Mitogenomes reveal multiple evolutionary units and low genetic diversity of the critically endangered pancake tortoise Malacochersus tornieri
Chuan Jiang, Nassoro Mohamed, Rudolf Mremi, Xuda Liu, Gabriel Mayengo, Yang Liu, Reginald T. Mwaya, Wenwen Zhu, Yiming Gao, Bo Li

TL;DR
Pancake tortoises have low genetic diversity and two distinct evolutionary groups, which suggests the need for targeted conservation efforts.
Contribution
The study reveals cryptic speciation and population divergence in pancake tortoises using mitogenome data.
Findings
Pancake tortoises have extremely low mitogenome nucleotide diversity.
Northern populations are expanding while central populations are contracting, with poor connectivity.
A deep genetic split of ~5.74 million years exists between Kenyan and Tanzanian clades.
Abstract
Pancake tortoise (Malacochersus tornieri), a critically endangered East African endemic, is threatened by habitat destruction and illegal collection. Its fragmented range and isolated populations may have driven genetic differentiation that could inform its conservation. We infer evolutionary histories and population structure of M. tornieri using mitochondrial genomes (mitogenomes) of 60 free-ranging individuals from 11 localities across northern and central Tanzania. M. tornieri has rearranged mitogenome structure, translocation mutations of protein-coding genes, and extremely low mitogenome nucleotide diversity. Northern and central Tanzanian populations exhibit shallow divergence and contrasting demographic histories, with recent expansion in the north and contraction in the central population. This, combined with isolation-by-distance patterns found, suggests poor population…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
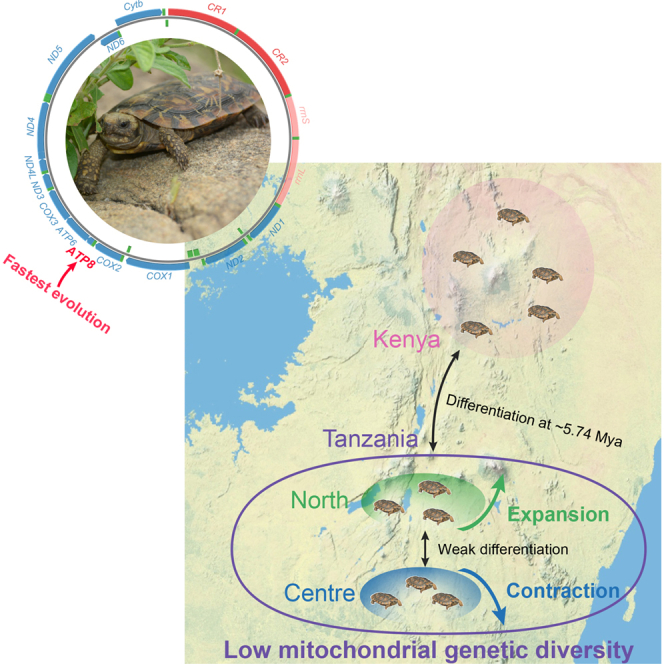 Figure 1
Figure 1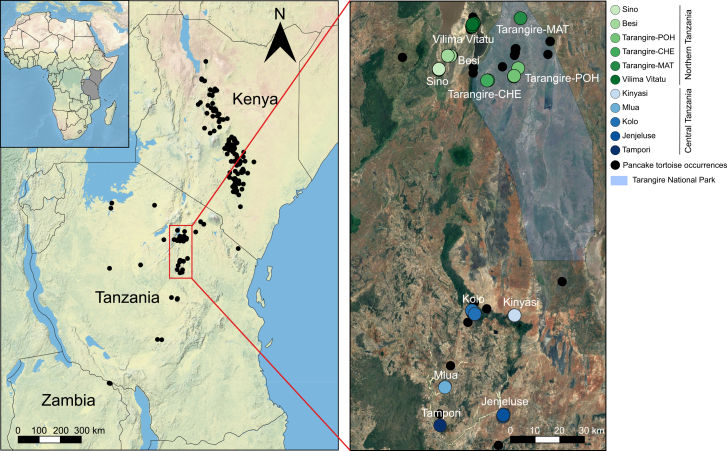 Figure 2
Figure 2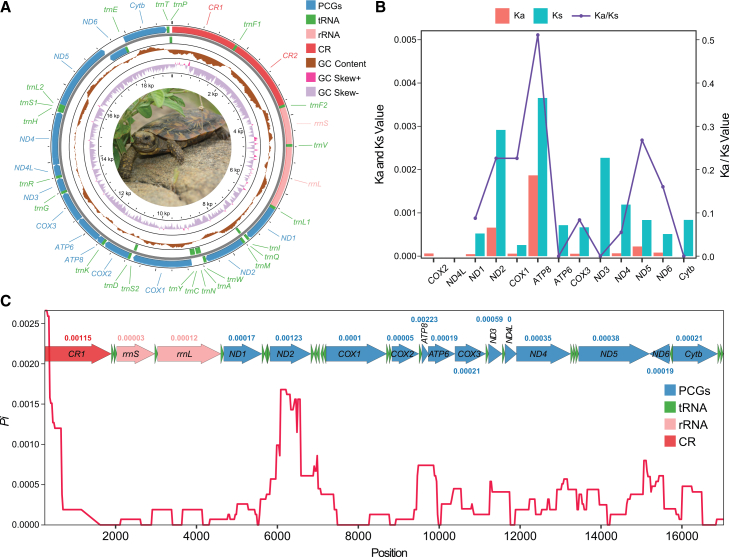 Figure 3
Figure 3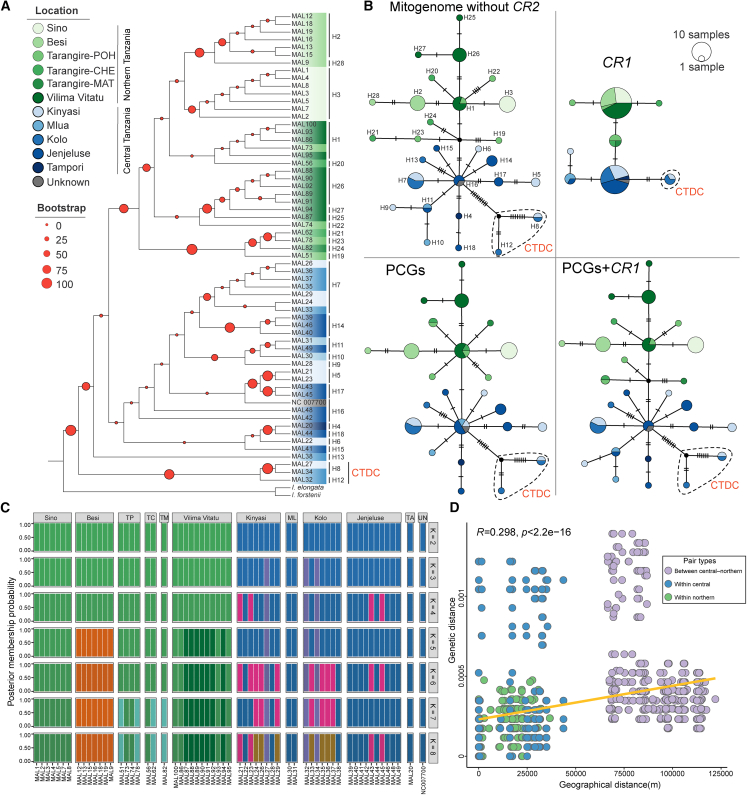 Figure 4
Figure 4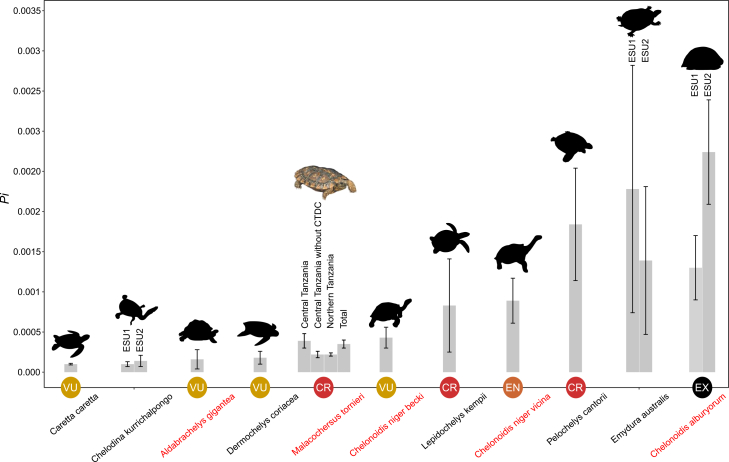 Figure 5
Figure 5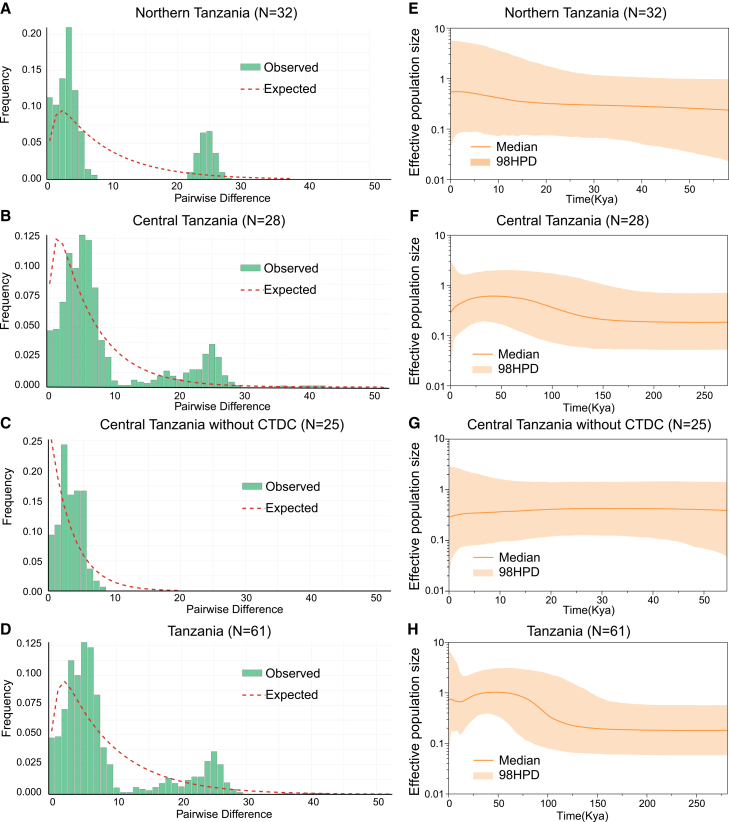 Figure 6
Figure 6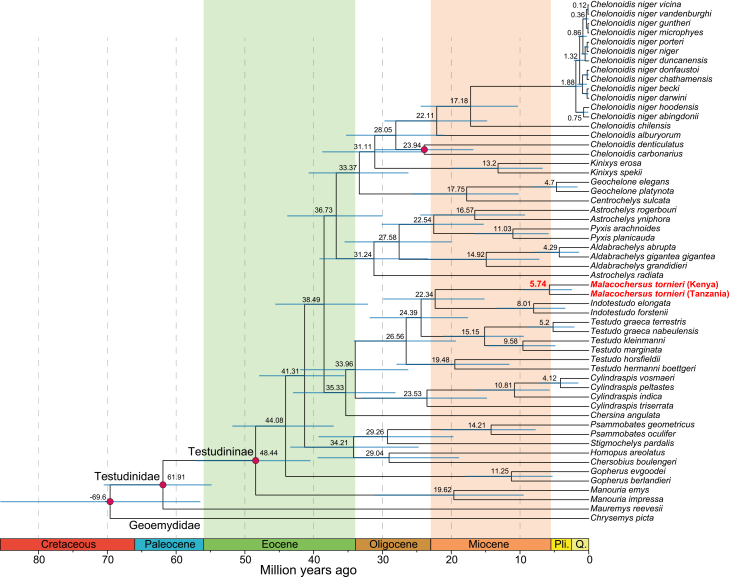 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTurtle Biology and Conservation · Genetic diversity and population structure · Evolution and Paleontology Studies
Introduction
Human activities have triggered the sixth mass extinction, causing unprecedented decline in global biodiversity.1^,^2^,^3 Many species have already gone extinct, while numerous others have experienced drastic population reductions and are on the brink of extinction.4 Major drivers include habitat loss, climate change, spread of invasive species, pollution, and overexploitation of biological resources.5^,^6 As the foundation for adaptation and resilience, greater genetic diversity leads to greater population adaptability and viability.7 However, genetic diversity is being lost worldwide, representing one of the greatest challenges to biodiversity.8 Most endangered species with restricted habitats exhibit low genetic diversity, which reduces reproductive success and adaptive capacity, thereby elevating their extinction risk.9^,^10^,^11 Thus, assessing the genetic diversity of endangered species is crucial for evaluating the status of their populations. On the other hand, conservation efforts should prioritize the preservation of genetic diversity using tools such as evolutionarily significant units (ESUs) and management units (MUs). ESUs emphasize long-term evolution, while MUs emphasize short-term management.12^,^13 These units help identify and protect unique lineages and locally adapted populations, ensuring that conservation strategies target biologically meaningful groups.14^,^15
With the tortoises being among the most threatened vertebrates worldwide,16 understanding and preserving their genetic diversity demand urgent attention. Pancake tortoise Malacochersus tornieri (Siebenrock, 1903) is one of the most threatened chelonian species and is listed as a critically endangered species by IUCN.17^,^18 The species is endemic to East Africa (Figure 1) and is currently recognized as the sole extant species within its genus.17 Genetics studies have shown that M. tornieri is closely related to Asian Indotestudo and Eurasian Testudo.19^,^20^,^21 Pancake tortoises exhibit unparalleled morphological, behavioral, and ecological uniqueness among extant Testudinidae tortoises.22^,^23^,^24 Its shell shows remarkable variation in both keratinous scutes and bony elements, particularly in the number of peripheral and suprapygal bones, and the bony shell is highly fenestrated, likely reflecting adaptation to a rock-dwelling lifestyle.25 These specialized traits, including the tortoise’s soft, flattened shell, have also made it a target in the international pet trade.17^,^18 Illegal exploitation for the exotic pet trade has significantly reduced M. tornieri populations in the wild and has resulted in local extirpation of some populations.17^,^18 To control trading in this species, its status was elevated to the “Appendix I” category in the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) in 2019 (CITES, https://www.cites.org). With only 23% of M. tornieri suitable habitat protected, the species is under continued threats from habitat destruction, agriculture expansion, and climate change.26^,^27^,^28 Moreover, M. tornieri distribution is not continuous, and the subpopulations are scattered across a few rocky outcrops in Kenya and Tanzania, with one population in only one locality in northern Zambia,17^,^22^,^23 which may have resulted in undetected genetic differentiation (i.e., multiple ESUs).17^,^29 Despite the ecological uniqueness of M. tornieri, the species remains largely understudied, with limited understanding of genetic diversity and connectivity among disjunct populations hindering effective conservation efforts.17^,^30Figure 1. Distribution range of the M. tornieri and sampling sites in this studyThe left map shows the natural distribution of the M. tornieri in East Africa (Tanzania, Kenya, and Zambia), based on occurrence data obtained from various sources iNaturalist and Global Biodiversity Information Facility (GBIF), as well as relevant literature and field surveys. The right map is an enlarged satellite image of the area outlined by the red rectangle in the left map, showing 11 sampling sites for M. tornieri in northern and central Tanzania.
The application of molecular techniques in genetic studies allows conservation practitioners to precisely determine the status of endangered species, providing important insights for species management.31 Among the molecular markers, the mitochondrial genome (mitogenome) has become an important marker in conservation genetics, taxonomy, and phylogenetics due to its conserved gene organization, relatively fast and predictable evolutionary rate, maternally linked inheritance, lack of recombination, and relative ease of amplification, although it also has some well-recognized limitations.32^,^33^,^34 For example, studies across multiple species demonstrate that mitogenomes offer essential data on genetic diversity, directly informing conservation strategies and supporting targeted conservation.10^,^35^,^36 In addition, because the mitogenomes have more copy numbers than nuclear genes, they typically offer an advantage in non-invasive sampling studies of endangered species, although modern nuclear DNA methods can also work with minimal tissue input. Therefore, research on mitogenomes contributes to the preliminary assessment of the genetic structure, diversity, and demographic history of endangered species, thus revealing population connectivity and identifying MUs.31
In this study, we sequenced and assembled mitogenomes of 60 M. tornieri individuals from 11 localities across northern and central Tanzania (Table S1) and performed population genetic analyses in combination with one published mitogenome. We first characterized the population evolutionary features of these mitogenomes and then revealed the genetic structure, genetic diversity, and recent demographic history of M. tornieri. We also assessed the divergence between Tanzanian and Kenyan populations based on the mitochondrial gene COX1. The findings of this study will help to inform conservation strategies and population management for this critically endangered species.
Results
Distinct mitogenomic configuration
Mitogenomes were assembled from quality-controlled reads. Sample MAL100 yielded identical circular mitogenomes from both HiFi long-read and PE150 short-read assemblies. The remaining 59 samples also yielded circular assemblies, with sequencing depth plots confirming structural integrity (Figure S1). All mitogenomes exhibited a conserved structure, consisting of 13 protein-coding genes (PCGs), 2 rRNAs, 2 CRs, and 23 tRNAs (Figure 2), consistent with previous reports for this species.19 This structure features an additional CR and tRNA-Phe (trnF) compared to other Testudinidae mitogenomes, which can be explained by the tandem duplication and random loss (TDRL) model.37^,^38Figure 2. Mitogenome structure and evolutionary features of M. tornieri(A) The circle map illustrates the unique mitogenome structure of M. tornieri. Genes encoded on the H or L strand are shown on the outside or inside of the map, respectively. The GC content (500 bp window size) indicates deviations from the GC content across mitogenome. The GC skew (500 bp window size) is plotted using a red and purple sliding window, representing positive and negative values, respectively.(B) The Ka, Ks, and Ka/Ks values of the 13 PCGs suggest their evolution under purifying selection.(C) The Pi values of the mitogenome (excluding CR2) plotted using a sliding window (500 bp window size with 25 bp step size) reveal higher variability in the CR1 and ND2. The arrows above the line graph represent genes, with their direction indicating gene orientation. The numbers above or below the arrows (color-matched) correspond to the Pi values of the respective genes, with only the longer genes’ Pi values listed.
Mitogenome lengths ranged from 18,136 to 19,634 bp (Table S2), primarily due to variation in repeat sequences in CRs. This was further validated by sequencing depth plots of reads from each sample mapped to MAL100 mitogenome, where some samples exhibited no read coverage in certain CR regions (Figure S2). The lengths of the 13 PCGs were conserved across all samples, with no insertions or deletions detected, ranging from 297 bp (ND4L) to 1,803 bp (ND5). Start and stop codons were also conserved, with initiation codons including ATG, GTG, and CTG and termination codons including TAG, AGG, TAA, and T (Table S3). Consistent with previous findings,19 adenine insertions were identified at position 175 of ND3 and at position 172 of ND4, conserved in all samples and likely tolerated through RNA editing or a specialized mitochondrial translation system.19^,^39 Except for COX2 (no non-synonymous substitutions) and ND4L (no substitutions), all PCGs had Ka/Ks ratios <1 (Figure 2B), indicating purifying selection, possibly related to the high metabolic demands of their rapid mobility. ATP8 exhibited the highest synonymous and nonsynonymous substitutions, consistent with previously reported evolutionary rates in birds and reptiles.40 Sliding window analysis of Pi, excluding poorly aligned CR2, revealed heterogeneous polymorphism, with CR1, ND2, and ATP8 showing markedly elevated Pi values, ATP8 being the most variable (Figure 2C).
Shallow divergence between central and northern Tanzanian populations and low genetic diversity
Phylogenetic analyses using three methods, together with MJ haplotype networks constructed from different genes, consistently revealed a clear yet shallow divergence between northern and central Tanzanian populations without mixing (Figures 3A and 3B), despite topological conflicts in some low-support nodes among the phylogenetic trees (Figures 3A and S3). Within the northern Tanzanian and central Tanzanian populations, individuals from different localities showed slight mixing. Notably, three central Tanzanian individuals (MAL27 from Kinyasi; MAL32 and MAL34 from Kolo) carried more mutations and formed a distinct clade (central Tanzania divergent clade [CTDC]) in all results (Figures 3A, 3B, and S3). Discriminant analysis of principal components (DAPC) further corroborated these findings, indicating independent ancestral components between northern and central populations regardless of K, while some localities within each area were composed of more than one ancestral component at K > 2 (Figure 3C). The CTDC individuals consistently displayed unique ancestral components at K ≥ 3. Sequences of unknown origin clustered with northern Tanzanian individuals in all analyses, indicating a probable origin in this region.Figure 3. Genetic structure and Mantel test of the M. tornieri population(A) Individual ML phylogenetic tree constructed based on mitogenomes without CR2. Haplotypes are listed on the right side of the sample labels. The red-marked CTDC represents three highly divergent samples.(B) MJ haplotype networks based on different mitogenome regions. The haplotypes in the mitogenome without the CR2 network correspond to those in the phylogenetic tree (A). The dashed circle encloses the haplotype group CTDC corresponding to the three highly divergent individuals.(C) DAPC result with K set to 2–8. TP represents Tarangire-POH, TC represents Tarangire-CHE, TM represents Tarangire-MAT, ML represents Mlua, TA represents Tampori, and UN represents unknown.(D) Mantel tests revealed a significant positive correlation between genetic and geographic distances in M. tornieri. Green and blue dots denote sample pairs from northern and central Tanzania, respectively, while purple dots represent interregional pairs.
Pairwise Fst calculations indicated minimal genetic differentiation among most localities, with only a few pairs showing significant divergence (Table S4). Pairwise Fst analysis revealed significant differentiation between several populations (p < 0.01), with the maximum value (0.00794) occurring between Kolo and Jenjeluse. Notably, no significant differentiation was detected between northern and central Tanzanian populations (Fst = 0). Analysis of molecular variance (AMOVA) for the 11 localities indicated that variation within localities (52.98%) was greater than variation among localities (47.02%), whereas AMOVA for northern and central Tanzanian populations indicated a more pronounced difference (57.08% within compared to 42.92% among; Table S5). These results suggest weak genetic structure among localities within the northern and central Tanzanian areas.
Mantel tests on georeferenced samples revealed a significant isolation-by-distance (IBD) pattern (R = 0.298; Figure 3D), which became stronger after excluding outlier CTDC individuals (R = 0.658; Figure S4A). IBD was also significant among northern Tanzanian individuals (R = 0.474; Figure S4B). For central Tanzanian individuals, the test was not initially significant (Figure S4C) but became so after exclusion of CTDC individuals (R = 0.121; Figure S4D).
Genetic diversity in M. tornieri, calculated from the mitogenome without CR2, revealed 28 haplotypes among 61 individuals, with total values of h = 0.887, Pi = 0.00035, and K = 5.80 (Table 1). Among the 11 localities, Jenjeluse exhibited the highest h (0.889), while Kolo showed the highest Pi (0.00052) and K (9.05). Central Tanzania displayed higher diversity (h = 0.952, Pi = 0.00035, and K = 5.80) than northern Tanzania (h = 0.887, Pi = 0.00018, and K = 3.05). However, after excluding the outlier CTDC from central Tanzania, its diversity indices decreased (h = 0.907, Pi = 0.00017, and K = 2.97), becoming comparable to northern Tanzania. Pi calculated from 13 PCGs yielded similar results: northern Tanzania = 0.00022, central Tanzania = 0.00039, central Tanzania without CTDC = 0.00022, and total Pi = 0.00035 (Table S6). These values are relatively low compared with other turtles calculated using the same genes (Figure 4; Table S6).Table 1. Statistics on maternal genetic diversity indices and neutrality tests of M. tornieri based on mitogenome without CR2PopulationNsNhHPiKDF_S_Sino71000––Besi720.286 ± 0.0390.00003 ± 0.000020.571−1.240.856Tarangire-POH441.000 ± 0.0760.00028 ± 0.000074.66667−0.49−0.615Tarangire-CHE221.000 ± 0.5000.00024 ± 0.000124.00000––Tarangire-MAT11000––Vilima Vitatu1140.709 ± 0.0990.00008 ± 0.000011.381820.043−0.053Kinyasi850.857 ± 0.1080.00042 ± 0.000177.25−1.311.62Mlua221.000 ± 0.5000.00006 ± 0.000031.00––Kolo740.714 ± 0.1810.00052 ± 0.000179.05−0.4323.16Jenjeluse1060.889 ± 0.0750.00015 ± 0.000032.53−0.992−1.61Tampori11000––Northern Tanzania32130.887 ± 0.0300.00018 ± 0.000023.04839−1.54∗−3.87∗Central Tanzania28150.923 ± 0.0350.00033 ± 0.000085.76984−1.51∗−3.10Central Tanzania without CTDC25130.907 ± 0.0430.00017 ± 0.000022.96667−1.47−5.60∗Unknown11000––Total61280.952 ± 0.0110.00035 ± 0.000045.80−1.86∗−10.4Note: Ns, number of samples; Nh, number of haplotypes; Pi, nucleotide diversity; h, haplotype diversity; K, average of nucleotide differences; D, Tajima’s D; Fs, Fu’s Fs; ∗p < 0.05.Figure 4. Comparison of genetic diversity among M. tornieri and other chelonians based on 13 mitogenome PCGsFor species with genetic substructures, the Pi of different ESUs is shown. EX, extinct; CR, critically endangered; EN, endangered; VU, vulnerable. Species of the family Testudinidae are highlighted in red.
Contrasting demographic histories between central and northern Tanzanian populations
Except for a few localities with insufficient sample sizes for neutrality tests, all others showed non-significant results (Table 1). In contrast, significantly negative values of Tajima’s D, Fu’s Fs, or both were observed in the northern Tanzania, central Tanzania (with and without CTDC), and the total dataset, suggesting recent demographic expansion. The mismatch distributions for northern Tanzania, central Tanzania, and total datasets were multimodal (Figures 5A, 5B, and 5D), indicating a complex demographic history in M. tornieri, yet all shared a dominant peak at pairwise differences below 10 (Figures 5A–5D), supporting recent expansion. Furthermore, the unimodal distribution of central Tanzania without CTDC confirms that high-difference peaks in the full central dataset were attributable to CTDC individuals (Figures 5B and 5C).Figure 5. The mismatch distribution and Bayesian skyline plots reveal different demographic histories between the Tanzanian central and northern populations(A–D) Mismatch distribution based on the mitogenomes without CR2.(E–H) Bayesian skyline plots based on the mitogenomes without CR2.
The Bayesian skyline plot (BSP) reveals a more detailed demographic history, particularly showing distinct trends between northern and central Tanzania (Figures 5E–5H). The northern population, dating back to ∼58 kya (thousand years ago), shows slow and steady growth (Figure 5E). In contrast, the demographic history of central Tanzania, which includes individuals with additional mutations in CTDC, can be traced back to ∼273 kya (Figure 5F). It initially grew slowly, began rapid expansion from ∼150 kya, and then declined ∼40 kya. Central Tanzania without CTDC traces back to a similar time as northern Tanzania, approximately 55 kya (Figures 5E and 5G), with population trends mirroring those of central Tanzania during the same period (Figures 5F and 5G). For all individuals, the demographic history dates back to ∼273 kya, remaining nearly stable initially, then experiencing rapid growth starting ∼150 kya, followed by a decline starting ∼40 kya, and a renewed increase starting ∼10 kya (Figure 5H).
Deep divergence between Tanzanian and Kenyan populations
The COX1 sequences of 63 M. tornieri (Table S7) were trimmed to 660 bp based on homologous sequences, resulting in three haplotypes: H1 for Tanzania without CTDC, H2 for CTDC, and H3 for Kenya (Figures S5A and S5B). Phylogenetic trees constructed using maximum likelihood (ML) and Bayesian inference (BI) methods revealed a deep split between the Kenyan and Tanzanian populations, which was further supported by the haplotype network (Figure 6C).Figure 6. Phylogenetic tree with divergence times inferred from partial COX1 sequencesEstimated mean ages and 95% highest posterior density (HPD) intervals are shown at the nodes. Four fossil calibration points are indicated by red circles. Geological periods are indicated below the tree, where Pli. denotes the Pliocene and Q. denotes the Quaternary. The labels for M. tornieri from Kenya and Tanzania, along with their divergence time, are highlighted in bold red.
Divergence time estimation based on four fossil calibration points for 56 species/subspecies (Table S8) produced a time framework similar to previous studies,41 wherein M. tornieri from Tanzania and Kenya diverged in the Late Miocene, at 5.74 mya (95% highest posterior density [HPD] 2.41–9.45 mya) (Figure 6). Notably, this divergence predates some species-level differentiations, such as that between Cylindraspis vosmaeri and C. peltastes at 4.12 mya (95% HPD 1.51–7.17 mya) and between Geochelone elegans and G. platynota at 4.7 mya (95% HPD 1.63–8.26 mya).
Discussion
Our findings indicate that the unique mitogenome organization reported previously for M. tornieri, characterized by gene rearrangements and translocation mutations of PCGs, is consistent across all sampled individuals. Despite relatively high h in these mitogenomes, the overall Pi is extremely low, which may be associated with slow mitochondrial mutation rates commonly observed in turtles, as reported in previous studies.40^,^42 However, the Pi remains relatively low compared to other turtles (Figure 4; Table S6), even though the M. tornieri may exhibit a higher mutation rate.19 This may result from either a historical population bottleneck, particularly during the last glacial maximum (LGM), or long-term small effective population size (Ne). Supporting this, the Ne of M. tornieri, particularly in central Tanzania, declined at ∼40 kya, roughly coinciding with the LGM, when cooler and drier climates contracted suitable habitats43 reducing Ne. Although patterns of high h and low Pi, significantly negative neutrality tests, low nucleotide mismatch peaks, and BSP trends indicate a recent slight expansion potentially linked to post-glacial environmental improvement,44 the persistently low genetic diversity (primarily Pi) still suggests potential threats to the long-term survival of the Tanzanian population. This raises concerns about the species’ resilience in the face of ongoing population declines in parts of its distribution range.17
We also found shallow divergence without mixing and distinct demographic histories between northern and central Tanzanian populations, suggesting their poor connectivity and that they warrant status as two ESUs. Furthermore, the substructure within both areas, combined with the IBD patterns observed, further indicates poor connectivity among localities and highlights the important role of geographical distance in shaping population structure. Potential barriers—such as mountains, rift valleys, rivers, roads, and human-modified landscapes—likely continue to impede dispersal and promote population differentiation45^,^46 particularly given the species’ limited vagility.47^,^48 Additionally, three individuals from central Tanzania (CTDC) exhibited significantly more mutations than others, which notably influenced overall Pi value and spatial genetic patterns. They may have resulted from a past population bottleneck that caused the loss of intermediate haplotypes.49^,^50^,^51 Alternatively, they may have been caused by ancient divergence events, with lineages persisting in these regions or originating from migration from unsampled areas, or from historic introgression after hybridization,52^,^53^,^54 implying the existence of highly differentiated ESUs in unsampled areas of Tanzania. Moreover, the region of central Tanzania, where Kolo and Kinyasi are located, is home to a variety of ancient rock paintings by hunter-gatherers who lived in the area for over two millennia.55 If the CTDC in these localities originated via migration or introgression, it could also be linked to translocation practices of these hunter-gatherers (and later, pastoralists).
Interestingly, despite the use of a single gene (COX1), which has a limited resolution among Tanzanian populations, it nonetheless clearly indicates a deep genetic divergence between Tanzanian and Kenyan populations during the Late Miocene (5.74 mya). The deep divergence temporally coincides with major geological and climatic events in East Africa during the Late Miocene, a period characterized by extensive tectonic activity associated with the formation of the East African rift and a shift toward increased aridity.43^,^56 Although no direct tests of climatic or geological causation were performed, the timing of these divergence events suggests that the emergence of large-scale landscape features and associated ecological transitions may have contributed to the isolation of the two ESUs. Similar temporal congruence between diversification and Late Miocene environmental change has been reported in other reptiles.57^,^58^,^59 Importantly, the observed divergence predates some other species-level divergences in Testudinidae, highlighting the evolutionary distinctiveness of the Kenyan and Tanzanian lineages. This raises the question of whether the Tanzanian and Kenyan populations of M. tornieri represent separate species.
While habitat management and control of illegal tortoise trade remain critical to enhance long-term survival of the M. tornieri, there is an urgent need for proactive measures to preserve the existing genetic diversity. Given the observed low genetic diversity within Tanzanian populations, the subtle but present internal population structure, and the deep divergence from Kenyan populations, we recommend integrating our findings to the region-specific conservation strategies. With the insights from our study, we recommend that, at a minimum, three MUs should be considered: the central Tanzanian population, the northern Tanzanian population, and the Kenyan population. These groups should be treated as separate MUs to preserve their evolutionary potential and uniqueness. Since the central Tanzanian population has recently faced a continuous decline in population size and possesses unique mitochondrial clades such as CTDC, this population should be given priority for conservation. In addition, given the presence of IBD patterns, conservation efforts should also consider maintaining habitat connectivity to prevent potential inbreeding depression and local extinction within localities. Furthermore, conservation actions such as protecting the breeding sites and key microhabitats, improving enforcement to protect the species, and limiting habitat loss from quarrying and wildfires are critically important. Additionally, because a large portion of the pancake tortoise population occurs outside protected areas, effective conservation will depend on effective collaboration with local communities and stakeholders to reduce illegal collection and habitat destruction.
Limitations of the study
Despite the insights gained from this study, some limitations should be acknowledged. First, sampling coverage remains incomplete across the species’ distribution range. Based on available M. tornieri occurrence data, most populations in Kenya and several small, discrete populations in Tanzania have not been studied due to sampling limitations. The evolutionary uniqueness and genetic diversity of these populations remain unknown, and they may harbor cryptic ESUs or potentially be related to CTDC, with implications for conservation prioritization. Second, the molecular clock analysis is based on a single mitochondrial gene, COX1. While it alone may be limited by its relatively low information content and potential saturation of nucleotide substitutions, the divergence time estimates presented in our study align with those reported in a recent study using complete mitogenomes,41 giving credibility to our results. Furthermore, the Kenyan population was represented by only two individuals, potentially limiting its representativeness. Finally, the inherent properties of mitochondrial genes confer both strengths and limitations as a molecular marker in population genetics and phylogenetic inference.60^,^61 With the rapid advancement of high-throughput sequencing and declining costs, future studies will likely prioritize whole-genome data to achieve a more comprehensive understanding of species’ genetic backgrounds.62^,^63 Nonetheless, we provide a valuable genetic foundation for M. tornieri, revealing threats from low genetic diversity and complex demography in central and northern Tanzania, offering conservation insights based on the observed genetic structure and poor population connectivity within Tanzanian populations, and revealing deep divergence between Tanzanian and Kenyan populations. We recommend broader geographic sampling covering more regions of Tanzania and Kenya, as well as the use of genomic data, to fully understand the genetic structure, diversity, load, and potential cryptic species within M. tornieri. In addition, comparative evaluations of morphological, behavioral, and other traits, particularly between Tanzanian and Kenyan populations, are needed to determine whether these lineages represent cryptic species.
Resource availability
Lead contact
Requests for further information and resources should be directed to and will be fulfilled by the lead contact, Bo Li ([email protected]).
Materials availability
This study did not generate new or unique reagents.
Data and code availability
- •The mitochondrial genome data of 60 Malacochersus tornieri generated in this study have been uploaded to NCBI under the accession numbers PX394300–PX394359.
- •This paper does not report original code.
- •Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This research was supported by the 10.13039/100017131National Supercomputer Center in Guangzhou and the High-performance Computing Public Platform (Shenzhen Campus) of 10.13039/501100002402Sun Yat-sen University. B.L. was supported by the National Basic Resources Investigation Program (2023FY100405). We are grateful to the College of African Wildlife Management, Mweka, for funding and supporting field data collection logistics. We also thank the Chinese Government Scholarship Council for their essential funding of this research. We acknowledge Godwin Nyerere, Joseph Naibala, and Augustino Mwageni for their assistance with field data collection.
Author contributions
Conceptualization, C.J., N.M., R.T.M., and B.L.; data collection, N.M. and R.T.M.; investigation, C.J., N.M., and B.L.; formal analysis, C.J.; writing – original draft, C.J. and N.M.; writing – review and editing, R.M., X.L., G.M., Y.L., R.T.M., W.Z., and Y.G.; funding acquisition, B.L.; supervision, B.L.
Declaration of interests
The authors declare no competing interests.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
STAR★Methods
Key resources table
REAGENT or RESOURCESOURCEIDENTIFIERDeposited dataThe mitochondrial genome data of 60 Malacochersus tornieri generated in this studyThis paperNCBI under the accession numbers PX394300-PX394359Published M. tornieri mitogenomeParham et al.19GenBank: DQ080042/NC_007700COX1 sequences of I. elongataParham et al.19GenBank: DQ080043/NC_007695COX1 sequences of I. forsteniiParham et al.19GenBank: DQ080044/NC_007696COX1 sequences of Kenyan M. tornieriNCBIGenBank: MK545068COX1 sequences of Kenyan M. tornieriNCBIGenBank: MK545069Software and algorithmsPMAT v2.1.5Han et al.64https://github.com/aiPGAB/PMAT2fastp v0.23.4Chen65https://github.com/OpenGene/fastpGetOrganelle v1.7.5Jin et al.66https://github.com/Kinggerm/GetOrganelleNOVOPlasty v4.3.5Dierckxsens et al.67https://github.com/ndierckx/NOVOPlastyBWA-MEM v0.7.18Jung & Han.68https://github.com/kaist-ina/BWA-MEME/SAMtools v1.21Danecek et al.69https://github.com/samtools/samtoolsMITOS v2.1.9Zea et al.70https://gitlab.com/Bernt/MITOStRNAscan-SE v1.21Lowe & Chan71http://trna.ucsc.edu/tRNAscan-SE/CGViewStothard72http://wishart.biology.ualberta.ca/cgview/MAFFT v7.313Katoh&Standley73https://mafft.cbrc.jp/alignment/software/DnaSP v6.0.7Rozas et al.74http://www.ub.edu/dnasp/IQ-TREE v2.2.2Minh et al.75https://github.com/iqtree/iqtree2MrBayes v3.2Ronquist et al.76http://www.mrbayes.netMEGA v11.0.13Tamura et al.77https://www.megasoftware.net/PartitionFinder v2.0Lanfear et al.78https://github.com/brettc/partitionfinderPOPART v1.7Leigh&Bryant79http://popart.otago.ac.nzArlequin v3.5Excoffier&Lischer80http://cmpg.unibe.ch/software/arlequin35/BEAST v1.8.4Drummond et al.81https://github.com/beast-dev/beast-mcmcLogCombiner v2.6.4Bouckaert et al.82https://github.com/beast-dev/beast-mcmcTracer v1.6.1Rambaut et al.83https://github.com/beast-dev/tracerTreeAnnotator v2.6.4Bouckaert et al.82https://github.com/beast-dev/beast-mcmcR v4.4.3R Core Teamhttps://www.rproject.org/adegenet v2.1.11Jombart et al.84https://github.com/thibautjombart/adegenetvegan v2.7.1Dixon85https://github.com/vegandevs/vegangeosphere v1.5.20Karney86https://github.com/rspatial/geosphere
Experimental model and study participant details
Sample collection
The necessary ethical approvals and research permits were obtained from the Laboratory Animal Management and Ethics Committee of Northeast Forestry University (No. 2024054). In Tanzania, the research was permitted by the Tanzania Wildlife Research Institute (TAWIRI) and was granted approval by the Tanzania Commission for Science and Technology (COSTECH) (Permit No: 2023 -726 -NA -2023 - 750) and Tanzania National Parks (TANAPA).
We collected 60 samples from M. tornieri across 11 localities in northern and central Tanzania. Among these, 59 were skin tissue samples collected between 2010 and 2011, and one blood sample was collected in 2024 at Vilima Vitatu in northern Tanzania (Figure 1 and Table S1).
Method details
DNA extraction and sequencing
High-throughput sequencing was performed on all individual samples. First, total genomic DNA was extracted from skin tissue and blood using an extraction kit (UElandy, Suzhou, China). High-quality DNA was then fragmented by ultrasonication and a sequencing library with an average insert size of approximately 350 bp was constructed using the MGIEasy Universal DNA Library Preparation Kit (BGI) on the DNBSEQ platform, following the manufacturer’s instructions. Paired-end sequencing (150 bp reads) was subsequently carried out on the DNBSEQ-T7 sequencer (MGI, China).
In addition, Pacific Biosciences (PacBio) sequencing was performed on the blood samples. High-molecular-weight DNA was extracted from blood using the SDS method. A SMRTbell library (average insert size = 15 kb) was prepared with the SMRTbell Template Prep Kit 1.0 (Pacific Biosciences), size-selected using Blue Pippin (Sage Science) and polymerase-bound with the Revio Polymerase Kit. After purification (SMRTbell Cleanup Beads), the library was sequenced on a Revio system (Pacific Biosciences).
Mitogenome assembly, annotation and characterization
For the HiFi data, we first assembled the mitogenome of sample MAL100 (Table S2) using PMAT v2.1.5.64 For the PE150 data, we initially performed quality filtering using fastp v0.23.465 retaining only reads with a minimum length of 100 bp while keeping all other parameters at their default settings. We then assembled the mitogenomes from the cleaned reads of each sample (Table S2) using GetOrganelle v1.7.5.66 Additionally, NOVOPlasty v4.3.567 was used for assembly to verify structural accuracy. To further validate the completeness of the assemblies, we aligned each sample's reads to its respective assembled mitogenome using BWA-MEM v0.7.1868 and calculated base depth using Samtools v1.21.69 Due to length variations observed among different samples, we also aligned all individuals’ PE150 reads to the MAL100 HiFi-assembled mitogenome using BWA-MEM v0.7.18 to assess sequencing depth and confirm the presence of structural variations.
The assembled mitogenomes were annotated using MITOS v2.1.970 with tRNA predictions cross-referenced with tRNAscan-SE v1.21.71 The annotation of all mitochondrial genes was manually verified by comparison with the published M. tornieri mitogenome (DQ080042/NC_007700),19 whose geographic origin is unknown.
The circular mitogenome map was generated using CGView.72 We incorporated a published M. tornieri mitogenome (NC_007700) with the 60 newly assembled mitogenomes, yielding 61 in total and aligned them using MAFFT v7.313.73 Because CR2 contains numerous repetitive sequences that complicate alignment, this region was excluded from downstream analyses. Nonsynonymous (Ka) and synonymous (Ks) substitution rates of the 13 protein-coding genes (PCGs) were estimated with DnaSP v6.0.7.74 Nucleotide diversity (Pi) across the mitogenome was assessed by sliding window analysis in DnaSP v6.0.7, using a window size of 500 bp and a step size of 25 bp.
Genetic structure, IBD test and genetic diversity in Tanzania
The mitogenomes of Indotestudo elongata and I. forstenii19 were downloaded from NCBI to use as outgroups for the phylogenetic analysis of the 61 M. tornieri sequences. After alignment using MAFFT v7.313, phylogenetic trees were constructed from the mitogenome without CR2 using maximum likelihood (ML) in IQ-TREE v2.2.2,75 Bayesian inference (BI) in MrBayes v3.2,76 and maximum parsimony (MP) in MEGA v11.0.13.77 The optimal partitioning and nucleotide substitution models for ML and BI (Table S9) were selected with PartitionFinder v2.078 under the corrected Akaike information criterion (AICc). Node support was assessed using posterior probabilities (PP) for BI and 1,000 bootstrap (BP) resampling for ML and MP. Haplotype networks were constructed based on alignment of different genes of 61 M. tornieri via the median-joining (MJ) method in POPART v1.7.79^,^87 Discriminant analysis of principal components (DAPC) was performed using the R package adegenet v2.1.1184 based on alignment of 61 M. tornieri mitogenomes without CR2, testing K-values from 2 to 8. Based on the same alignment, analysis of molecular variance (AMOVA) and pairwise Fst calculations were performed between samples from different localities and areas in Arlequin v3.5.80
Mantel tests were performed using the R package vegan v2.7.185 on 60 georeferenced samples to assess isolation by distance (IBD), where great-circle geographic distances were calculated using the R package geosphere v1.5.20,86 and genetic p-distances based on the alignment of mitogenomes without CR2 were computed using MEGA v11.0.13. Additionally, we performed tests on samples from northern and central Tanzania separately. As three individuals from central Tanzania (central Tanzania divergent clade, CTDC) showed considerable divergence, potentially biasing the results, we repeated the analyses excluding these individuals for both Northern+Central Tanzania and Central Tanzania datasets.
We used DnaSP v6.0.7 to calculate haplotypes, haplotype diversity(h), nucleotide diversity (Pi), and average nucleotide differences (K) for all localities and area samples based on alignment of mitogenomes without CR2. To facilitate comparisons, Pi was also calculated for all samples and for the northern and central Tanzania samples based on the 13 PCGs. Given that CTDC may cause the overestimation of Pi in central Tanzania, we recalculated Pi for this area excluding CTDC. For comparison, we computed Pi of the 13 PCGs for ten other chelonian species using NCBI data, selected for their larger number of individual sequences. For sequences lacking annotation, PCGs were annotated using the same procedure as described above for M. tornieri. To account for potential bias from genetic substructure, we calculated Pi separately for each ESU in species with genetic substructure.
Maternal demographic history in Tanzania
To infer the maternal demographic history of the M. tornieri, we performed neutrality tests (Tajima’s D and Fu’s Fs) using Arlequin 3.5 and estimated the nucleotide mismatch distribution under a constant population size model using DnaSP v6.0.7, based on alignment of the 61 M. tornieri mitogenomes without CR2.
We also conducted Bayesian skyline plot (BSP) analysis using the same alignment. After determining the optimal nucleotide substitution model (Table S9) under the AICc criterion using PartitionFinder v2.0, we performed two independent runs in BEAST 1.8.481 under a strict molecular clock, assuming a mutation rate of 1.005 × 10^-9^ per site per year for the turtle mitogenome.40 Each run consisted of 20,000,000 generations, with sampling every 1,000 generations. The results were combined using LogCombiner v2.6.482 and convergence was assessed in Tracer v1.6.183 by confirming that all parameters had an effective sample size >200, after discarding the first 25% as burn-in. The final BSP plots were generated in Tracer v1.6.1.
BSP analyses were also repeated separately for individuals from Northern and Central Tanzania to investigate the demographic history of M. tornieri in different areas. In addition, to avoid potential bias in Central Tanzania caused by CTDC individuals, we performed an analysis excluding this group.
Differentiation analysis between Tanzania and Kenya populations
We further downloaded COX1 sequences of two Kenyan M. tornieri (MK545068, MK545069) and two outgroups, I. elongata (DQ080043/NC_007695) and I. forstenii (DQ080044/NC_007696) (Table S7), to investigate whether the Kenyan population is genetically distinct from the Tanzanian population. After alignment using MAFFT v7.313, all COX1 sequences were trimmed based on sequence homology to ensure equal length, and ML and BI phylogenetic analysis were conducted using the same methods previously applied to the mitogenome without CR2 within the Tanzanian population. Haplotype networks were constructed based on alignment of COX1 of 63 M. tornieri via the MJ method in POPART v1.7.
Given the considerable genetic divergence inferred between Tanzanian and Kenyan populations of M. tornieri, we further estimated their divergence time. To improve accuracy and provide time references, mitochondrial sequences from 54 turtle species/subspecies were retrieved from NCBI based on the time framework established in a previous study,41 with only one representative individual of M. tornieri from each of the Tanzanian and Kenyan populations being selected (Table S8). Using alignment and model selection methods as previously described, trimmed homologous sequence alignments and optimal substitution models were obtained (Table S9). Divergence time was inferred via BI in BEAST v1.8.4, employing a Yule tree prior and a relaxed molecular clock with a lognormal distribution. Four fossil calibration points validated by previous study41 were applied, with lognormal prior distributions parameterized by offset, mean, and standard deviation (SD): 1) an offset of 50.3 million years ago (Mya) for the divergence between Testudinidae and Geoemydidae (Mean=25.4, SD=0.5); 2) 33.9 Mya for the crown Testudinidae (Mean=16, SD=0.5); 3) 33.9 Mya for the crown Testudininae (Mean=6, SD=0.6); and 4) 11.8 Mya for the divergence between Chelonoidis carbonarius and C. denticulatus (Mean=10.75, SD=0.5). Two independent MCMC runs were performed, each for 200 million generations with sampling every 1,000 generations. Convergence was assessed by confirming that all parameters had ESS greater than 200 and the first 20% of trees were discarded as burn-in using Tracer v1.6.1. The two independent runs were combined using LogCombiner v2.6.4, and the final maximum clade credibility tree was generated with TreeAnnotator v2.6.4.82
Quantification and statistical analysis
In this study, samples sizes from 11 localities across northern and central Tanzania (Figure 1) were as follows: Sino (n = 7), Besi (n = 7), Tarangire-Poachers Hide (n = 4), Tarangire-Chemchengeu (n = 2), Tarangire-Matete (n = 1), Vilima Vitatu (n = 11), Tampori (n = 1), Kinyasi (n = 8), Mlua (n = 2), Kolo (n = 7), Jenjeluse (n = 10). Mitogenomes were assembled using GetOrganelle v1.7.5,66 NOVOPlasty v4.3.5,67 and PMAT v2.1.5,64 with structural validation through read mapping using BWA-MEM v0.7.1868 and Samtools v1.21.69 The assembled mitogenomes were annotated using MITOS v2.1.970 with tRNA predictions cross-referenced with tRNAscan-SE v1.2171 (Figure 2). Pi, h, K, and Ka/Ks ratios were calculated in DnaSP v6.0.7.74 Phylogenetic trees were constructed using maximum likelihood in IQ-TREE v2.2.275(1,000 bootstraps for node support), Bayesian inference in MrBayes v3.276 (posterior probabilities for node support) (Figure 3), with optimal partitioning and substitution models selected by PartitionFinder v2.0.78 Phylogenetic tree was also constructed using maximum parsimony method in MEGA v11.0.1377 (Figure S3). Haplotype networks were generated using the median-joining method in POPART v1.7.79 DAPC was conducted in adegenet v2.1.184 (testing K = 2–8). AMOVA, pairwise Fst, and neutrality tests (Tajima’s D and Fu’s Fs) were performed in Arlequin v3.580 (significance tested with 10,000 permutations, p < 0.01 for Fst) (Table 1). IBD was assessed via Mantel tests in vegan v2.7.185 (significance based on 999 permutations). Demographic history was inferred using mismatch distributions in DnaSP v6.0.774 and BSP in BEAST v1.8.481 under a strict molecular clock (Figure 5). Divergence times between Tanzanian and Kenyan populations were estimated in BEAST v1.8.481 using a relaxed clock with lognormal distribution, Yule prior, and four fossil calibrations (Figure 6). The convergence of all MCMC used in the analyses was verified with Tracer v1.6.1,83 ensuring ESS of parameters > 200. All statistical analyses incorporated multiple independent runs where applicable, and significance levels were set at p < 0.05 (indicated by ∗) unless otherwise specified.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Barnosky A.D.Matzke N.Tomiya S.Wogan G.O.U.Swartz B.Quental T.B.Marshall C.Mc Guire J.L.Lindsey E.L.Maguire K.C.Has the Earth’s sixth mass extinction already arrived?Nature 4712011515710.1038/nature 0967821368823 · doi ↗ · pubmed ↗
- 2Tilman D.Clark M.Williams D.R.Kimmel K.Polasky S.Packer C.Future threats to biodiversity and pathways to their prevention Nature 5462017738110.1038/nature 2290028569796 · doi ↗ · pubmed ↗
- 3Cowie R.H.Bouchet P.Fontaine B.The Sixth Mass Extinction: fact, fiction or speculation?Biol. Rev.97202264066310.1111/brv.1281635014169 PMC 9786292 · doi ↗ · pubmed ↗
- 4De Vos J.M.Joppa L.N.Gittleman J.L.Stephens P.R.Pimm S.L.Estimating the normal background rate of species extinction Conserv. Biol.29201545246210.1111/cobi.1238025159086 · doi ↗ · pubmed ↗
- 5Early R.Bradley B.A.Dukes J.S.Lawler J.J.Olden J.D.Blumenthal D.M.Gonzalez P.Grosholz E.D.Ibañez I.Miller L.P.Global threats from invasive alien species in the twenty-first century and national response capacities Nat. Commun.720161248510.1038/ncomms 12485 PMC 499697027549569 · doi ↗ · pubmed ↗
- 6Maxwell S.L.Fuller R.A.Brooks T.M.Watson J.E.M.Biodiversity: The ravages of guns, nets and bulldozers Nature 536201614314510.1038/536143 a 27510207 · doi ↗ · pubmed ↗
- 7Teixeira J.C.Huber C.D.The inflated significance of neutral genetic diversity in conservation genetics Proc. Natl. Acad. Sci. USA 1182021 e 201509611810.1073/pnas.2015096118 PMC 795843733608481 · doi ↗ · pubmed ↗
- 8Shaw R.E.Farquharson K.A.Bruford M.W.Coates D.J.Elliott C.P.Mergeay J.Ottewell K.M.Segelbacher G.Hoban S.Hvilsom C.Global meta-analysis shows action is needed to halt genetic diversity loss Nature 638202570471010.1038/s 41586-024-08458-x 39880948 PMC 11839457 · doi ↗ · pubmed ↗