Molecular epidemiology and genomic analysis of bulbul coronavirus in Guangdong, China
Lina Zhang, Shuting Chen, Huihua Li, Lixia Li, Hao Liu

TL;DR
This study investigates the prevalence and genetic characteristics of bulbul coronavirus in Guangdong, China, revealing insights into its evolution and potential zoonotic risks.
Contribution
The study provides the first detailed genomic and epidemiological analysis of bulbul coronavirus in southern China.
Findings
BuCoV was detected exclusively in Shenzhen, with all positive samples from bulbuls.
The GD2411 strain shows high nucleotide identity with HKU11 strains but low similarity to other delta coronaviruses.
Recombination and amino acid substitutions in the spike protein suggest adaptive evolution and potential host adaptation.
Abstract
Bulbul coronavirus (BuCoV), a delta coronavirus recently identified in passerine birds, remains poorly characterized regarding its ecology and evolutionary dynamics. This study aimed to determine the prevalence of BuCoV in wild avifauna, clarify its evolutionary relationship with other delta coronaviruses, and identify genetic signatures potentially relevant to host adaptation and cross-species transmission in southern China. From 2023 to 2024, we conducted molecular surveillance across 12 regions in Guangdong Province, China. A total of 2,145 avian fecal samples were collected and screened for BuCoV using real-time quantitative PCR. The complete genomes of representative strains were obtained using next-generation sequencing. Subsequent analyses included phylogenetic reconstruction using maximum likelihood methods, recombination detection using RDP4 and SimPlot, and comparative amino…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
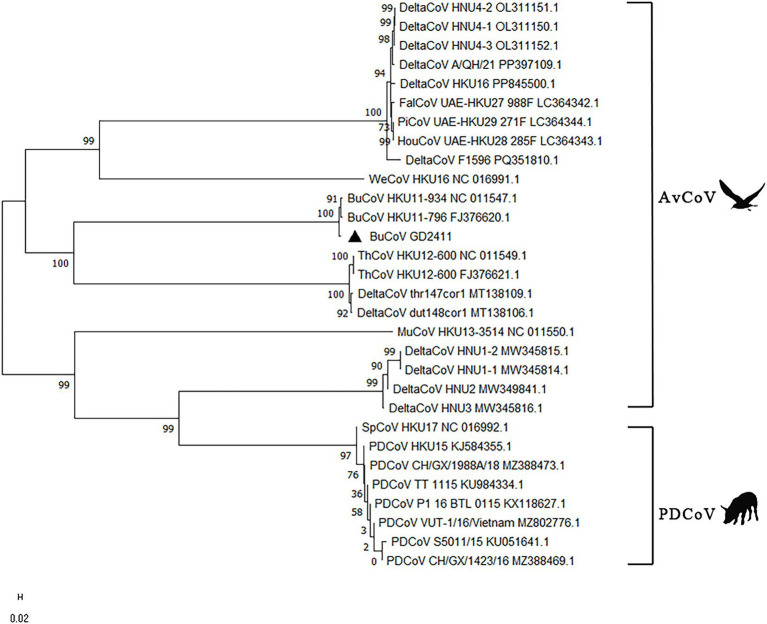 Figure 1
Figure 1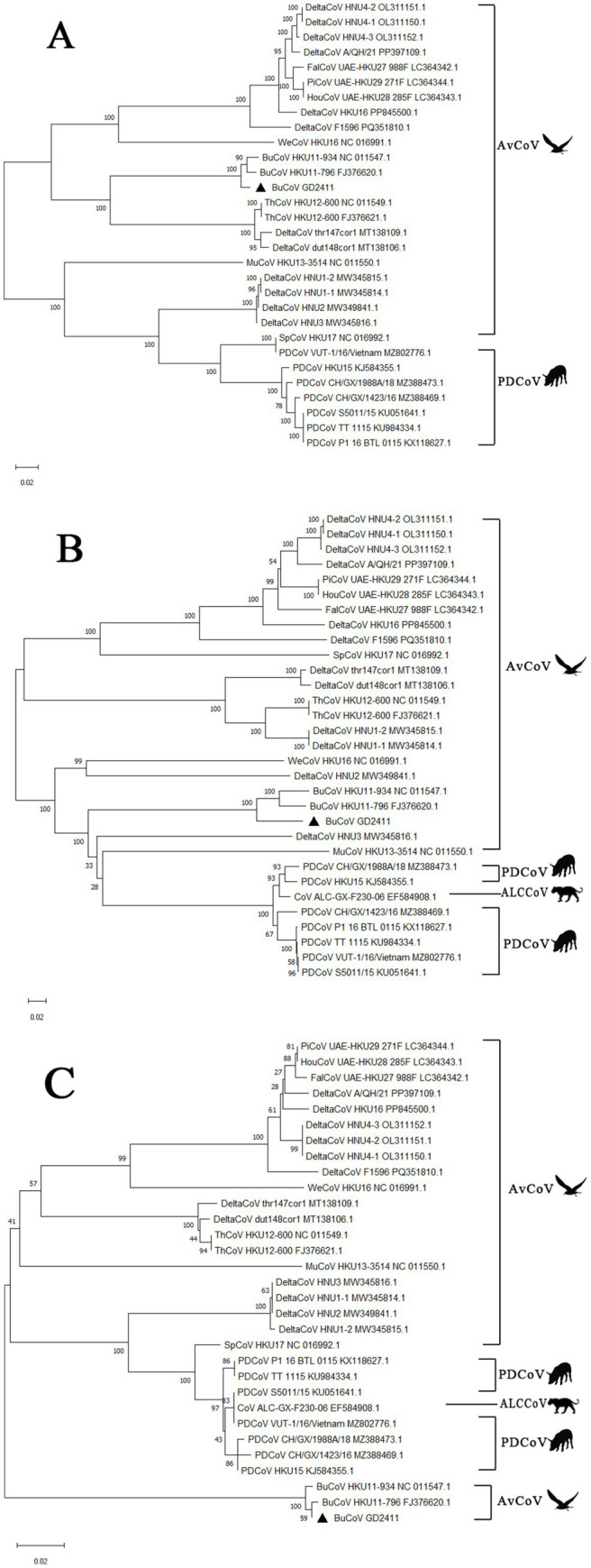 Figure 2
Figure 2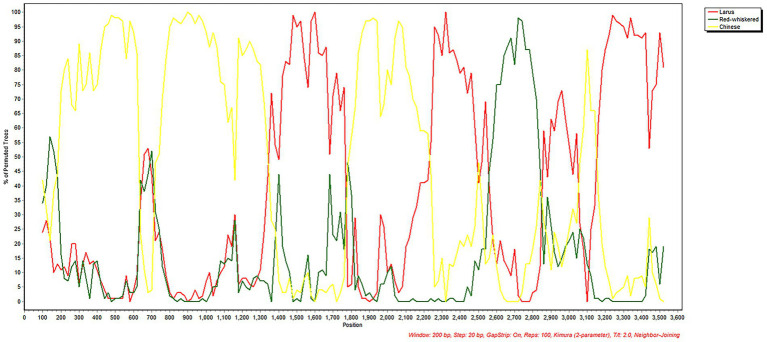 Figure 3
Figure 3| Region | Habitat type | Number of samples | BuCoV positive samples |
|---|---|---|---|
| Guangzhou | Haizhu wetland | 93 | 0 |
| Nansha wetland | 90 | 0 | |
| Shenzhen | Shenzhen mangrove reserve | 168 | 3 |
| Huizhou | Elephant Head Mountain Reserve/West Lake Park | 66 | 0 |
| Shanwei | Haifeng Nature Reserve | 144 | 0 |
| Jieyang | Great Beishan Forest Park/Binjiang Park | 90 | 0 |
| Shantou | South Australia Island Reserve | 87 | 0 |
| Chaozhou | Western Australia egret Paradise | 149 | 0 |
| Zhanjiang | Taurus Island mangrove forest | 89 | 0 |
| Maoming | Shuidong Bay Park | 131 | 0 |
| Yangjiang | Yuanyang Lake Park | 92 | 0 |
| Jiangmen | Xinhui Rock Creek Park | 60 | 0 |
| Foshan | Foshan Xianxi Reservoir | 886 | 0 |
| Total | 2,145 | 3 | |
| Protein | HKU11-796/HKU11-934 identity (%) | DeltaCoV thr147cor1/dut148cor1/HKU12-600 identity (%) |
|---|---|---|
| 3CLpro | 97.7–98.8 | 78.6–79.2 |
| RdRp | 96.9–97.9 | 83.8–84.0 |
| Hel | 98.1–98.9 | 87.3–87.7 |
| S | 92.3–94.5 | 51.5–52.2 |
| N | 98.2–98.6 | 73.7–73.8 |
| Reference strains | Amino acid position | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RdRp | 3Clpro | N | S | |||||||||
| 3,764 | 3,376 | 3,836 | 4,352 | 2,742 | 154 | 433 | 453 | 489 | 512 | 518 | ||
| GenBank number | Name | V | D | S | D | S | S | V | L | D | T | Y |
| GD2411 | I | G | H | N | N | A | I | D | N | E | N | |
| HKU11-796 | E | N | A | H | S | R | ||||||
| NC011547 | HKU11-934 | E | N | H | S | R | ||||||
| dut148cor1 | N | P | S | P | I | K | ||||||
| thr147cor1 | N | P | S | P | I | K | ||||||
| A/QH/21 | A | P | H | R | ||||||||
| HKU16 | A | P | Y | R | ||||||||
| HNU4-1 | A | P | H | R | ||||||||
| HNU4-2 | A | P | H | R | ||||||||
| HNU4-3 | A | P | H | R | ||||||||
| HNU1-1 | D | T | P | I | S | |||||||
| HNU1-2 | D | T | P | I | S | |||||||
| HNU3 | D | S | E | D | H | |||||||
| HNU2 | D | K | S | Y | T | |||||||
| UAE-HKU28 285F | A | P | Y | R | ||||||||
| UAE-HKU29 271F | A | P | Y | R | ||||||||
| UAE-HKU27 988F | A | P | P | H | R | A | ||||||
| HKU12-600 | N | P | S | P | I | S | ||||||
| NC011549 | HKU12-600 | N | S | P | I | S | ||||||
| F1596 | A | P | Q | K | ||||||||
| HKU15 | D | I | P | E | E | T | ||||||
| S5011/15 | D | I | P | E | E | T | ||||||
| CH/GX/1423/16 | D | I | P | E | E | T | ||||||
| CH/GX/1988A/18 | D | I | P | E | E | T | ||||||
| NC016992 | HKU17 | D | Y | K | E | |||||||
| VUT-1/16/Vietnam | D | I | P | E | E | T | ||||||
| NC016991 | HKU16 | I | N | N | H | S | Y | N | ||||
| NC011550 | HKU13-3514 | Q | P | D | L | S | V | L | R | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Virus Infections Studies · SARS-CoV-2 and COVID-19 Research · SARS-CoV-2 detection and testing
Introduction
1
Coronaviruses (CoVs) are enveloped, positive-sense single-stranded RNA viruses belonging to the family Coronaviridae within the order Nidovirales. They infect a wide range of hosts, including mammals and birds, and are classified into four genera: Alpha coronavirus, Beta coronavirus, Gamma coronavirus, and delta coronavirus. The White-eyed Bulbul (Pycnonotus sinensis), widely distributed in the Yangtze River basin and southern China, serves as an important biological control agent in agricultural and forest ecosystems owing to its extensive consumption of insect pests. BuCoV, first identified in Hong Kong in 2009, was taxonomically assigned to the genus Delta coronavirus (DeltaCoV), primarily infects White-eyed Bulbuls and other avian species. Infection may manifest as clinical signs involving the intestinal, respiratory, and reproductive systems, posing potential threats to avian health (1). Despite BuCoV’s demonstrated pathogenicity in birds, research on this virus remains limited both domestically and internationally, leaving critical knowledge gaps regarding its epidemiology and zoonotic potential. The ongoing SARS-CoV-2 pandemic has dramatically underscored the imperative for proactive surveillance of coronavirus diversity in wildlife reservoirs, particularly given the demonstrated capacity for cross-species transmission among delta coronaviruses (1–4). Notably, these avian coronaviruses exhibit unique RNA-dependent RNA polymerase template-switching capabilities during replication, driving frequent recombination events (5–7). In this study, we conducted large-scale molecular surveillance of wild birds across Guangdong from 2023 to 2024, followed by metagenomic sequencing and genomic characterization of representative BuCoV strains. By integrating phylogenetic, recombination, and mutational analyses, we aimed to determine the prevalence of BuCoV in wild avifauna, clarify its evolutionary relationship with other delta coronaviruses, and identify genetic signatures potentially relevant to host adaptation and cross-species transmission.
Materials and methods
2
Sample collection
2.1
A spatially stratified sampling design was implemented across major avian habitats in Guangdong Province. Fresh fecal samples were collected from March 2023 to November 2024. Specimens were immediately preserved in viral transport medium (Hank’s Balanced Salt Solution supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 2.5 μg/mL amphotericin B) and cryopreserved at −80 °C within 24 h to ensure RNA integrity.
Virus detection
2.2
RNA extraction and reverse transcription
2.2.1
For each avian fecal sample, total RNA was extracted from 200 μL of supernatant using a commercial extraction kit (Fast Pure® Viral DNA/RNA Mini Kit, Vazyme Biotech Co., Ltd., China). The extracted RNA was subsequently reverse transcribed into complementary DNA (cDNA) using a reverse transcription kit (HiScript® II 1st Strand cDNA Synthesis Kit, Vazyme Biotech Co., Ltd.). The synthesized cDNA was stored at −20 °C pending further analysis.
Real-time quantitative PCR detection
2.2.2
Based on available BuCoV gene sequences in GenBank, specific qPCR primers were designed using Primer Premier 5 software and synthesized by Sangon Biotech (Shanghai) Co., Ltd. The primer pair (YG-BuCoV-F1: 5’-ACCGAGCCTAGTACGGATAA-3′; YG-BuCoV-R1: 5’-GCAAAGTGATTCGGTGAGATAAG-3′) yielded a 113-bp amplicon. The qPCR reaction was performed in 20-μL volumes containing: 10 μL of 2 × SGE, 1.0 μL each of YG-BuCoV-F1 and YG-BuCoV-R1 primers, 4.0 μL of template cDNA, and 4.0 μL of ddH_2_O. Thermal cycling conditions comprised: initial denaturation at 95 °C for 3 min; 40 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 20 s. The specificity of the qPCR assay was validated by melting curve analysis.
Whole genome sequencing
2.3
Viral metagenomic detection was performed using the HiPure Viral RNA Kit (Magen) for viral RNA extraction. Following extraction, the VAHTS Universal V10 RNA-seq Library Prep Kit for Illumina (Novogene) was employed for library construction. High-throughput sequencing was performed on the Illumina HiSeq 2,500 platform to obtain viral genome sequence information. After sequencing, Raw sequencing reads were initially processed for quality control and filtering using the fastp software to enhance the accuracy and reliability of the data. Subsequently, the MEGAHIT assembler was utilized to assemble the filtered reads into complete viral genome sequences.
Homology and phylogenetic evolutionary analysis
2.4
Homology analysis of genomic sequences from 29 publicly available coronavirus strains (retrieved from GenBank) was performed using DNAStar software. This analysis encompassed key viral proteins-including ORF1ab (containing 3CLpro, and RdRp domains), nucleocapsid (N), and spike (S) proteins-for both nucleotide and amino acid sequence comparisons. Additionally, potential recombination events in the S protein gene of the GD2411 strain were investigated using RDP4 software. Phylogenetic analysis was conducted using MEGA 11 software, employing the Maximum Likelihood (ML) method with 1,000 bootstrap replicates under the Kimura 2-parameter model to infer evolutionary relationships (8).
Results
3
Detection of BuCoV in wild birds
3.1
Among the 2,145 fecal samples collected from 12 regions in Guangdong Province (Supplementary Figure S1), BuCoV RNA was detected exclusively in Shenzhen. Of the 168 samples obtained from this site, three tested positive, yielding a prevalence rate of 1.78% (3/168). Melting curve analysis demonstrated a single sharp peak at 80.8 °C for all positive samples, with no primer-dimer artifacts observed in negative controls (Supplementary Figure S2). No BuCoV-positive samples were identified in the remaining regions. The three positive samples originated from bulbuls (Pycnonotus spp.), confirming their role as the natural host reservoir in this region (Table 1).
Genomic features of BuCoV GD2411
3.2
High-throughput sequencing successfully recovered near-complete genomes from the three positive samples. One representative strain, designated GD2411, was selected for detailed analysis owing to its superior coverage and completeness. The genome of GD2411 comprised 26,482 nucleotides, with a typical delta coronavirus genome organization: 5′-ORF1ab-S-E-M-N-3′ (7). ORF1ab (18,791 bp) encodes a 6,262-amino acid polyprotein harboring PLpro, 3CLpro, RdRp, and other nonstructural proteins. Structural genes include the S gene (nt 19,382-22,876, 3,495 bp) and N gene (nt 24,121–25,171, 1,051 bp). Moreover, GD2411 retains the conserved Delta CoV transcriptional regulatory sequence 5′-ACACCA-3′, confirming its taxonomic placement within the genus delta coronavirus (7).
Homology and phylogenetic relationships
3.3
Pairwise sequence comparisons revealed that GD2411 shared the highest nucleotide identity with BuCoV strains HKU11-796 (97.26%) and HKU11-934 (96.79%). In contrast, nucleotide similarity with other delta coronaviruses-including porcine delta coronavirus (PDCoV), sparrow coronavirus HKU17, and night heron coronavirus HKU19-was substantially lower (78.9%–82.4%). Maximum-likelihood phylogenetic analysis based on complete genomes clustered GD2411 within a monophyletic clade together with HKU11 strains, distinctly separated from other delta coronaviruses (Figure 1). Furthermore, whole-genome comparison revealed that GD2411 shared 81.30–81.38% nucleotide identity with other delta coronavirus strains, including thr147cor1 (MT138109, 81.38%), dut148cor1 (MT138106, 81.35%), and ThCoV HKU12 (FJ376621, 81.30%), while identity with porcine delta coronavirus (PDCoV) strains ranged from 78.9% to 82.4% (Table 2) (Supplementary Figure S3).
The phylogenetic tree was constructed using the complete genome sequence of BuCoV GD2411 and 29 other coronavirus strains from various animal hosts in MEGA 11 software. Bootstrap analysis with 1,000 replicates was performed to assess the robustness of the tree topology. Bootstrap values are indicated at each node of the tree. The triangle symbol represents the isolate BuCoV GD2411.
Gene-specific homology and evolutionary divergence
3.4
Consistent with whole-genome findings, ORF1ab and its encoded domains (3CLpro, RdRp, and Hel) as well as the N protein exhibited the highest similarity with HKU11-796 and HKU11-934 (99.4% and 99.1%, respectively), with markedly reduced identity (75.6%–83.7%) compared to other delta coronaviruses (Table 2; Figures 2A, 2C). In contrast, the spike (S) protein gene demonstrated greater divergence, displaying the highest identity to HKU11-796 (94.5%) and HKU11-934 (92.3%), yet notably higher similarity to porcine delta coronavirus (PDCoV; 66.8%–67.2%) than to other avian strains (51.6%–66.0%) (Figure 2B). This pattern suggests distinct evolutionary trajectories for the S gene (Supplementary Figure S3).
Phylogenetic trees were constructed using the ORF1ab, S, and N gene sequences of BuCoV GD2411 and other coronaviruses in MEGA 11 software. The analysis employed the Bootstrap method with 1,000 replicates to assess the reliability of the phylogenetic trees. The numerical values at each node of the tree represent Bootstrap values, which indicate the statistical support for the corresponding branches. Triangular symbols denote the positions of different proteins of BuCoV GD2411 in the trees. (A) Shows the phylogenetic tree of ORF1ab. (B) Represents the phylogenetic tree of the S protein. (C) Illustrates the phylogenetic tree of the N protein.
Recombination events in the spike gene
3.5
Recombination analysis using RDP4 and SimPlot identified significant recombination breakpoints within the S gene of GD2411. These recombination events likely originated from homologous recombination among HKU11-796, HKU11-934, and the avian coronavirus Fiocruz-F1596, supporting the critical role of recombination in the evolution and diversification of BuCoV (Figure 3).
The figure illustrates the genetic recombination events involving two BuCoV strains, HKU11-796 and HKU11-934, as well as the avian coronavirus Fiocruz-F1596.
Amino acid substitutions
3.6
Comparative amino acid analysis, based on the aligned translation frames of viral open reading frames, identified 37 substitutions in GD2411 relative to HKU11-796/HKU11-934. As illustrated in the schematic translation map, the majority of mutations were localized within the S protein (n = 31), with 18 mutation sites densely clustered within a 92-bp region (nt 431–523), including V433I, L453D, D489N, T512E, and Y518N. Additional substitutions were identified in RdRp (n = 4: V3764I, D3776G, S3836H, and D4352N), 3CLpro (n = 1: S2742N), and the N protein (n = 1: S154A) (Table 3) (Supplementary Figure S4). These mutations may potentially influence viral replication efficiency, host receptor binding affinity, and immune evasion mechanisms.
Discussion
4
The detection of BuCoV exclusively in Shenzhen Mangrove Nature Reserve (1.78% positivity) represents a significant geographic extension of this virus from Hong Kong to the southern coastal region of mainland China (1). Shenzhen Mangrove Reserve constitutes a critical node in the East Asian-Australasian Flyway, serving as a wintering ground for migratory birds from Southeast Asia and northern China, where avian species from Siberia, Australia, and Southeast Asia converge during wintering periods (9). The high genetic similarity (97.26%) between GD2411 and Hong Kong strains HKU11-796/934 strongly suggests that migratory birds function as vectors, facilitating viral introduction to this specific locality.
The focal distribution of BuCoV, detected exclusively in Shenzhen despite sampling across 12 regions throughout Guangdong Province, warrants careful consideration. Several factors may account for this spatially restricted pattern. First, the Shenzhen Mangrove Nature Reserve represents a unique ecological niche characterized by high biodiversity, dense vegetation coverage, and year-round warm and humid conditions (average temperature 25 °C, relative humidity >80%). These environmental conditions may favor coronavirus persistence and transmission among avian hosts. In contrast, other sampled habitats, including inland wetlands (Haizhu, Nansha), mountain reserves (Elephant Head Mountain), and freshwater parks, present different microclimatic conditions that may be less conducive to BuCoV survival outside hosts.
Second, field observations during sampling revealed that Shenzhen Mangrove Reserve hosts the highest density of white-eyed bulbuls (Pycnonotus sinensis) in Guangdong Province. The local bulbul population comprises year-round residents, facilitating continuous virus circulation. Higher host density promotes increased contact rates and more efficient viral transmission, potentially sustaining endemic circulation that may not be achievable in areas with lower population densities. In contrast, other regions exhibited lower host densities and higher proportions of transient individuals, thereby reducing transmission opportunities.
Whole-genome sequencing and phylogenetic analyses demonstrated that BuCoV GD2411 shares the highest nucleotide identity (97.26% and 96.79%, respectively) and closest evolutionary relationship with strains HKU11-796 and HKU11-934. Furthermore, GD2411 exhibited 81.30%–81.38% nucleotide identity with Delta CoV strains including thr147cor1, dut148cor1, and ThCoV HKU12 (Table 2), with which it clustered within the same clade, indicating its classification as a novel Delta CoV member. Phylogenetic analysis of GD2411-specific genes, including 3CLpro, RdRp, and Hel within ORF1ab, as well as N genes-corroborated the whole-genome results, confirming its close genetic linkage to HKU11-796/HKU11-934. The spike (S) gene of GD2411 showed the highest identity with HKU11-796 (94.5%) and HKU11-934 (92.3%), but markedly lower identity (66.8%–67.2%) with other DeltaCoV members (e.g., PDCoV strains MZ388473, MZ388469, KU051641, KX118627, KU984334, MZ802776, and KJ584355) and Asian leopard cat coronavirus (ALC/GX/F230/06; 68.8%). This pronounced divergence suggests host-driven differential selection pressure on the S protein during coronavirus evolution, resulting in significant intra-genus divergence.
Recombination represents a hallmark of coronavirus evolution, particularly in the spike gene, which mediates host entry and tropism (10–12). GD2411 emerged through inter-lineage recombination involving HKU11-796, HKU11-934, and the avian coronavirus Fiocruz-F1596. This mosaic structure of the S gene likely confers altered antigenic or receptor-binding characteristics, potentially expanding BuCoV’s host range, which serves as a key driver of cross-species transmission, particularly through S gene recombination (13–19). These recombination events have been increasingly reported in avian delta coronaviruses and are considered a principal mechanism underlying cross-species transmission. The identification of a viable recombinant strain in a wild bird population underscores the importance of active surveillance at the wildlife-livestock-human interface.
The majority of non-synonymous mutations identified in GD2411 were localized to the spike protein, particularly within regions associated with receptor binding and membrane fusion. These substitutions may modulate viral entry efficiency, immune evasion capacity, or cross-species transmissibility. Although fewer mutations were observed in RdRp and 3CLpro, these enzymes are essential for viral replication and proteolytic processing, respectively, and even single amino acid changes can significantly impact viral fitness. Notably, the S154A substitution in the nucleocapsid protein may influence viral RNA packaging or interactions with host cellular factors.
To further elucidate the functional implications of these variations, we conducted in silico structural analysis. For the RdRp protein, the mutation V3764I (located in the viral polymerase domain) involves a substitution between two hydrophobic amino acids, suggesting a conservative change that likely maintains the structural integrity of the catalytic core while potentially fine-tuning polymerase fidelity. However, the mutations observed in the Spike protein, particularly D489N and T512E, map to the surface-exposed loops of the S1 subunit. Structural modeling suggests that these substitutions alter the local electrostatic potential, which may influence receptor recognition or antigenicity, consistent with the high recombination rate observed in this region. These structural insights support the hypothesis that GD2411 is undergoing adaptive evolution. Functional studies are warranted to determine whether these mutations confer adaptive advantages.
Cross-species transmission events mediated by coronaviruses have occurred frequently (20–22). While BuCoV has not yet been associated with zoonotic transmission, its genetic plasticity, as demonstrated by frequent recombination and adaptive mutations, raises concerns regarding its potential to breach the species barrier. The white-eyed bulbul is a common urban and peri-urban bird in southern China, frequently interacting with human-modified environments. Should BuCoV acquire mutations facilitating mammalian cell entry—as observed in other delta coronaviruses—it could pose a spillover risk. Our findings highlight the necessity for expanded surveillance of avian delta coronaviruses, particularly in regions characterized by high biodiversity and anthropogenic pressure.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Woo P-C-Y Lau S-K-P Lam C-S-F Lai KKY Huang Y Lee P . Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J Virol. (2009) 83:908–17. doi: 10.1128/jvi.01977-08, 18971277 PMC 2612373 · doi ↗ · pubmed ↗
- 2Brian D-A Baric R-S. Coronavirus genome structure and replication. Curr Top Microbiol Immunol. (2005) 287:1–30. doi: 10.1007/3-540-26765-4_115609507 PMC 7120446 · doi ↗ · pubmed ↗
- 3Ma M Ji L Ming L Xu Y Zhao C Wang T . Co-circulation of coronavirus and avian influenza virus in wild birds in Shanghai (2020-2021). Transbound Emerg Dis. (2022) 69:3985–91. doi: 10.1111/tbed.1469436054919 PMC 9538215 · doi ↗ · pubmed ↗
- 4Ye X Chen Y Zhu X Guo J Xie D Hou Z . Cross-species transmission of delta coronavirus and the origin of porcine delta coronavirus. Evol Appl. (2020) 13:2246–53. doi: 10.1111/eva.12997, 32837537 PMC 7273114 · doi ↗ · pubmed ↗
- 5Pasternak AO Willy-J-MS Eric-JS. Nidovirus transcription: how to make sense? J Gen Virol. (2006) 87:1403–21. doi: 10.1099/vir.0.81611-0, 16690906 · doi ↗ · pubmed ↗
- 6Lai M-M. RNA recombination in animal and plant viruses. Microbiol Rev. (1992) 56:61–79. doi: 10.1128/mr.56.1.61-79.1992, 1579113 PMC 372854 · doi ↗ · pubmed ↗
- 7Lai M-M. Genetic recombination in RNA viruses. Curr Top Microbiol Immunol. (1992) 176:21–32. doi: 10.1007/978-3-642-77011-1_2, 1600753 · doi ↗ · pubmed ↗
- 8Kumar S Stecher G Li M Knyaz C Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy 096, 29722887 PMC 5967553 · doi ↗ · pubmed ↗