Uncovering genetic variation in humoral inborn errors of immunity in African populations: insights from the African genome variation database
Luyanda Hlongwa, Ayton Meintjes, Nicola Mulder, Elizabeth Mayne

TL;DR
This study explores genetic variations linked to immune disorders in African populations using a genome database, revealing new insights into antibody-related immune conditions.
Contribution
The study identifies population-specific genetic variants associated with humoral IEIs in African populations, many of which are not previously documented in ClinVar.
Findings
815 variants were identified in 23 genes, with 219 unique to African populations.
Four pathogenic variants were found in TCF3 and RAG1/2 genes.
144 variants (43%) were not listed in ClinVar, with 53 predicted to be deleterious.
Abstract
Inborn Errors of Immunity (IEIs) are rare genetic disorders affecting immune function. Humoral IEIs, the most commonly diagnosed subtype, are characterised by antibody deficiencies. There is limited data regarding the variation of genes associated with humoral IEI in Africa with implications for designing genetic assays for testing for these diseases. This study aimed to assess the genetic variation in genes which are commonly associated with humoral IEI in African populations. Using the African Genome Variation Database (AGVD) which contain genotype frequencies from African populations. We analysed genotype frequency data from the African Genome Variation Database (AGVD), covering diverse African populations, to identify variants in 23 genes associated with humoral IEIs. Variants were annotated using Ensembl’s Variant Effect Predictor and classified for clinical significance with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
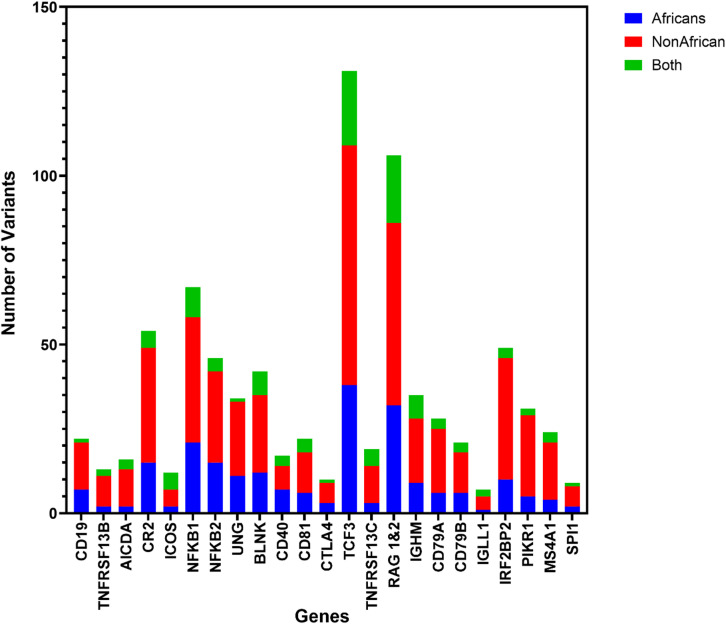 Figure 1
Figure 1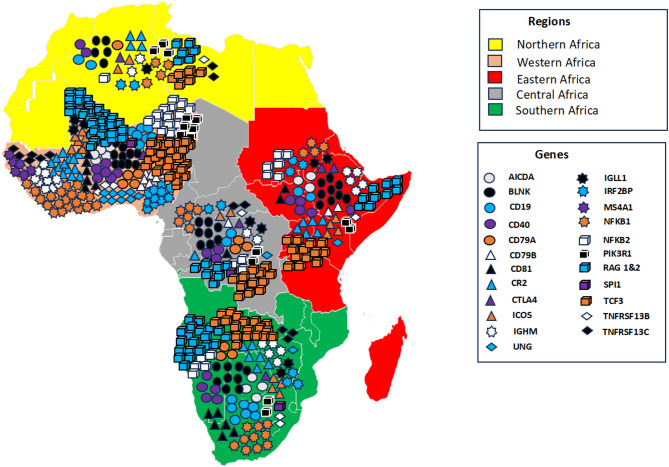 Figure 2
Figure 2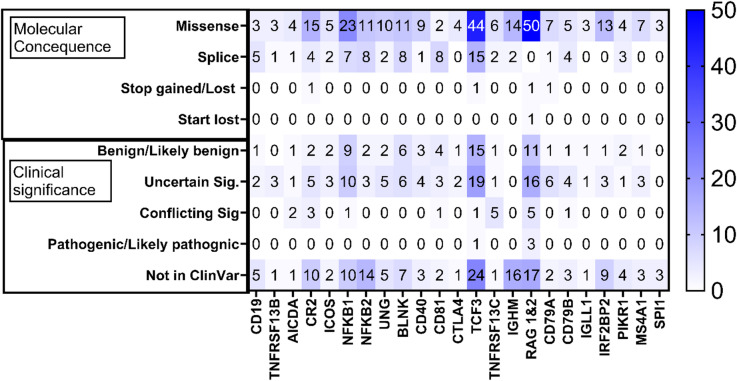 Figure 3
Figure 3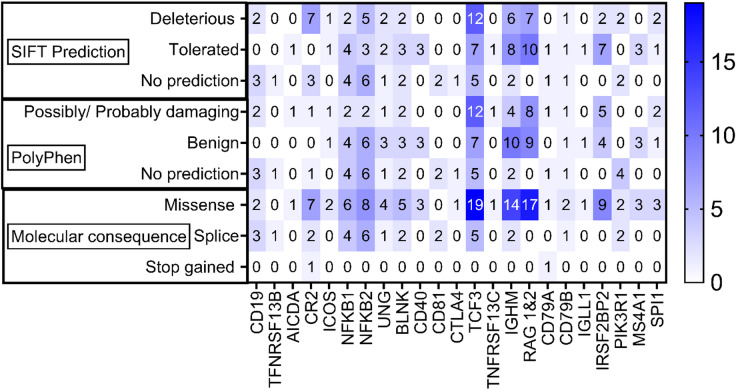 Figure 4
Figure 4- —National Institutes of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmunodeficiency and Autoimmune Disorders · Genomics and Rare Diseases · Blood disorders and treatments
Introduction
Inborn Errors of Immunity (IEIs), also known as primary immunodeficiencies, are disorders caused by genetic defects affecting either the innate or adaptive immune function or both^1,2^. Clinically, IEIs present mainly as recurrent or severe infections, immune dysregulation (autoimmune or autoinflammatory disorders), and lymphoproliferation with or without dysmorphic features^3^. IEIs are often misdiagnosed or diagnosed only after the development of serious or life-threatening complications^4^. The prevalence of IEIs is still poorly understood for several reasons, including under-diagnosis, under-reporting, and lack of physician awareness. Although IEIs are considered rare^5^, systematic reviews of IEIs patient registries suggest that IEIs may be present in as many as 1:5000 to 1:1000 live births^6^. In Africa, one million individuals are estimated to be suffering from IEIs, with nearly 42 000 of these cases in South Africa^7,8^. The commonest disorders reported in South Africa are humoral IEIs, characterised by antibody deficiencies^6^. In low- and middle-income countries (LMIC) such as South Africa, there is a lack of diagnostic capacity for IEIs diagnosis, which is further compounded by the high infectious disease burden^9^. Diagnostic testing in South Africa, including sequencing for causative mutations, is not widely available especially in the State sector.
There are 485 individual IEIs as of 2022 which are primarily identified by the underlying genetic abnormality^10^. The International Union of Immunological Societies (IUIS) classify IEIs according to the different components of the immune system involved. Humoral IEIs, also called primary antibody deficiencies, are a sub-group characterised by impaired antibody production^11^ and represent nearly 70% of all diagnosed IEIs with a global prevalence of 1:100 000–1:8 500^6^ although in certain populations and for certain disorders the prevalence may be higher^12,13^. Patients with IEIs may present with recurrent infections, autoimmunity, autoinflammation and lymphoid malignancies which may complicate the diagnosis^14^ by masking infection. The severity of humoral IEIs varies which may present with isolated immunoglobulin subclass deficiencies or with absent or virtually absent B-cell responses. Patients with severe humoral IEIs require life-long immunoglobulin replacement therapy and in some cases prophylactic antibiotic treatment^14^.
Table 1. Humoral IEI with commonly affected genes^10^. ConditionGeneFunctionOMIM numberAgammaglobulinemiaIGHMConstant region of immunoglobulin mu-chain147020IGLL1One of the surrogate chains forming the pre-B cell receptor, important for the development of B cells from pro-B cells to pre-B cells.146770BLNKCytoplasmic linker important in B-cell development604515BTKPlays a crucial role in B cell development300300CD79AIg-alpha, part of the B cell receptor. Important for B cell development112205CD79BIg-beta, part of the B cell receptor. Important for B cell development.147245PAX5B cell-specific activator protein, a transcription factor binding to promoters of the CD19 gene167414PIK3CDClass 1 PIK3 protein, which binds to p85 proteins and GPT-bound RAS602839PIK3R185 kDa subunit of the phosphatidylinositol 3-kinase enzyme171833SLC39A7Responsible for transporting Zinc from the Golgi and reticulum to the cytoplasm. Important for the activation of tyrosine kinases.601416SPI1ETS domain transcription factor which activates gene expression during myeloid and B cell development.165170TCF3Two basic helix-loop-helix transcription factors (E12 & E47) involved in regulation of immunoglobulin gene expression147141TOP2BDNA topoisomerase II, an enzyme that relieves topological stress during DNA replication.126431LRRC8AFacilitates the transport of cyclic GMP-AMP across membranes, binds identical proteins, and functions as a volume-regulated ion channel.608360FNIP1The protein is involved in the regulation of cellular metabolism and nutrient sensing by influencing AMPK and mTOR signalling pathways.Common variable immunodeficiency (CVID)ARHGEF1Member of the guanine nucleotide exchange factor family, specific for GTPase RhoA601855ATP6AP1Subunit of the vacuolar (V)-ATPase protein pump300197CD19Member of the immunoglobulin superfamily, earliest marker of B cells.107265CD20B cell surface marker that plays a role in the differentiation of B cells into plasma cells.112210CD21 (CR2)Receptor of complement component C3d, EBV binds to this receptor during infection.120650CD81Member of the transmembrane 4 superfamily, which mediates signal transduction events that regulate cell development, activation, growth and motility.186845IKZF1Transcription factor of the zinc finger family, DNA-binding proteins associated with chromatin remodelling603023IRF2BP2Protein that interacts with the C-terminal of the transcriptional repression domain of IRF2615332KARS1Lysyl-trna synthase, which catalyses the aminoacylation of trna-lys in both the cytoplasm and mitochondria601421MOGSFirst enzyme of the N-linked oligosaccharide processing pathway.601336NFkB1Precursor of the DNA-binding subunit of the transcription regulator NFkB164011NFkB2Subunit of the transcription complex, NFkB, can be an activator or a repressor, depending on the dimerisation partner615577PIK3CDClass 1 PIK3 protein, binds to p85 proteins and GPT-bound RAS602839PIK3R1Codes for the 85 kDa subunit of the phosphatidylinositol 3-kinase enzyme171833POU2AF1Involved in positive regulation of transcription by RNA polymerase II601206PTENPhosphatidylinositol-3,4,5-triphosphate-3-phosphatase, dephosphorylates phosphoinositide substrates.601728RAC2GTPase belonging to the RHO family induce the interferon gamma promoter.TNFRSF13BLymphocyte-specific member of the tumour necrosis factor receptor family, induces activation of transcription factors NFAT, AP1, NFkB604907TNFRSF13CB cell activating factor receptor involved in B cell survival and development606269TNFSF12Cytokine belonging to the TNF ligand family602695TRNT1CCA-adding enzyme belonging to the tRNA nucleotidyl transferase/ Poly(A) polymerase family612907CTLA4Part of the immunoglobulin superfamily whose function is transmitting inhibitory signals to T cells.123890ICOSFrom homodimers and functions in cell to cell signalling, immune responses, and regulation of cell proliferation.604558IL21Plays a role in the immune system by inducing differentiation, proliferation, and activity of multiple immune cell targets.605384IL21RReceptor to IL21 cytokine, transduces growth promoting signals and functions in the proliferation and differentiation of T, B and NK cells.605383LRBAInvolved in the formation intracellular vesicles to activated receptor complexes.606453MS4A1Functions in the development and differentiation of B cells into plasma cells.112210PRKCDInvolved in B cell-mediated signalling.176977PLCG2Plays a role in transmitting signals from growth factor receptors and immune system receptors across the cell membrane.600220SEC61A1Essential for insertion of secretory and membrane proteins into the endoplasmic reticulum.609213VAV1Important in haematopoiesis, playing a role in the development and activation of T and B cells.164875BLKPlays a role in B cell receptor signalling and the development of B cells.191305ZBTB20Functions as a transcriptional repressor and contributes to processes such as neurogenesis, regulation of glucose levels and postnatal growth.606025Hyper IgM syndromeAICDARNA editing deaminase involved in somatic hypermutation, gene conversion and class-recombination.605257CTNNBL1Component of the pre-mRNA processing factor 19 - cell division cycle 5-like protein complex, activates pre-mRNA splicing. Binds to AICDA.611537INO80Catalytic ATPase subunit of the INO80 chromatin remodelling complex.610169MSH6heterodimerize with MSH2 form a complex that exchanges ADP and ATP as DNA mismatches are bound, and dissociate.600678TNFSF13APRIL, a proliferation-inducing ligand of the TNF ligand superfamily604472UNGOne of the several uracil-DNA glycosylases, prevents mutagenesis by eliminating uracil from DNA molecules191525CD40LGProtein encoded by this gene is expressed on T cells and regulates B cell function by engaging CD40 on the surface of the B cell.300386CD40Important in a number of immune responses including T cell-dependent class-switching, memory B cell development and germinal centre formation.109535IKBKGRegulatory subunit of the IKK complex regulating the activity of nuclear factor kappa B.300248Isotype, light chain, or functional deficienciesCARD11Protein that acts as a scaffold for NFkB, controlling peripheral B-cell differentiation607210IGKCConstant region of the kappa light chain of immunoglobulins147200
Most IEIs are associated with well-characterised genetic mutations (Table 1). Humoral IEIs are linked to abnormal B-cell development which may affect B-cell maturation and B-cell receptor rearrangement. Common variable immunodeficiency (CVID), characterised by deficiencies in antibody isotypes, typically results from gene mutations affecting downstream signalling following B-cell-T-cell interactions. X-linked agammaglobulinemia, the most severe form of humoral IEI, is most commonly caused by mutations in a tyrosine kinase (BTK), which allows signalling through the B-cell receptor following heavy chain rearrangement^15^. Genetic testing is increasingly used to diagnose humoral IEI, although there is significant heterogeneity in underlying abnormalities and the genes which are involved, which may complicate the interpretation of these studies^16^.
In response to the lack of reporting on IEI in Africa, many countries, including South Africa, have established IEI registries. The South African Primary Immunodeficiency Registry (SAPIDR) contains data from 446 individual patients. African patients comprise 17% of the patients in the registry, but 81.4% of the population^17^. Data on gene mutations causing IEIs in South African and other African populations are lacking^17^. It is likely, given genetic diversity in Africa, that disease-causing variants may differ in the African population from populations elsewhere^8^.
In preparation for the development of targeted gene panels to diagnose IEIs in African populations, we aimed to investigate the variation in genes associated with humoral IEI in a database containing genotype frequencies from African populations.
Methods
Ethics
Ethics approval for this project were granted by the Human Research Ethics Committee at the University of Cape Town (Ethics number: 538/2022). Additionally, AGVD has its own ethics approval for analysis of variants from existing aggregate data in target genes (R037/2020). AGVD data were collected from various H3 Africa projects, participants provided informed consent and agreed to data sharing and future use. The University of Cape Town Human Research Ethics Committee provides this ethics. This study was conducted in accordance with the guidelines, including those of the University of Cape Town Human Research Ethics Committee and the Declaration of Helsinki.
A retrospective analysis of data obtained from the African Genome Variation Database (AGVD) (https://nyame.h3abionet.org/accounts/login/?next=/refresh ) of the H3ABioNet Consortium was performed. The AGVD is currently in its beta phase and contains genotype frequency data from individuals representing diverse African populations, compiled from various research projects under the H3Africa initiative^18^. The database provide genotype frequencies for African populations in the following African regions: Central Africa, Eastern Africa, Southern Africa and Western Africa (Table 2)^19^. The data base was made from 1658 samples from individuals of African ancestry and 2552 non-African individuals. The read depth of projects included in the database ranged between 25X to 65X.
Table 2. African regions.RegionCountriesCentral AfricaRepublic of Burundi, Republic of Cameroon, Central African Republic, Republic of Chad, Republic of the Congo, Democratic Republic of Congo, Republic of Equatorial Guinea, Gabonese Republic, Democratic Republic of São Tomé and PríncipeEastern AfricaUnion of the Comoros, Republic of Djibouti, State of Eritrea, Federal Democratic Republic of Ethiopia, Republic of Kenya, Republic of Madagascar, Republic of Mauritius, Republic of Rwanda, Republic of Seychelles, Federal Republic of Somalia, Republic of South Sudan, Republic of the Sudan, United Republic of Tanzania, Republic of UgandaNorthern AfricaPeople’s Democratic Republic of Algeria, Arab Republic of Egypt, Libya, Islamic Republic of Mauritania, Kingdom of Morocco, Sahrawi Arab Democratic Republic, Republic of TunisiaSouthern AfricaRepublic of Angola, Republic of Botswana, Kingdom of Eswatini, Kingdom of Lesotho, Republic of Malawi, Republic of Mozambique, Republic of Namibia, Republic of South Africa, Republic of Zambia, Republic of ZimbabweWestern AfricaRepublic of Benin, Burkina Faso, Republic of Cabo Verde, Republic of Côte d’Ivoire, Republic of the Gambia, Republic of Ghana, Republic of Guinea, Republic of Guinea-Bissau, Republic of Liberia, Republic of Mali, Republic of Niger, Federal Republic of Nigeria, Republic of Senegal, Republic of Sierra Leone, Togolese Republic
Search for variants in humoral IEI-associated genes
A systematic search was conducted in the AGVD to identify variants in genes commonly associated with humoral IEIs^10^. Each target gene was manually queried using the AGVD search function to retrieve all recorded variants. The resulting variant data were exported to Microsoft Excel for further processing. Variants were annotated using Ensembl VEP (version 112)^20^ by querying rsIDs against the GRCh38 human reference genome. Annotation was performed using MANE Select transcripts where available; if not available, canonical transcripts were used. For downstream analysis, only variants classified as missense, stop-gained, stop-lost, start-lost, or splice-site mutations were retained. Identified variants were categorised based on the population in which they were observed (African or non-African). Variants identified in African populations were further evaluated for clinical significance using the ClinVar database.
Variant significance
ClinVar (National Institutes of Health, United States of America, http://www.ncbi.nlm.nih.gov/clinvar/) provides a freely and publicly available archive of many medically significant gene variants. ClinVar clinical significance is recorded as part of the details provided by the VEP tool for each variant where available. Variants identified in the AGVD were classified according to their clinical significance as recorded in ClinVar. ClinVar classifies variants as pathogenic, likely pathogenic, benign, likely benign and of uncertain significance. The terms are described in detail in the guidelines published by the American College of Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP)^21,22^. Briefly, variants are classified as pathogenic if there is strong evidence to support their association with disease, as likely pathogenic if there is a high probability of them being disease-causing but with lower levels of evidence, as benign if they are not associated with disease and as likely benign if they are unlikely to cause disease. Variants are classified as variants of uncertain significance (VUS) if there is insufficient evidence to classify them as either benign or pathogenic. Using the VEP tool, variants can be classified as deleterious (low/high confidence) or tolerated (low/high confidence) using Sorting Intolerant From Tolerant (SIFT) score (version 6.2.1.)^23^. A SIFT score of 0.0 to 0.05 are predicted to be deleterious with the lowest scores (closest to 0) predicting deleterious effects with high confidence. Variants with a score between 0.05 and 1.0 are predicted to be tolerated with the highest scores (closest to 1.0) predicting tolerated effects with high confidence. Variants can also be classified as possibly damaging, probably damaging or benign using PolyPhen-2^24^.
Results
A systematic search on the AGVD was conducted for variants in genes known to be associated with humoral IEIs. A total of 815 gene variants were identified in 23 genes associated with humoral IEI of which 335 were identified in African populations; 219 gene variants were identified in African populations only, 480 gene variants in Non-African populations only and 116 gene variants identified in both populations (Fig. 1, Supplementary Table S1). The geographic distribution of gene variants across African populations was analysed, revealing distinct regional patterns. Genetic variants were predominantly clustered in Western and Southern Africa, while Northern Africa exhibited comparatively lower genetic diversity (Fig. 2).
Fig. 1. Variant distribution across African and non-African populations. Variants that were identified were grouped according to the population in which they were identified. African populations – 219, non-African populations – 480, identified in both populations – 116.
Fig. 2. Geographic distribution of gene variants across African regions. Each region is color-coded according to its geographic classification (Northern, Western, Eastern, Central, and Southern Africa). Distinct shapes and colours represent specific genes, as indicated in the legend. The number of symbols displayed within each region corresponds to the number of identified gene variants for that gene in that region.
Fig. 3. Clinical significance and Molecular consequence. Variants were grouped according to their clinical significance and molecular consequence. There were 4 categories under Molecular consequence. Missense – 256, splice – 74, Stop gain/loss – 4, Start loss – 1. There were 5 categories under clinical significance. Benign/Likely benign – 67, Uncertain significance – 101, Conflicting significance – 19, Pathogenic/likely pathogenic – 4 and Not in ClinVar (for variants not recoded in ClinVar) – 144.
Molecular consequence and clinical significance
The 335 variants that were identified in the African population, including variants identified in both African and non-African populations were classified according to the predicted molecular consequence. The majority of the variants identified were classified as missense mutations, followed by splice mutations. Only 3 variants (in the CR2, TCF3 and RAG genes) were identified as stop-gain or -loss, while 1 variant (in the RAG gene) was identified as start-lost. Identified variants were classified according to their clinical significance as recorded in ClinVar. Sixty-six (65) variants were classified as benign or likely benign, 95 variants as uncertain, 19 variants as conflicting significance and 4 variants as pathogenic or likely pathogenic (details of pathogenic or likely pathogenic variants supplementary Table S2). Of the variants classified as pathogenic or likely pathogenic, 1 was from the TCF3 gene (frequency of 0.00396 in West African population) and 3 from the RAG 1 & 2 genes (frequency of 0.000495 in West African population) (Fig. 3).
Variants not in ClinVar
A total of 144 variants from African populations were not recorded in ClinVar (43%). To assess the potential impact of these variants, SIFT and PolyPhen-2 predictions were considered. Of the 144 variants, 53 (36.8%) were predicted to be deleterious, 57 (39.6%) were predicted to be tolerated and 34 (23.6%) were without SIFT predictions. The variants were also grouped according to their PolyPhen-2 predictions as possibly/probably damaging, benign, or no prediction. Of the total Variants, 46 variants were predicted to be possibly/probably damaging, 56 were predicted to be benign, and 34 variants had no PolyPhen-2 predictions. 111 variants were identified as missense mutations, 31 as splice mutations, and 2 as stop-gained mutations. The stop-gained mutations were found in the CR2 and CD79A genes (Fig. 4) with a genotype frequency of 0.000495 in the West African population. The genotype frequency of variants not in ClinVar and predicted to be deleterious by either SIFT or PolyPhen-2 varied in the different African Populations. Genotype frequencies for these variants ranged between 0.076692 and 0.6692 in Central African populations, 0.004237 to 0.5424 in Eastern African populations, 0.0125 to 0.45 in Northern African populations, 0.001866 to 0.4086 in Southern African populations and 0.000495 to 0.5376 in Western African populations. While most of the variants were low frequency variants, several variants had notably high genotype frequencies in different African populations. These include variants in CR2, PIKR3, RAG 1 and 2, and IGHM (Table 3).
Fig. 4SIFT and PolyPhen-2 predictions and Molecular consequence of variants not recorded in ClinVar. Clinical significance for variants not recorded in ClinVar could not be determined. SIFT predictions, PolyPhen predictions and Molecular consequence were used to assess potential impact of these variants.
Table 3. Genotype frequencies of variants not in clinvar predicted to be deleterious.Variant IDrsIDGeneCentral AfricaEastern AfricaNorthern AfricaSouthern AfricaWestern Africa12_8606914_C_Trs759799595AICDA00000.00049510_96216721_A_Trs587723641BLNK00000.00049510_96209853_G_Ars781845656BLNK00000.00049510_96227501_C_Trs17111469BLNK0.076920.033900.031720.0148516_28937781_C_Trs200792237CD1900.00423700016_28937025_C_Trs113797527CD190.007692000.003731019_41879058_G_Ars557160710CD79A000.01250017_63930335_G_Ars375597674CD79B00000.00099011_207468893_A_Trs368883636CR200000.0004951_207454465_C_Grs375649712CR20000.00186601_207469989_A_Grs551427271CR200000.00099011_207468703_C_Grs769415969CR20000.00186601_207472823_G_Trs144075435CR20.015380.00847500.0018660.025251_207480019_A_Grs17618CR20.20770.029660.06250.13620.077231_207473553_T_Crs4308977CR20.66920.54240.450.40860.53762_203955686_G_Crs539709751ICOS00000.0004952_203936854_C_Trs77411896ICOS00.00847500.0018660.0306914_105854656_G_Ars372061764IGHM00000.00352114_105854644_G_Ars530414987IGHM00.00434800014_105854360_C_Grs549809721IGHM00000.000505614_105854618_C_Trs782070553IGHM0000.00188014_105855558_C_Trs1059216IGHM0.42240.40620.26250.37970.406614_105856143_C_Trs372261472IGHM00000.0030331_234607828_T_Crs1012466215IRF2BP200.0042370001_234608450_C_Trs1052817071IRF2BP200000.0004951_234609136_G_Ars1195336113IRF2BP200000.00050451_234608663_C_Grs1360414372IRF2BP200000.0004951_234608614_C_Grs755757862IRF2BP200000.0004954_102610685_G_Trs1578834273NFKB10000.00559704_102613462_T_Crs200451929NFKB100000.00049510_102403342_G_Ars572223815NFKB200.00847500010_102405018_T_Crs200053412NFKB200.00423700010_102405248_G_Ars759227476NFKB200000.00049510_102405451_T_Grs2135450097NFKB200000.00049510_102405463_C_Trs752053696NFKB200000.0004955_68295452_A_Grs1218229603PIK3R100000.0004955_68293360_C_Trs3730090PIK3R10.061540.18640.08750.12690.132211_36573364_C_Trs138801620RAG1, RAG20000.013060.00049511_36573401_T_Ars572993073RAG1, RAG200.00423700011_36574837_G_Trs533793542RAG1, RAG200000.00049511_36575125_C_Trs183806098RAG1, RAG20000.001866011_36575763_A_Grs2227973RAG1, RAG20.046150.038140.20.10820.0920811_36593297_G_Ars112927992RAG2, RAG10000.003731011_36594157_C_Grs1851109380RAG2, RAG10000.001866011_36593015_C_Trs1157290414RAG2, RAG100000.00049511_36593979_T_Crs535374501RAG2, RAG100000.00049511_47359926_C_Trs1370001180SPI100000.00049511_47359927_G_Crs537934199SPI10.007692000019_1646390_G_Ars1340859555TCF300000.00049519_1615692_C_Trs141432924TCF300000.0029719_1611790_C_Grs146047446TCF300000.00049519_1632351_G_Crs1487181959TCF300000.00049519_1622220_C_Ars2146137895TCF300000.00049519_1621889_C_Trs374898725TCF300000.00049519_1621865_G_Ars531068508TCF300000.00049519_1619348_G_Trs532539524TCF300000.00049519_1619800_C_Trs535113552TCF300000.00049519_1611813_C_Trs538013937TCF300000.00049519_1619351_C_Trs1052692TCF30.092310.06780.08750.05410.050519_1619793_C_Trs117006898TCF30000.001866019_1620976_C_Trs148928381TCF30.0076920000.0019819_1615376_T_Crs149166972TCF300000.00792122_41925453_G_Ars1303856630TNFRSF13C00000.000495
Discussion
Genetic diagnosis is increasing in importance in IEIs although in certain populations, genetic information regarding genes associated with disease is incomplete. IEIs are predicted to be higher in prevalence than the numbers included within the registries^17^. This project examined the variation of genes associated with humoral IEIs within an African population through an analysis of the AGVD, a database housing genotype frequency information from countries across Africa collected by projects of the H3Africa consortium. Africa has high genetic diversity with a corresponding paucity of clinical genetics facilities^9,25^ and for this reason, there is a lack of genetic information from African populations regarding humoral IEIs and IEIs in general^17^. We found 815 gene variants within regions of interest for humoral IEI of which 335 were present in individuals of African descent and 219 were only found within African individuals.
Gene function
The gene variants identified are associated with all aspects of B cell function including B cell development and signalling (CD19, CD79A, CD79B, MS4A1, PIK3R1, IGHM, IGLL1, TCF3 and SPI1), class switch and somatic hypermutation (AICDA and UNG). B-cell co-stimulation and survival (CD40, ICOS, CTLA4, TNFRSF13B, TNFRSF13C, CD81 and CR2) and immune regulation and transcription factors (NFKB1, NFKB2 and IRF2BP2). We also found variants in the RAG1 and 2 genes that could affect V(D) J recombination (RAG1 and RAG2) and hence potentially impact both T-cell and B-cell receptor rearrangement. A comprehensive analysis of genes impacting T-cell development was outside the scope of this study but represents an important consideration when developing diagnostic strategies for IEIs.
Pathogenic gene variants
The AGVD provides access to genotype frequency information mainly from participants used as controls in different studies and therefore defined as ‘healthy’. Several of the variants described here were classified as benign although some variants were classified as either of uncertain significance or conflicting significance. Unexpectedly, four variants were detected that were classified as pathogenic or likely pathogenic. Several factors may have influenced the clinical presentation of these variants in ostensibly healthy individuals, including the inheritance pattern of the gene (dominant or recessive), penetrance, mosaicism and homozygosity for the deleterious variant. Environmental factors may also ameliorate clinical presentation of inborn errors of immunity^26^. Considering the variable presentation of humoral IEIs^27^, it is possible that the H3Africa criteria for healthy individuals may have included some affected individuals. In one of the variants in the TCF3 gene, a stop-gain mutation was identified, which was predicted to lead to loss-of-function (LOF), an established cause of agammaglobulinemia. Biallelic LOF and monoallelic dominant-negative LOF result in disease, but there are cases of monoallelic loss-of-function resulting in milder forms of immunodeficiency with increased susceptibility to infections^28–30^. RAG 1 and RAG 2 proteins mediate the V(D)J recombination process in both T and B cells^31^ and LOF is associated with severe combined immunodeficiency (SCID) with T-cell and B-cell dysfunction^10^. Three pathogenic or likely pathogenic gene variants were identified in this gene.
Variants not in ClinVar
Several variants were identified that were not recorded in ClinVar. To assess their potential impact, SIFT and PolyPhen predictions were considered alongside their molecular consequences. Among the variants absent from ClinVar, two stop-gain mutations were identified in the CR2 and CD79A genes, both variants were predicted to be deleterious and possibly damaging by SIFT and PolyPhen. CR2 encodes a key receptor involved in immune function, particularly in Epstein–Barr virus interactions, and its deficiency has been associated with CVID, which is characterised by decreased serum IgG levels accompanied by a reduction in either IgA or IgM^32^.Ideally, the pathogenicity of this variant should be assessed in functional validation. CR2 mutations in CVID exhibit an autosomal recessive inheritance pattern, suggesting that pathogenic variants may be present in healthy individuals. CD79A forms part of the B cell antigen receptor; mutations in this gene have been linked to autosomal recessive agammaglobulinemia, which have been shown to be more severe than the X-linked form.
High frequency variants
The frequencies of variants varied both intra and inter-regionally. The majority of the variants found were rare (genotype frequency less than 0.001) however, several variants predicted to be deleterious by SIFT or PolyPhen-2 were more common (genotype frequencies up to 0.7). Variant effect predicting tools such as SIFT and PolyPhen-2 predict whether the variant will impact protein function or not, but may not necessarily have a negative impact. Variants with higher frequencies in a population (often above 5%) are often either beneficial (adaptive) or neutral^33^. We observed a variant from the CR2 gene (not recorded in ClinVar) which was predicted to have a deleterious effect by SIFT and PolyPhen-2. This specific variant had higher genotype frequencies in several African populations, 0.67 in Central African populations and 0.54 in Western and Eastern African populations. CR2 or CD21 complexes with CD19, CD81 and CD225 to form the B cell coreceptor complex, lowering the B cell receptor’s activation threshold^34^. Epstein-Barr Virus (EBV) binds CD21 during B-cell infection because the EBV envelope protein is homologous to C3d^35,36^. It is therefore possible that mutations in this receptor may affect susceptibility to EBV in affected individuals. Africa is one of the regions with significantly high incidences of Burkitt’s lymphoma and is almost always linked to EBV infection^37^. The potential impact of other high-frequency variants predicted to be deleterious remains to be determined, and it would be important to correlate the genotype with the phenotype including levels of the production of functional protein.
Clinical implications
Findings of this study have important clinical implications, specifically in the diagnosis and management of IEIs in individuals of African ancestry. The identification of both known and novel variants in genes involved in B-cell development, class-switch recombination, and immune regulation highlights the need to incorporate African genomic data into diagnostic reference frameworks. The presence of pathogenic or likely pathogenic variants in healthy individuals the absence of many potentially clinically relevant African variants in ClinVar suggests that current variant interpretation strategies, largely derived from European-ancestry datasets, may misclassify clinically relevant mutations in African populations. Furthermore, IEI of immunity range in symptomatic presentation and may be asymptomatic and present later in life, but these individuals remain at risk of related complications. This underlines the urgent need to develop population-specific diagnostic gene panels and variant annotation pipelines to enhance diagnostic yield, reduce the burden of variants of uncertain significance (VUS), and avoid false negatives. Our findings underscore the complexity of this task, revealing that many variants present in African populations remain poorly characterized. Moving forward, expanding the analysis to include all genes implicated in humoral IEIs will provide a more comprehensive view of candidate genes for inclusion in diagnostic panels. The detection of potentially adaptive, high-frequency deleterious variants in genes such as CR2 and IGHM may point to evolutionary pressures shaped by regional infectious disease burdens—an insight that could influence future research into immune resilience and vaccine response. Integrating these findings into clinical practice could significantly improve early detection, genetic counselling, and personalized treatment strategies for IEIs across diverse African populations.
Limitations
The findings presented in this study have limited clinical applicability, as the data came from healthy participants. Despite this limitation, these data provide valuable insight that can inform the design of future clinical studies. Importantly, the data reported here is a the population level rather than the individual levels. This restricts the ability to track specific variants and determine their zygosity (i.e. whether individuals were homozygous or heterozygous for a gene variant). Furthermore, the study included only 23 genes, whereas more that 60 genes have been implicated in humoral IEIs (Table 1).
Conclusions
The substantial genetic diversity found in African populations is highlighted in this study, along with its connection to humoral inborn errors of immunity (IEIs). We found 815 gene variants through AGVD analysis, including 335 present in individuals of African descent. Many of these variants impact important genes involved in immune regulation, signalling, and B-cell development. Unexpectedly, a number of variants that were deemed pathogenic or likely pathogenic were discovered in apparently healthy people, possibly indicating variable penetrance, recessive inheritance, or environmental modulation. Several variants not recorded in ClinVar were predicted to have potential functional impact and, in some cases, occurred at genotype frequencies similar to known pathogenic variants. These observations warrant further investigation to clarify their clinical relevance. These findings highlight the significance of using African genomic data in variant interpretation and the requirement for population-specific methods in the diagnosis of IEIs.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary Material 2
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tangye, S. G. et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J. Clin. Immunol.42, 1473–1507 (2022).10.1007/s 10875-022-01289-3PMC 924408835748970 · doi ↗ · pubmed ↗
- 2Edwards, E. S. J. et al. Predominantly Antibody-Deficient patients with Non-infectious complications have reduced Naive B, Treg, Th 17, and Tfh 17 cells. Front. Immunol. 10, 2593 (2019).10.3389/fimmu.2019.02593 PMC 687323431803177 · doi ↗ · pubmed ↗