of 3 g. | [ | |
| Linezolid | 600 mg BID | 600 mg TID or 900 mg BID | In obesity, the typical dose of 600 mg BID of the moderately lipophilic linezolid is insufficient to reach the target time above MIC for MRSA. The clinical relevance is debatable with few high-quality studies available. | [ | |
| Voriconazole | Dosed based on ABW | Dose based on adjBW or IBW | Use of ABW is associated with significantly higher trough levels manifesting clinically as neurotoxicity. Limited data available for empiric dosing with adjBW vs IBW. TDM recommended. | [ | |
| Cystic fibrosis | Ceftaroline | 600 mg q12h, 1-h infusion | 600 mg q8h, 2-h infusion | Since ceftaroline half-life is shorter in CF patients, a higher dose and longer infusion will optimize | [ |
| HSCT/SOT | Cefotaxime | 1 g q6h infused over 20 min | 4 g as continuous infusion | Based on a small study of patients undergoing orthotopic liver transplants. Both arms had adequate biliary exposures and were able to reach serum target concentrations above the MIC for at least 60% of the dosing interval. However, some patients in the intermittent arm developed undetectable levels or insufficient levels during the reperfusion phase of the transplant surgery. | [ |
| Pregnancy | Darunavir/ritonavir | 800 mg/100 mg daily if treatment naive | 600 mg/100 mg q12h with food | Pregnant individuals experience low darunavir exposures and do not qualify for daily dosing even if treatment naive. Instead, BID dosing is recommended. The use of the higher dose (800 mg/100 mg) does not increase darunavir exposures and is not recommended by current guidelines. | [ |
| Amoxicillin | 500–875 mg PO every 8–12 h | Increase frequency (eg, every 6–8 h) or dose | Increased renal clearance during pregnancy reduces drug exposure, requiring dose adjustments to maintain therapeutic concentrations. | [ | |
| Burn injury | Piperacillin-tazobactam | 4.5 g every 6–8 h, infused over 30 min | 4.5 g q6h, prolonged infusion (≥3 h) or continuous infusion | Burn patients may have augmented renal clearance, increasing drug clearance and reducing systemic exposure for highly renally eliminated ABX. Some studies suggest higher doses beyond 18 g/d, but safety and tolerability should be considered. | [ |
| Meropenem | 500 mg, 1 g, or 2 g IV q8h, infused over 30 min | 1 g IV at 0, 4, and 8 h then 1 g q8h; consider prolonged or continuous infusion | Assuming standard MIC targets (ie, not an MDRO) and infected tissue that is not an anatomically protected site (eg, CNS), then this dosing strategy provides a higher total loading dose to accommodate the larger Vd of a hydrophilic drug in burn victims with prolonged infusions to optimize the time over MIC PD parameter. | [ | |
| Ciprofloxacin | 400 mg IV BID or 500 mg PO BID | 400 mg IV q8h or 600 mg PO q12h | As a concentration-dependent antimicrobial, larger doses may be needed to get adequate bactericidal concentrations when patients have a larger than normal Vd. Minimal clinical data are available to support this dosing, but there are some safety data in burn victims up to 600 mg q8h. | [ |
| Patient Population | Resources or Reviews [Reference] | Notable Content |
|---|---|---|
| Renal dysfunction | Antibiotic dosing for critically ill adult patients receiving intermittent hemodialysis, prolonged intermittent renal replacement therapy, and continuous renal replacement therapy: an update (2020) [ |
PK considerations specific to the renal replacement modality ABX dosing recommendations in critically ill patients receiving intermittent hemodialysis, prolonged intermittent renal replacement therapy, and CRRT |
| An ex vivo model to determine transmembrane clearance of antimicrobials during continuous renal replacement therapy (2025) [ |
An ex vivo CRRT model of cefepime, meropenem, levofloxacin, and micafungin to determine the adsorption and transmembrane clearance across various hemofilters, modes, and effluent flow rates These models were clinically validated and can be used to develop appropriate dosing regimens | |
| Critically ill | Optimization of the treatment with beta-lactam antibiotics in critically ill patients—guidelines from the French Society of Pharmacology and Therapeutics (2019) [ |
French guideline with recommendations on renal function calculations, plasma protein measurements, β-lactam PK/PD targets, and administration techniques |
| Pharmacokinetics of commonly used antimicrobials in critically ill adults during extracorporeal membrane oxygenation: a systematic review (2021) [ |
Summary of available literature and dosing recommendations for ABX in ECMO patients (vancomycin, β-lactams, amikacin, linezolid, caspofungin) | |
| Pharmacokinetics, pharmacodynamics, and dosing considerations of novel β-lactams and β-lactam/β-lactamase inhibitors in critically ill adult patients: focus on obesity, augmented renal clearance, renal replacement therapies, and extracorporeal membrane oxygenation (2022) [ |
PK/PD exposures of ceftolozane-tazobactam, ceftazidime-avibactam, cefiderocol, ceftobiprole, imipenem-relebactam, and meropenem-vaborbactam | |
| Antibiotic dose optimisation in the critically ill: targets, evidence and future strategies (2024) [ |
Challenges related to inadequate antibiotic dosing in critically ill patients Summary of limitations of currently available data | |
| A narrative review on antimicrobial dosing in adult critically ill patients on extracorporeal membrane oxygenation (2024) [ |
Comprehensive review of the PK/PD impact ECMO poses on various antimicrobials | |
| Obesity | Antimicrobial dosing in obese patients (1997) [ |
Historical review of medications with narrow therapeutic windows from before the obesity epidemic |
| Demystifying drug dosing in obese patients (2015) [ |
Pathophysiologic changes in obesity in general Drug class chapters with literature review and drug-specific examples of clinical implementation | |
| Comprehensive guidance for antibiotic dosing in obese adults (2017) [ |
Obesity-specific dosing guidance | |
| Updated antimicrobial dosing recommendations for obese patients (2024) [ |
Comprehensive review of PK in obesity Obesity-specific dosing guidance | |
| Sanford Guide (Yearly) [ |
Obesity dose adjustments table with recommended dosing and citations Table includes drugs that do not need to be adjusted Updated regularly | |
| The pharmacokinetics of antibiotics in patients with obesity: a systematic review and consensus guidelines for dose adjustments (2025) [ |
Systematic review that found modest β-lactam PK changes in obesity without evidence to support routine dose adjustment Aminoglycosides and glycopeptides demonstrate clinically relevant PK alterations warranting weight-based dosing Evidence quality across antibiotic classes was low to very low Therapeutic drug monitoring is recommended as a pragmatic approach when obesity-specific PK data are limited | |
| Cystic fibrosis | The pharmacokinetics of antibiotics in cystic fibrosis (2021) [ |
Discussion on how CF-related physiological changes can alter the PK of various antibiotics |
| Population pharmacokinetic modeling of cefepime, meropenem, and piperacillin-tazobactam in patients with cystic fibrosis (2025) [ |
PTA of common β-lactam antimicrobials in PwCF Table with suggested dose and infusion time based on age | |
| HSCT/Transplant | Clinical pharmacokinetics in organ transplant patients (1989) [ |
PK overview and differences by organ |
| Pharmacokinetics of drugs in adult living donor liver transplant patients: regulatory factors and observations based on studies in animals and humans (2016) [ |
PK-based approach to alterations in liver transplant Summary tables of PK changes Nonantimicrobial drug-specific review | |
| Peri- and postsurgical evaluations of renal transplant (2017) [ |
Review of posttransplant complications and physiologic alterations that ultimately impact PK | |
| Pregnancy | Physiologic and pharmacokinetic changes in pregnancy (2014) [ |
Systems-based review of physiologic changes in pregnancy and their PK implications |
| Pregnancy-associated changes in pharmacokinetics: a systematic review (2016) [ |
Systematic review with rigorous quality assessment Summary tables with drug-specific examples and citations | |
| Gestation-specific changes in the anatomy and physiology of healthy pregnant women: an extended repository of model parameters for physiologically based pharmacokinetic modeling in pregnancy (2017) [ |
Review of physiologic changes in pregnancy-specific PK equation modifications based on the literature | |
| Drugs in pregnancy: pharmacologic and physiologic changes that affect clinical care (2020) [ |
PK approach to reviewing PK changes Enzyme-specific summaries for metabolic changes in pregnancy | |
| The pharmacokinetics and target attainment of antimicrobial drugs throughout pregnancy: parts I and III (2023) [ |
Drug class–based review of the literature Dedicated paper to penicillins with illustration summary of PK changes Part III has Table 10, which reviews pregnancy risk in animal and human studies | |
| A review of antibiotic safety in pregnancy—2025 update (2025) [ |
Review of antibiotic safety data in pregnancy. Focus of the review is on newly approved antibiotics since prior 2015 publication | |
| Burn injury | Influence of burns on pharmacokinetics and pharmacodynamics of drugs used in the care of burn patients (2008) [ |
Overview of physiologic changes Drug class–based review |
| Intravenous antibiotic and antifungal agent pharmacokinetic-pharmacodynamic dosing in adults with severe burn injury (2016) [ |
PK changes and drug review Drug dosing suggestions based on PTA for specific MICs | |
| The biochemical alterations underlying post-burn hypermetabolism (2017) [ |
Review of biochemical mechanisms driving PK alterations | |
| The effects of major burn related pathophysiological changes on the pharmacokinetics and pharmacodynamics of drug use: an appraisal utilizing antibiotics (2018) [ |
Overview of physiologic changes with illustration for PK implications PK-based review of changes Alternative dosing strategies | |
| Pharmacokinetics and pharmacodynamics of antimicrobial agents in burn patients (2021) [ |
PK changes overview Drug class–based review |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotics Pharmacokinetics and Efficacy · Cystic Fibrosis Research Advances · Antibiotic Use and Resistance
Pharmacokinetics (PK) and pharmacodynamics (PD) are key determinants of antimicrobial exposure, but they can vary widely across different patient populations. Optimization of antimicrobial dosing based on PK/PD principles can have important implications in the clinical treatment of infections. Historically, drug development programs for novel or established antimicrobials generated limited PK data and neglected important subpopulations. This narrative review highlights special populations at risk for clinically relevant alterations in PK and calls for proactive steps to generate actionable PK/PD data in these patients.
OVERVIEW OF PK/PD PRINCIPLES
PK describes the absorption, distribution, metabolism, and elimination of an agent through the body. PK properties, such as half-life, clearance, and volume of distribution (V_d_), determine the drug's concentration in the body over time. In infectious diseases, these concentration-time profiles correlate antimicrobial exposure with efficacy and/or toxicity, also known as PD. Antimicrobial PK/PD is traditionally categorized into 1 or more of the following PK/PD indices: (1) duration of time that the free drug concentration remains above the minimum inhibitory concentration (fT > MIC), (2) the ratio of the maximal free drug concentration to the MIC (fC_max_/MIC), and (3) the ratio of the 24-hour free area under the concentration time-curve (AUC) to the MIC (fAUC/MIC) [1, 2]. Quantifying the magnitude of antimicrobial exposure to maximize efficacy and minimize toxicity aids in dose determination, a key step in preclinical drug development [3, 4].
The significance of PK/PD data in preclinical studies and regulatory submissions has evolved over time, in concert with our understanding of this science [4, 5]. Historically, PK studies were conducted in healthy male volunteers, which can underestimate human variability in drug exposures due to the homogeneous population [6]. It is now appreciated that differences in sex, body weight, organ impairment, genetic polymorphisms, and food/drug interactions can alter a drug's expected PK properties. Several of these variables can be evaluated using population PK models; however, more robust PK data are needed from subpopulations of interest to more accurately predict human drug exposures associated with efficacy and toxicity [7]. Both the US Food and Drug Administration (FDA) and the European Medicines Agency now emphasize the need for clinical PK studies across diseases states and age groups at various stages of clinical development to support PK/PD analyses [8, 9].
Sole reliance on in vitro, animal, or healthy volunteer PK/PD studies for dose design and execution in later phase clinical studies has resulted in the failure of drug development programs. For example, daptomycin's lung-specific PK/PD was not studied in humans prior to clinical trials assessing safety and efficacy for the treatment of acute bacterial community-acquired pneumonia. Rather, clinical utility was assumed based on animal pneumonia models and in vitro activity against common bacterial pneumonia organisms (eg, Streptococcus pneumoniae and Staphylococcus aureus). Only after clinical studies failed was it discovered that human pulmonary surfactants drastically decreased the activity of daptomycin, explaining the poor clinical outcomes [10]. Similar stories of limited PK/PD data driving flawed decision-making in clinical trials include tigecycline for bloodstream infections, ceftobiprole for hospital- or ventilator-acquired pneumonia, doripenem for ventilator-associated pneumonia, and oral eravacycline for urinary tract infections [11–14]. The availability of robust PK/PD data to inform drug dosing and development could have mitigated some of these high-profile clinical failures. Instead, patients suffered increased clinical failures, and the antimicrobials received black-box warnings, failed to achieve approval for specific indications, or never made it to the market.
Unfortunately, the story of poor investment in PK/PD studies continues. Regulatory approved doses may not be optimized against specific infections or patient populations—an issue that is neither identifiable nor correctable until clinical failures begin to emerge. Higher doses than initially recommended are now standard for several antimicrobials (Table 1) [15].
This raises the question: How many patients are undertreated before we get the “right dose”? Available literature identifies several patient populations at risk for altered PK, including, but not limited to, patients with obesity, renal dysfunction, critical illness, or cystic fibrosis (CF). Identifying how antimicrobial PK differs in these populations compared with the general population is essential in anticipating risks of reduced efficacy and toxicity.
RENAL DYSFUNCTION AND HYPERFUNCTION
Renal function is a major determinant of antimicrobial PK because of its central role in drug clearance. Chronic kidney disease (CKD) affects a substantial proportion of the US population, ranging from 6% to 38% of people. Older adults and populations with comorbidities such as diabetes and hypertension have higher rates of CKD [21]. Hyperfunction of the kidney, known as augmented renal clearance (ARC), is observed in approximately 40% of patients in intensive care units (ICUs) [22]. Acute infection-related changes in renal function may arise from direct renal involvement, immune-mediated injury, or hypoperfusion during septic shock [23].
Antimicrobials with substantial renal elimination (eg, >30% unchanged drug excreted in urine) are particularly sensitive to changes in kidney function [24]. Hydrophilic agents, such as aminoglycosides and most β-lactams, are greatly impacted by renal clearance, whereas lipophilic agents are more commonly eliminated hepatically [25]. ARC increases antimicrobial elimination, resulting in reduced systemic drug exposure, whereas CKD or acute kidney injury (AKI) decreases renal elimination, leading to increased systemic exposure and potential toxicity (Table 2).
Failure to achieve appropriate antimicrobial exposure in the setting of altered renal function has been associated with delayed bacterial clearance, treatment failure, and selection of resistant subpopulations [75–77]. Dose reductions in the setting of CKD and AKI are routine practice but are often informed by imprecise estimates of clearance. Dose increases or prolonged infusions in the setting of ARC are not standardized since ARC has only recently gained broader recognition [78]. Therapeutic drug monitoring (TDM) can improve dosing and exposures in patients with ARC, but access to routine TDM and the necessary infectious diseases expertise to interpret and act upon TDM data beyond vancomycin and aminoglycosides are limited [30, 79].
Conversely, upfront investment in a population PK model with ARC helped justify higher cefiderocol dosing and improved PK/PD target attainment [80]. This example highlights the need for additional research in populations with altered renal function. Accurate estimation of renal function remains a major challenge in antimicrobial dosing. There are multiple ways to calculate creatinine clearance (CrCl), such as the Modification of Diet in Renal Disease (MDRD) and Cockcroft-Gault formulas, which serve as surrogates for actual renal filtration but lead to inconsistencies in dosing recommendations [81]. Crude estimates of CrCl in patients with AKI and renal replacement therapies (RRTs) often dictate recommended antimicrobial doses [82]. In the setting of acute changes, a patient's serum creatinine may be significantly delayed in reflecting the degree of injury. Delayed recognition of renal injury may lead to drug accumulation and toxicity, whereas premature dose reduction risks inadequate exposure during the critical early phase of infection [83, 84]. Given the frequency and dynamic nature of renal dysfunction in hospitalized patients, renal-specific PK/PD considerations are essential for optimizing antimicrobial dosing and avoiding preventable treatment failure or toxicity.
CRITICAL ILLNESS
The complex pathophysiology of critical illness alters drug PK/PD parameters [85]. Drug distribution can change during critical illness due to changes in V_d_ and hypoalbuminemia. Increases in V_d_ result from endothelial dysfunction, capillary leak, altered protein binding, and aggressive fluid resuscitation. These changes disproportionately affect hydrophilic antimicrobials, such as β-lactams, which primarily distribute within the intravascular space and may require loading doses or alternative infusion strategies to achieve adequate early exposure [27, 28].
Renal dysfunction and utilization of RRTs are common in ICUs. To address impaired kidney function and volume status, RRT is prescribed with varying dialysate and filtrate flow rates [86]. Application of continuous RRT (CRRT) utilizes different modalities: convection (continuous veno-venous hemofiltration [CVVH]), diffusion (continuous veno-venous hemodialysis [CVVHD]), or both (continuous veno-venous hemodiafiltration [CVVHDF]) [87]. The impact on antimicrobial PK depends on the modality and dose of CRRT used, but in general, utilization of CRRT increases V_d_ and clearance of antimicrobials [88]. All modalities using convection more effectively eliminate antimicrobials with high molecular weights, whereas the combination of convection and diffusion (CVVHDF) results in augmented clearance of antimicrobials compared to convection or diffusion alone [88]. Additionally, the higher the CRRT dose or effluent flow rate, the greater the drug clearance. Other drug-specific characteristics, such as lipophilicity and ionization, impact clearance as well [86]. As CRRT only eliminates the free fraction of the antimicrobials, highly protein-bound antimicrobials are less impacted by CRRT (Table 2) [89]. Notably, cefiderocol represents the first antimicrobial with dose-optimized and clinically validated CRRT dosing recommendations, supported by population PK modeling and clinical data, highlighting the feasibility and value of proactive PK/PD evaluation in patients receiving CRRT [90, 91].
Critically ill patients with cardiorespiratory failure may require extracorporeal membrane oxygenation (ECMO) for adequate blood perfusion and organ oxygenation. Due to the large surface area of ECMO circuits, lipophilic and/or highly protein-bound antimicrobials bind the circuit components, resulting in subtherapeutic plasma levels [88]. The degree of sequestration depends on the type of tubing, pump, oxygenator, and priming solution. Furthermore, the addition of priming solution can increase the V_d_ of drugs [88]. Evaluation of antimicrobials under ECMO conditions (eg, cefepime, piperacillin-tazobactam, cefiderocol) has demonstrated variable target attainment with conventional and high-dose regimens, highlighting the limitations of standard dosing in this setting [92–94].
Given these complexities, many antimicrobials are ineffectively dosed in critical illness due to variable patient PK, intra- and interinstitutional variability in antimicrobial dosing, and extracorporeal therapies used [95]. Clinical outcomes in critically ill populations demonstrate that when PK/PD targets are not attained, there is an increased risk of microbiological failure, prolonged ICU length of stay, and mortality [95–97]. Key unmet needs include defining dosing recommendations that account for extracorporeal therapies, determining when TDM meaningfully improves outcomes, and clarifying the role of model-informed precision dosing tools in routine ICU practice [98].
OBESITY
Approximately 42% of US adults meet criteria for obesity based on body mass index. Despite this prevalence, antimicrobial PK data across obesity classes (class I–III) remain ill-defined, with most studies enrolling small cohorts that limit class-specific dosing conclusions [99–101]. Given the frequency of obesity among hospitalized patients, clarifying when obesity meaningfully alters antimicrobial exposure is increasingly relevant to routine clinical care.
The first consideration is administration route and tissue distribution. Despite a theoretical increase in intestinal perfusion and additional abdominal fat, the lack of significant bioavailability differences for oral drugs accentuates the imperative for additional PK and clinical data [101]. Concerns related to intralipomatous injections in obese patients that were intended as intramuscular injections have been raised since 1982 [101, 102]. Obesity may alter antimicrobial penetration into certain tissues, although the relationship between tissue concentrations and clinical outcomes remains poorly defined. A lipid partition coefficient can approximate the drug's ability to concentrate in adipose tissue but does not necessarily correlate to serum drug concentrations [33, 101]. Other drug distribution factors impacted by obesity include more lipoproteins to competitively bind albumin and alpha-1-acid glycoproteins [101]. As a result, weight-based dosing strategies vary by antimicrobial, with total, ideal, or adjusted body weight used (Table 3) [34].
The impact of obesity on metabolism is complex. Despite no substantial difference in hepatic blood flow, enzymatic damage due to excessive fatty acid deposits in obesity can reduce antimicrobial metabolism [101]. Phase I metabolism is mostly unaffected by obesity; phase II may be increased in obesity [33]. The degree to which predicted renal clearance is impacted in obesity varies by the specific equation used. Lean and ideal body weight appear more accurate than total body weight in CrCl and estimated glomerular filtration rate calculations, although neither has a perfectly linear correlation into the class III obesity range. Corrective equations for obesity such as the Salazar-Corcoran and 4-variable modification of MDRD (MDRD4) also lack precision and vary drastically by obesity classification [101].
In a retrospective cohort, obese patients receiving β-lactams experienced higher rates of clinical treatment failure and longer hospitalization compared with nonobese patients [132]. Toxicity considerations are also relevant, as data have shown obesity as a risk factor for vancomycin-associated nephrotoxicity [133]. Given PK changes in obesity, TDM may help optimize exposure and improve outcomes in high-risk infections; however, serum concentrations do not always correlate with tissue exposure [113, 120]. These gaps in knowledge related to the implications of obesity on antimicrobial dosing will have increasing clinical importance given its rising prevalence in the population.
CYSTIC FIBROSIS
People with CF (PwCF) experience more infections and consequently antimicrobial exposures than non-PwCF [134, 135]. Therefore, optimization of PK/PD in this population is essential to improve antimicrobial efficacy and safety. While information varies on PK changes in PwCF, we highlight key PK considerations; a more extensive review may be found in previous publications [136–138].
Prior studies have described PwCF PK alterations in drug absorption, V_d_, plasma protein binding, metabolism, and elimination [138]. Alterations are specific to the antimicrobial with select agents displaying similar PK to non-PwCF comparators, while other antimicrobials (eg, β-lactams and aminoglycosides) have quite distinct PKs with noted increases in V_d_ and clearance. Due to cystic fibrosis transmembrane conductance regulator (CFTR) channel modifications, diet, and antimicrobial exposure changing intestinal inflammation and gut microbiome, PwCF experience altered gut physiology that may impair the absorption of oral antimicrobials [138, 139]. Several studies have shown potential delayed time to maximum concentrations without subsequent changes in overall bioavailability (Table 2) [42, 43, 140].
Several strategies to optimize antimicrobial dosing have been explored (Table 3). The established practice of TDM for tobramycin and vancomycin may promote their safety and efficacy, but the optimal sampling approach remains poorly defined [45, 141, 142]. One recent study suggests that AUC-based vancomycin dosing may improve its safety profile in PwCF [143]. Extended and continuous infusions of β-lactams are commonly employed to promote better PK/PD target attainment, but limited evidence exists to suggest a clear benefit on clinical outcomes [44, 144]. In addition, continuous infusion approaches alone may lead to insufficient concentrations in approximately 50% of patients, lending further support for TDM [133]. A combination strategy of TDM with continuous infusion approach has been explored for ceftolozane-tazobactam [145]. More recent PK/PD studies of ceftolozane-tazobactam and ceftaroline suggest that more “aggressive” dosing strategies via higher doses (eg, 3 g for ceftolozane-tazobactam) and increased frequency of administration (eg, every 8 hours for ceftaroline) are necessary to optimize PK/PD in PwCF [125, 146].
The use of inhaled antimicrobials offers the potential to increase target pulmonary exposure and reduce systemic exposure, which could optimize target PK/PD and limit toxicity. Many factors influence the lung distribution of inhaled antimicrobials and variable exposures may occur [147–149]. Several antimicrobials are FDA approved (eg, tobramycin) and additional antimicrobials (eg, ceftazidime) are used “off-label” to manage pulmonary exacerbations [150]. However, further studies are needed to ensure optimal delivery methods, dosing efficacy, and safety in prevention and treatment strategies in PwCF [150].
Historically, PwCF are underrepresented or excluded from registrational trials, limiting our knowledge surrounding novel therapeutics. Fortunately, more recent trials have provided an understanding of novel antimicrobials’ PK in this population; however, several research gaps related to PK/PD remain [151, 152]. Notably, future novel antimicrobials with a spectrum inclusive of multidrug-resistant organisms (MDROs) commonly seen in CF should be addressed during the traditional approval process. Other key areas of focus include future PK studies of approved antimicrobials (eg, cefiderocol, meropenem-vaborbactam, and lefamulin), PK/PD and efficacy studies of oral and inhaled therapies, the role of TDM in PwCF, the relationship between CFTR modulators and antimicrobials (eg, drug–drug interactions and infection prevention/treatment), and the interplay between combination therapy and its effects on PK/PD optimization.
OTHER NOTABLE POPULATIONS
Other important populations with significant PK alterations include transplant candidates or recipients, pregnant persons, and patients with significant burns. Key high-level PK changes in each of these populations are reviewed below.
PK variations in solid organ transplant candidates and recipients are related to pathophysiologic changes from end-organ damage, graft function, immunosuppression, and several other factors. Chemotherapy and radiation therapy in hematopoietic stem cell transplant recipients introduce additional temporary PK alterations [153, 154]. Posttransplant PK changes are most consequential in the first month posttransplant, including decreased bioavailability and delayed absorption of oral antimicrobials [47, 155]. Transplant recipients also frequently encounter drug–drug interactions and medications susceptible to metabolism and elimination PK changes. Polymorphisms of metabolic alleles, particularly CYP3A5 and CYP2C19, have demonstrated significant PK alterations in transplant recipients [55, 156–158]. Additionally, the type of graft transplanted can impact the recipient's PK. For example, renal transplant recipients will have fluctuating antimicrobial concentrations as graft function recovery influences renal drug elimination [159–162]. Surgical complications, immune-mediated injury, and nephrotoxic concomitant therapies further contribute to highly variable renal clearance, complicating empiric antimicrobial dosing in this population [156, 159, 161].
Some of the more dramatic changes in PK occur during pregnancy and differ depending on trimester [163]. Absorption can be altered by the increased stomach pH and delayed gastric emptying that occur in pregnancy [58, 59, 63]. Antimicrobial distribution in pregnant women is also variable. For example, hypoalbuminemia is a common occurrence in pregnancy that changes the free drug availability of highly protein-bound medications [58, 59, 63]. Concentration-dependent antimicrobials are also diluted in pregnancy given the expansion of plasma volume and additional amniotic fluid. As the pregnancy progresses and during the postpartum period, fat tissue increases with differential tissue distributions, potentially altering lipophilic antimicrobial distribution to infected tissues. Increased antimicrobial elimination can occur through breastfeeding, with significantly increased blood flow to the mammary arteries [59, 63]. Furthermore, antimicrobial dosing in the setting of ARC, discussed in general above, represents an important consideration specifically in pregnancy [58, 163]. Pregnant women develop ARC to a CrCl of 150 mL/minute or more in the first trimester [58, 59, 63]. Conversely, hepatic metabolism and elimination in pregnancy are variable [59, 63]. The FDA has made substantial updates to pregnancy, lactation, and fertility drug labeling. However, these updates are mostly limited to animal data, limiting the ability to make exposure-based dosing recommendations [164].
Clinically relevant PK changes also occur when burns exceed 20% or more of the total body surface area [70]. In burn patients, oral absorption can be impaired by significant peripheral and gastric blood flow constriction while disrupted skin integrity alters topical absorption [70]. Burn-induced cytokine responses may trigger life-threatening hemodynamic shifts, increased capillary permeability, and myocardial depression contributing to extensive V_d_ changes [70, 71]. Burn management also requires aggressive fluid resuscitation, further diluting antimicrobials. Additional shifts of protein-rich fluids to the interstitial space increases the amount of free drug for highly protein-bound antimicrobials (eg, ceftriaxone) [70, 71]. Patients with burns more reliably exhibit a hypercatabolic state starting about 5 days post–thermal injury. Elimination is also variable with renal damage occurring in roughly a third of burn patients, with the remaining patients prone to ARC [70, 71].
Scrutiny of the PK heterogeneity in these special populations is essential to defining the therapeutic window of antimicrobials. There are insufficient PK studies in these select populations, significantly limiting available information to guide tailored antimicrobial dosing and forcing reliance on broader population PK and therapeutic indices. Inconsistent PK and drug interactions alone support the need for antimicrobial TDM and further research into its application.
CALL TO ACTION
In most historical clinical trials, standard antimicrobial doses and infusion rates were applied across all populations with little consideration of patient- or organism-specific factors [7]. As the special populations discussed above expand, there exists heightened concern that relying on traditional antimicrobial dosing alone will lead to suboptimal patient outcomes [165]. In the following sections, we propose several action items to address this challenge.
Encourage Broader Inclusion Criteria in Antimicrobial Registration Trials to Inform Optimal Dosing in Special Populations
While pharmacotherapy has made significant progress in characterizing optimal antimicrobial dosages and exposures, many of the applied recommendations in these groups are derived from small studies or extrapolated. As illustrated in Figure 1, early and intentional inclusion of select special populations within antimicrobial development programs represents a high-feasibility opportunity to generate actionable PK data before registrational trials are complete. Newer compounds, such as cefiderocol, meropenem-vaborbactam, and aztreonam-avibactam, have been studied as a prolonged infusion coupled with PK sampling, which provided additional data for dose optimization [166, 167]. Another complementary approach would be broader inclusion criteria for enrollment in clinical trials, particularly in special populations (eg, CF, ECMO) to gather actionable PK data [168]. Although direct links between PK/PD optimization and clinical outcomes remain limited for many special populations, established exposure–response relationships have been sufficient to inform dosing recommendations and regulatory labeling in select settings. However, when exposure–response relationships are uncertain or dosing strategies substantially alter clinical practice, prospective clinical outcomes data may be necessary to support broader adoption and guideline endorsement [169].
Prioritization roadmap for PK/PD evaluation in special populations during antimicrobial development. A framework for incorporating special population PK/PD data across the antimicrobial development lifecycle is shown. Early development focuses on populations with predictable physiologic alterations and high feasibility, while later phases expand to more complex or vulnerable populations. A combination of modeling, therapeutic drug monitoring, and real-world data approaches enables scalable and ethical data generation to inform dosing and labeling decisions. Abbreviations: ARC, augmented renal clearance; CRRT, continuous renal replacement therapy; ECMO, extracorporeal membrane oxygenation; HD, hemodialysis; PBPK, physiologically based pharmacokinetic (modeling); PK, pharmacokinetic.
Create Market Incentives for Drug Development to Generate PK and Outcomes Data in Special Populations
The financial costs and barriers of bringing a new antimicrobial through research and development are significant, and anti-infectives are not considered a highly profitable therapeutic class. In response to this, the US Congress enacted the Generating Antibiotics Incentives Now (GAIN) Act, which provides additional benefits to pharmaceutical companies for developing novel products that serve a critical need in treating life-threatening and MDROs. Antimicrobials can be placed under application for qualified infectious diseases product status, which grants an additional 5 years of market exclusivity as a financial incentive [170]. However, most antimicrobials enter the market based on trials in less serious, uncomplicated infections, rather than for the indications needed in actual clinical practice [171, 172]. One recent positive development is randomized trials of novel β-lactam/β-lactamase inhibitors and other compounds to generate best-practice guidance for treatment of infections caused by MDROs [166, 167]. The additional time and expense of studying the PK and outcomes in special populations carries similar challenges. Given the high morbidity and mortality these populations face, they deserve the same level of urgency for generating high-quality PK and outcomes data. Like the MDRO space, researchers will face significant enrollment challenges, and there are no market-entry incentives to evaluate infected PwCF, patients with ARC, or other special groups in antimicrobial development. However, the FDA does recommend separate PK studies in patients with renal dysfunction if it is suspected to be significantly altered. A major priority in the National Action Plan for Combating Antimicrobial Resistance (CARB) includes the advancement of in vitro, animal model, and PK studies to facilitate antimicrobial drug development [173, 174]. Furthermore, the Society of Infectious Diseases Pharmacists has also emphasized the importance of economic incentives to support antimicrobial drug development, calling for policy-driven strategies to ensure continued innovation in this field [175].
Organize Private–Academic Partnerships and Research Networks to Facilitate Patient Recruitment for Special Populations in Postmarketing PK Studies
To answer unmet needs in PK/PD and outcomes data, clinicians have created networks to quickly recruit and study patients in special populations [176]. Collaboratives from private–academic partnerships, corporations, and organizational research networks have generated important PK/PD safety and efficacy data that inform dosing [7, 177]. Limited funding, time, and resources along with recruitment challenges and clinical confounders are among the most difficult obstacles researchers face when studying rare patient groups at high risk for morbidity and mortality. However, support is available through government, nonprofit organizations, and industry to conduct PK and outcomes studies in these groups [178]. The use of real-world, pragmatic randomized designs to answer challenging clinical questions has also demonstrated critical value in their ability to rapidly scale high-quality data in large populations [179].
Utilize In Vitro and In Silico Modeling to Simulate PK Parameters in Special Populations or in Artificial Organ Support Settings
Another potential solution to the lack of readily available patients with rare conditions that can be efficiently sampled for PK analyses is to turn to in vitro and in silico modeling to simulate conditions at target sites or in the setting of organ support therapies [91]. Improvements in these models have substantially increased understanding of PK/PD interactions, without the risks associated with in vivo PK sampling [180–182]. However, in vitro effects do not necessarily predict in vivo efficacy [166, 183]. Furthermore, there are challenges in replicating and interpreting these models for human infections including, but not limited to, interpatient variability in clearance/distribution, presence of blood and blood proteins, and longer dosing periods.
Expand Access and Efficiency of TDM in Special Populations to Promote Bedside PK/PD Applications
Broader application of TDM should be pursued in patients with unpredictable PK, patients with serious infections, and patients receiving antimicrobials with actionable and narrow therapeutic and safety indexes. These groups certainly include patients with altered V_d_ and clearance such as those with CF, ARC, burns, and/or support from extracorporeal modalities [184]. Triazole antifungals, aminoglycosides, and glycopeptides have paved the way by demonstrating how defined safety and efficacy targets can improve patient care [185]. Clinical data continue to emerge for TDM in other classes such as β-lactams, fluoroquinolones, echinocandins, and linezolid [177, 184, 186, 187]. Unfortunately, most novel antimicrobials require high-performance liquid chromatography and mass spectrometry for TDM, which few US centers, let alone low- and middle-income countries, have direct access to on site. Advent of broader antibiotic assays with faster turnaround time and lower costs would facilitate precision dosing in special populations across diverse global healthcare settings [188].
Integrate Special Population Dosing Into Clinical Guidelines
Clinical practice guidelines are a readily available resource to many providers. While select guidelines acknowledge dosing considerations for certain special populations, PK/PD guidance across infections and patient populations remains limited [189–191]. Incorporation of antimicrobial dosing recommendations for special populations, including guidance on dose adjustment, infusion strategies, and use of TDM, is needed to drive adoption at the institutional level. Several high-quality reviews and consensus documents provide a framework for special population dosing, though their integration into routine practice and clinical guidelines remains inconsistent (Table 4).
CONCLUSIONS
A significant and increasing proportion of patients with serious infections fall into a population at risk for altered PK parameters. These special patient populations—including those with critical illness, CF, altered renal function, obesity, transplantation, burns, or pregnancy—are underrepresented in the available literature and clinical trials data that inform our current antimicrobial dosing. This lack of actionable clinical and PK/PD data is a major obstacle to optimization of antimicrobial therapy for these patients. To address this clinically important gap in knowledge, we call on regulatory agencies, the pharmaceutical industry, and the academic community to partner on a multipronged approach to remedy these research gaps to improve the care of these important populations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Craig WA . Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 1998; 26:1–10; quiz 11–2.9455502 10.1086/516284 · doi ↗ · pubmed ↗
- 2Ambrose PG, Bhavnani SM, Rubino CM, et al Pharmacokinetics–pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis 2007; 44:79–86.17143821 10.1086/510079 · doi ↗ · pubmed ↗
- 3Jorda A, Zeitlinger M. Preclinical pharmacokinetic/pharmacodynamic studies and clinical trials in the drug development process of EMA-approved antibacterial agents: a review. Clin Pharmacokinet 2020; 59:1071–84.32356105 10.1007/s 40262-020-00892-0PMC 7467913 · doi ↗ · pubmed ↗
- 4Palmer ME, Andrews LJ, Abbey TC, et al The importance of pharmacokinetics and pharmacodynamics in antimicrobial drug development and their influence on the success of agents developed to combat resistant gram negative pathogens: a review. Front Pharmacol 2022; 13:888079.35959440 10.3389/fphar.2022.888079 PMC 9359604 · doi ↗ · pubmed ↗
- 5Tuntland T, Ethell B, Kosaka T, et al Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front Pharmacol 2014; 5:174.25120485 10.3389/fphar.2014.00174 PMC 4112793 · doi ↗ · pubmed ↗
- 6Karakunnel JJ, Bui N, Palaniappan L, et al Reviewing the role of healthy volunteer studies in drug development. J Transl Med 2018; 16:336.30509294 10.1186/s 12967-018-1710-5PMC 6278009 · doi ↗ · pubmed ↗
- 7Alshaer MH, Goutelle S, Santevecchi BA, et al Cefepime precision dosing tool: from standard to precise dose using nonparametric population pharmacokinetics. Antimicrob Agents Chemother 2022; 66:e 0204621.34902271 10.1128/aac.02046-21PMC 8846452 · doi ↗ · pubmed ↗
- 8European Medicines Agency . Guideline on the use of pharmacokinetics and pharmacodynamics in the development of antimicrobial medicinal products. London: European Medicines Agency, 2016.