A high-throughput biocatalytic platform for screening isomeric kainoid natural products
Robert A. Shepherd, Manasa Ramachandra, Austin R. Hopiavuori, Melanie C. Jones, Conrad A. Fihn, Alex J. Tabag, Ananya Manjunath, Limar Y. Gad, Chloe R. Whipple, Shaun M.K. McKinnie, Laura M. Sanchez

TL;DR
The paper introduces a fast method using MALDI-TIMS-MS to screen isomeric products from engineered enzymes, enabling high-throughput discovery of improved kainoid synthases.
Contribution
A MALDI-TIMS-MS platform is developed for high-throughput screening of isomeric kainoid products from enzyme variants.
Findings
Screening of 1,054 KabC variants identified seven with improved KAL formation.
The workflow retains favorable expression and in vitro stability of enzyme variants.
MALDI-TIMS-MS is established as a promising tool for isomeric product screening.
Abstract
The growing field of biocatalysis relies on the generation of large, genetically diverse libraries for downstream colorimetric, fluorogenic, or chromatographic screening. However, challenges remain when assessing enzyme variants in a high-throughput manner whose transformations yield isomeric low-molecular-weight products. The kainoid synthase subfamily of Fe/αKG-dependent dioxygenases produces the isomeric neurochemicals kainic acid (KA) and KA lactone (KAL) in a range of yields and ratios. To enable an improved throughput screening of engineered kainoid synthases, we developed a matrix-assisted laser desorption/ionization-trapped ion mobility spectrometry-mass spectrometry (MALDI-TIMS-MS) platform to directly detect regioisomeric products with near-baseline resolution from bacterial colonies. Screening of 1,054 genetically diversified KabC variants identified seven with improved KAL…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
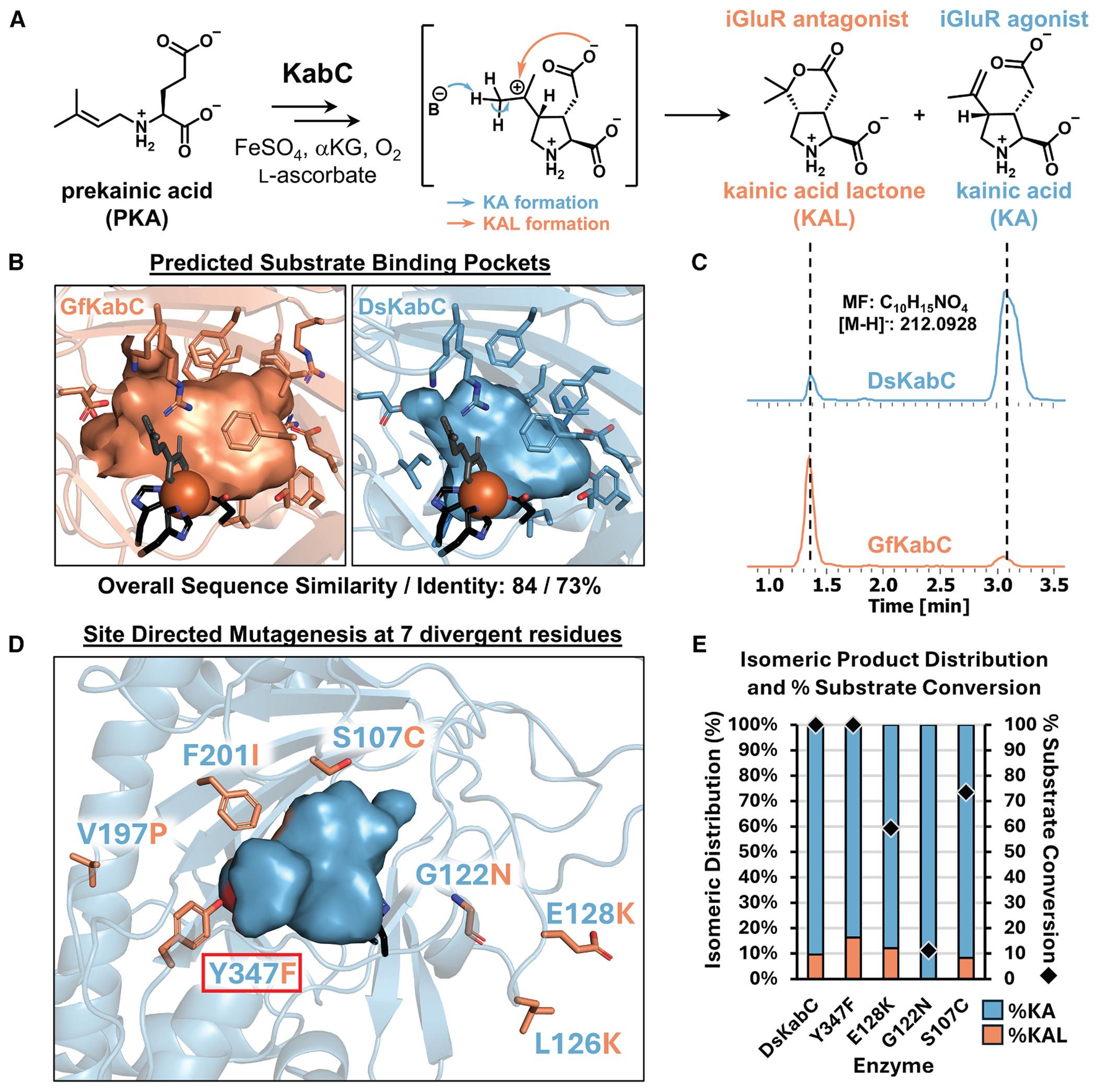 Figure 1
Figure 1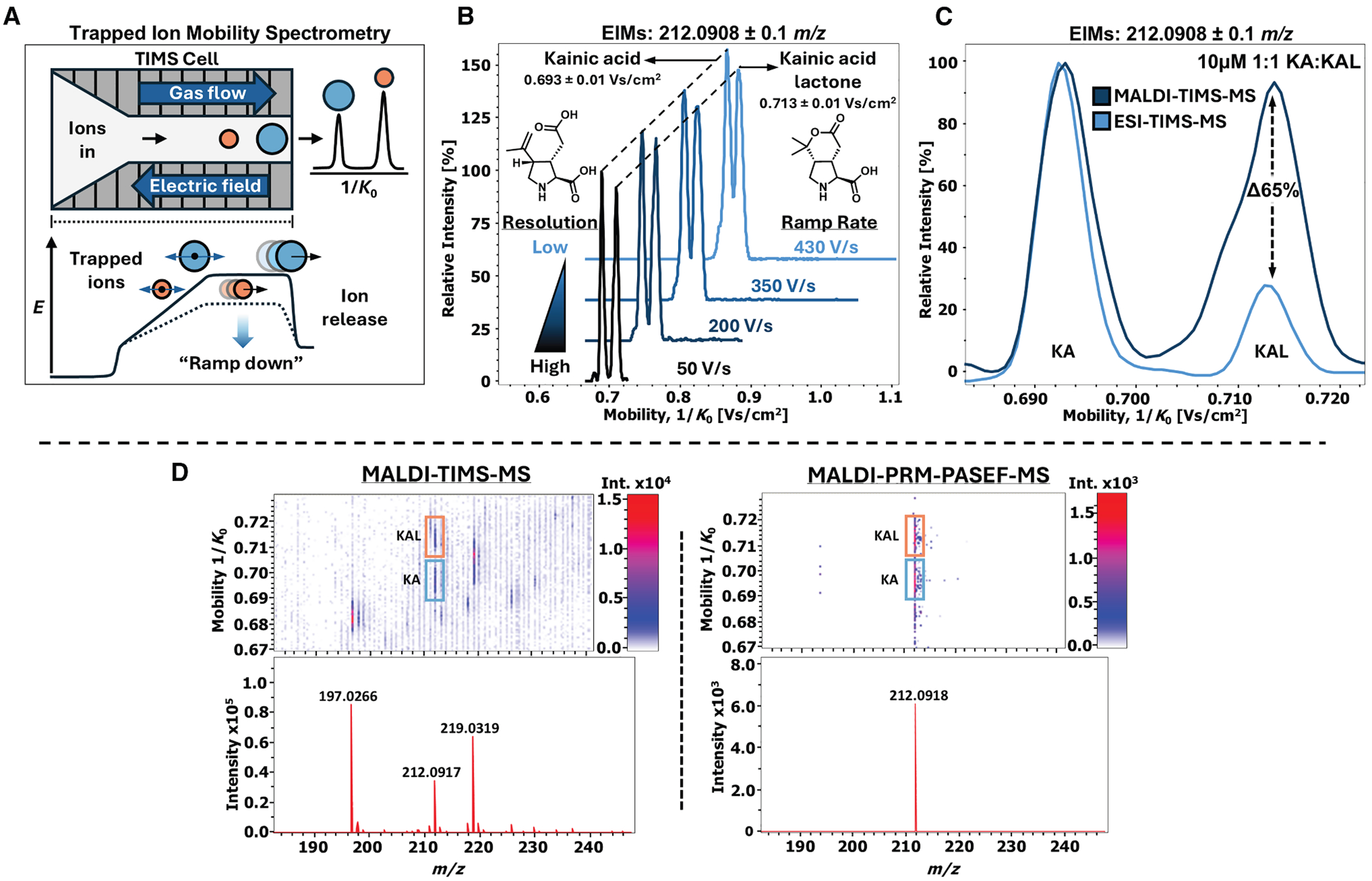 Figure 2
Figure 2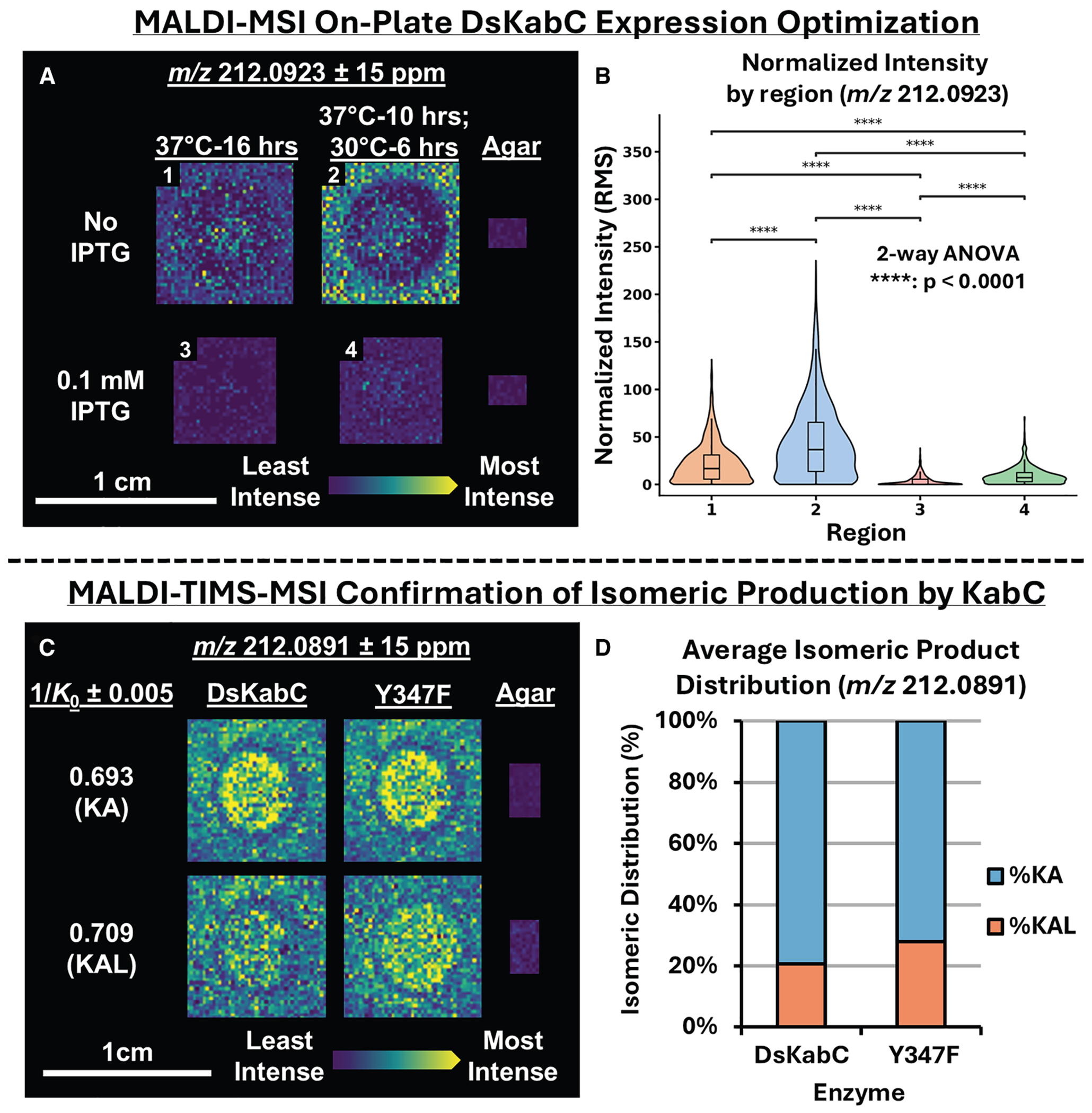 Figure 3
Figure 3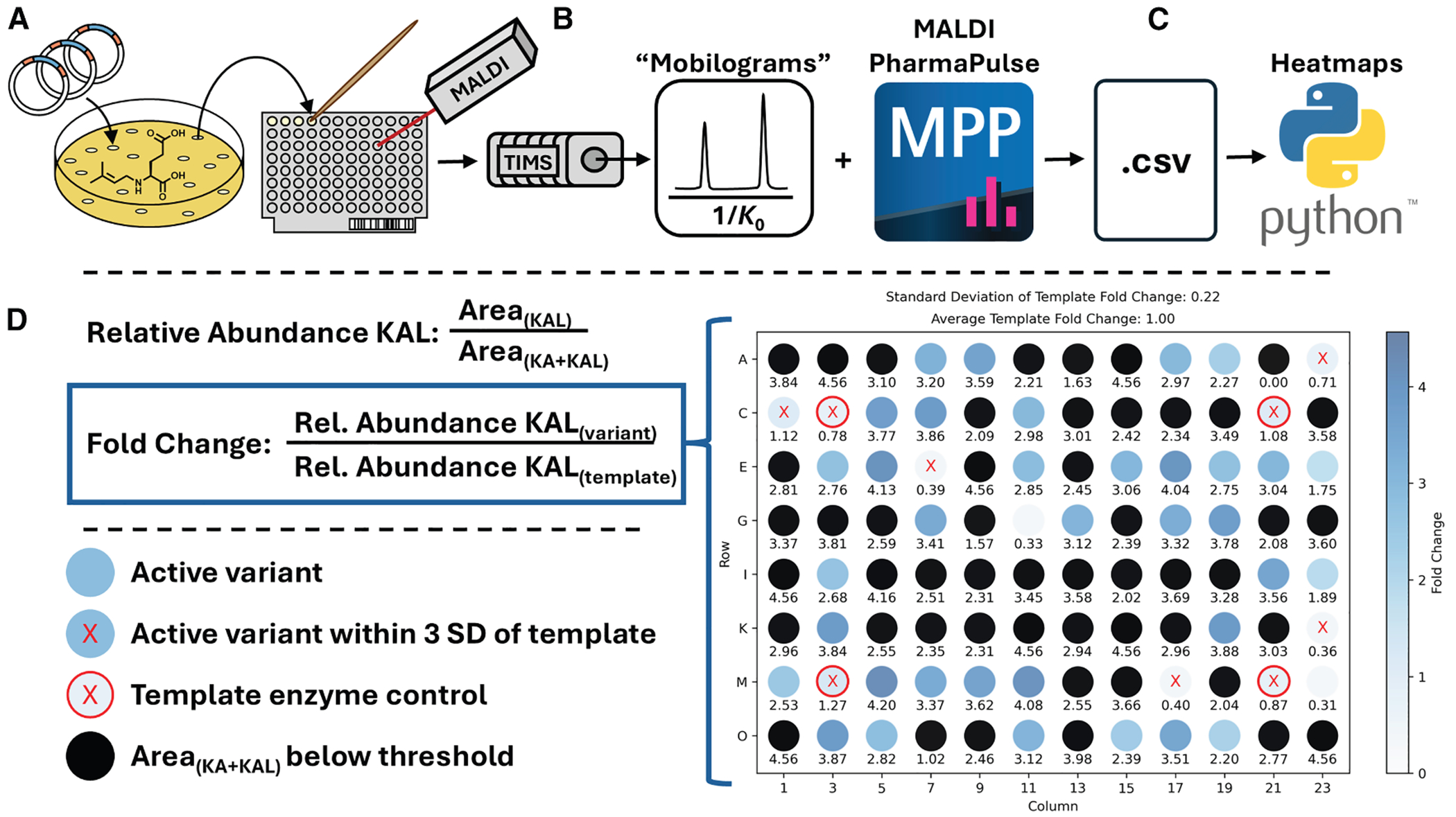 Figure 4
Figure 4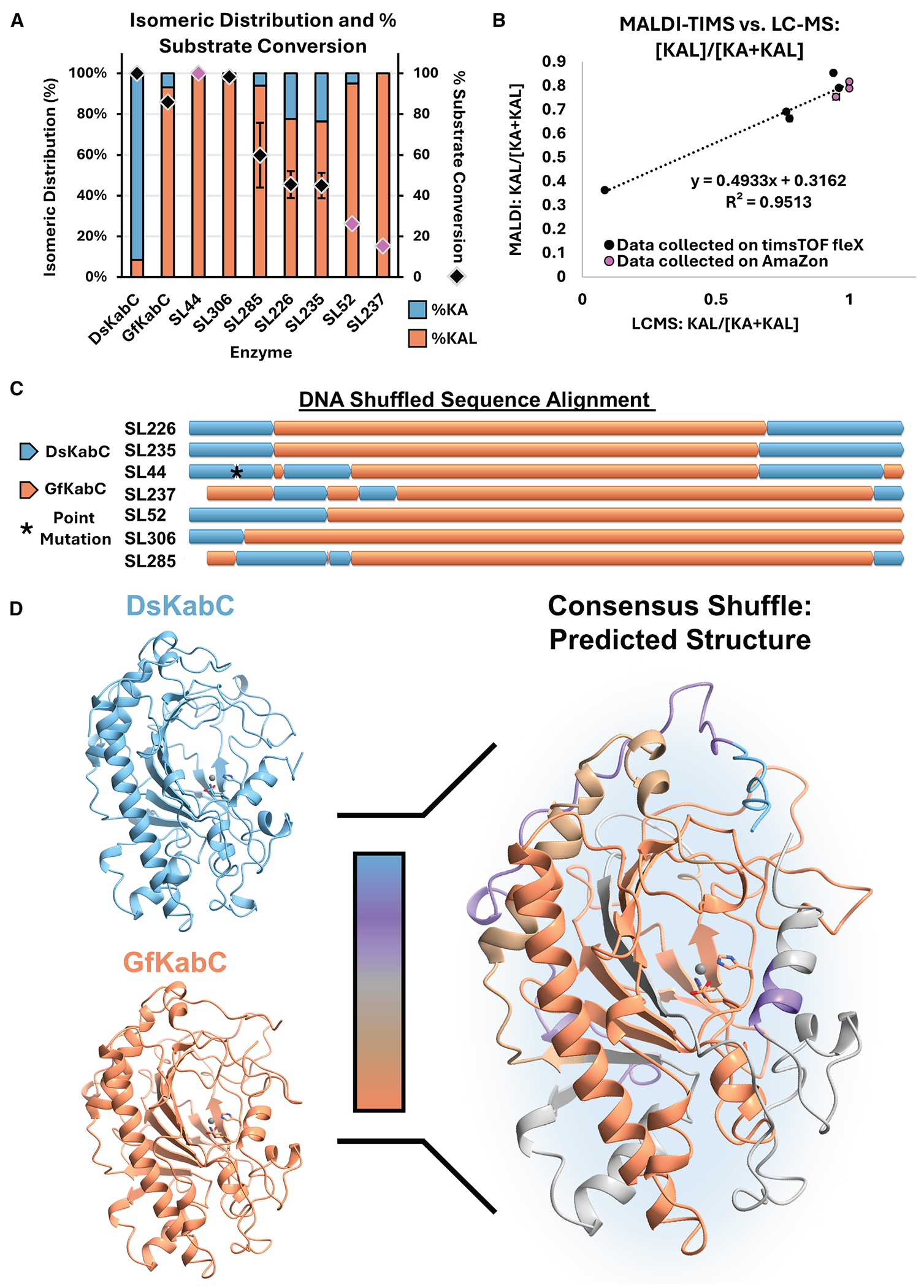 Figure 5
Figure 5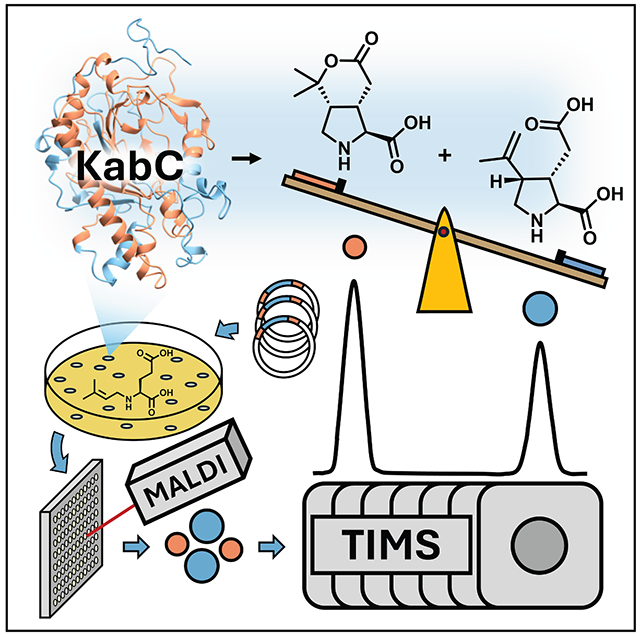 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEnzyme Catalysis and Immobilization · Microbial Natural Products and Biosynthesis · Cyclopropane Reaction Mechanisms
INTRODUCTION
Random mutagenesis techniques have significantly enhanced our ability to rapidly generate large libraries with unprecedented genetic diversity.^1,2^ For example, directed evolution of heme proteins has produced enzymes capable of catalyzing complex, new-to-Nature chemistries such as C–H amination and the formation of chiral organoboranes.^3,4^ However, random mutagenic libraries often contain hundreds to thousands of variants, making the identification of those variants capable of performing highly specialized chemical reactions a major bottleneck. Notable progress has been made using automated screening and selection technologies, such as microfluidic sorting,^5,6^ digital imaging,^7,8^ and microtiter plate workflows,^9–13^ paired with optical methods to enable high-throughput enzymatic screening. While these approaches effectively address throughput challenges, many depend on absorbent, fluorescent, or chromogenic components and require optimization for specific biochemical reactions, which limits their general applicability.
Mass spectrometry (MS)-based reaction screening approaches have gained traction over the past decade due to their label-free nature, making them highly adaptable to a wide range of screening scenarios.^14^ While MS generally sacrifices throughput, its broad applicability is a major advantage. However, MS is only suitable for transformations in which the substrate and/or products exhibit a detectable difference in mass. This limits the utility in biochemical systems that generate multiple isomeric or isobaric products, as MS alone may not have the resolution to distinguish between them. Orthogonal methods such as liquid chromatography (LC) are often employed to resolve isomeric or isobaric analytes prior to MS analysis, further reducing throughput. To address this challenge, we developed a chromatography-free high-throughput workflow to monitor isomeric products using the kainoid synthase family of enzymes as a model system.
Kainoid synthases belong to the broader superfamily of nonheme iron/α-ketoglutarate-dependent dioxygenases (Fe/αKGs), a large and versatile group of enzymes involved in both primary and secondary (specialized) metabolism.^15–18^ Fe/αKGs utilize a mononuclear Fe(II) cofactor, α-ketoglutarate, and molecular oxygen to catalyze oxidative transformations via a radical mechanism.^16–18^ These enzymes support a broad range of chemistries—including hydroxylation, halogenation, ring closure, epimerization, and epoxidation—making them ideal for diverse biocatalytic applications.^19–22^ A small subset of Fe/αKGs function as oxidative cyclases, forming new C–N^23^ or C–C bonds to construct ring systems. Isolated from various marine micro- and macroalgae, kainoid synthases catalyze the cyclization of various N-prenylated L-glutamic acid substrates to generate the pyrrolidine ring characteristic of “kainoid” marine neurotoxins.^18,24–27^ Among them, DsKabC, from the red alga Digenea simplex, oxidatively cyclizes its prekainic acid (PKA, N-dimethylallyl-L-glutamic acid) substrate to kainic acid (KA)—a potent ionotropic glutamate receptor (iGluR) agonist used extensively in neurological research and murine epilepsy models.^28–30^ DsKabC produces a minor secondary product, KA lactone (KAL) (Figures 1A and 1C), a bicyclic isomer with antagonistic iGluR activity.^31^ Intriguingly, GfKabC, an ortholog from the red alga Grateloupia filicina, produces primarily KAL in vitro, with KA being its minor secondary product (Figure 1C).^26^ Although DsKabC and GfKabC share high sequence homology, accept identical PKA substrates, and exhibit highly similar predicted substrate pocket compositions (Figure 1B), they display distinct product profiles (Figure 1C). The mechanistic branchpoint between KA and KAL formation likely occurs after pyrrolidine ring closure (Figure 1A); however, the structural basis underlying this divergence remains unclear.^26,32^
Screening KabC products at scale, however, presents significant challenges. KA and KAL are constitutional isomers that share the same chemical formula (C_10_H_15_NO_4_) but differ in structure (Figures 1A and 1C), making them indistinguishable by MS or MS/MS alone (Figure S1). Furthermore, their lack of distinguishable chromophores renders traditional absorption-based enzymatic screening methods ineffective for monitoring their relative abundance. To address this, liquid chromatography-mass spectrometry (LC-MS) is typically employed as mentioned above. While LC-MS is reliable for quantifying KA and KAL, its lengthy acquisition time—requiring up to 15 min per sample—makes it impractical for large-scale screening. We developed a matrix-assisted laser desorption/ionization MS (MALDI-MS) reaction screening platform due to its high-throughput, label-free nature, enabling the rapid analysis of a diverse range of chemical products.^33–38^ Notably, MALDI-MS has been widely applied to map mutagenic products in high-throughput, ranging from site-specific DNA modifications,^39^ to engineered microbial fatty acid composition^40^ and relative abundances of isomeric membrane lipids.^41^
Our platform was enhanced by the use of ion mobility spectrometry (IMS), which is a timescale-compatible method for gas-phase ion separation.^42–47^ A unique advantage of IMS is its ability to rapidly separate isomeric species based on their size and 3D shape.^48–50^ Here, we combine the simple sample preparation and rapid analysis of MALDI-MS with the isomeric separation capability of trapped ion mobility spectrometry (TIMS) to screen a mutant library of KabC enzymes for relative KA and KAL production. Using this MALDI-TIMS-MS platform, we evaluated a total of 1,054 random KabC variants with a total acquisition time of ~1.5 h. The results showed that random mutagenesis informed by the evolutionary similarity and functional diversity of dskabC and gfkabC is a powerful approach for rapidly probing how subtle sequence changes—particularly in second-shell or distal regions—influence active site geometry and control isomeric selectivity.
RESULTS AND DISCUSSION
Semi-rational design through site-directed mutagenesis and fusion enzyme construction
Given the absence of kainoid synthase crystal structures, we initiated our studies using AlphaFold 2^51^ to model both enzymes and select specific residues for mutation. Sequence homology across KabC orthologs highlighted residues that are generally conserved but differ between DsKabC and GfKabC. Orthologs from Palmaria palmata (PpKabC) and Rhodophysema elegans (ReKabC) also produce both KA and KAL, though with lower conversion and a consistent bias toward KA.^26^ Multiple sequence alignment further highlighted thirteen residues conserved in PpKabC, ReKabC, and uncharacterized PhKabC (Palmaria hecatensis). In contrast, DsKabC and GfKabC diverge at these positions relative to each other and to the broader ortholog set, suggesting these residues may shape kainoid isomer distribution (Figure S2). Given the reported solubility and stability constraints of GfKabC,^24,26^ the corresponding residues were introduced into the more robust DsKabC scaffold. Seven of the thirteen planned site-directed mutants (SDMs) were successfully generated and confirmed by Sanger sequencing (Figure 1D).
DsKabC SDM constructs were initially evaluated using whole-cell lysate reactions and monitored for relative product distribution via LC-MS (Figure S3). Four mutants (S107C, G122N, E128K, and Y347F) were prioritized for protein purification (Figure S4) and in vitro characterization based on either improved total conversion or increased relative KAL production (Figure S5). Of these, only E128K and Y347F showed measurable increases in KAL abundance—3% and 7%, respectively—relative to wild-type DsKabC (Figures 1E and S5). Counterintuitively, the G122N mutation eliminated KAL production and drastically reduced substrate conversion (Figure 1E). Notably, Y347F was the only mutant to retain the >99% substrate conversion seen in WT DsKabC. Prior studies in similar Fe/αKG-dependent enzymes have shown that even single mutations within the binding pocket can significantly impact activity due to the stringent geometric constraints required for radical-based catalysis.^52^ In the case of DsKabC, however, only modest changes in isomeric selectivity were observed from rational mutagenesis to their GfKabC counterparts.
Given the minimal changes in isomeric distribution observed from DsKabC SDM, we deliberately replaced larger regions with the corresponding GfKabC sequences. These “fusion” enzyme constructs were generated by targeting two regions with low sequence homology: a C-terminal alpha-helical region and an internal non-flexible loop, replacing the GfKabC sequence into the DsKabC scaffold (Figure S6). Following in vitro reactions with purified fusion KabC constructs, no substantial changes in the relative abundance of KA and KAL were observed, and substrate conversion was greatly reduced (Figures S7 and S8). This outcome underscored the complexity of kainoid synthase structure-activity relationships and motivated us to pursue random mutagenesis approaches. Given the throughput limitations of LC-MS for separating regioisomers, we prioritized developing a higher-throughput platform to enable screening of larger mutagenic KabC libraries.
Optimization of MALDI-TIMS-MS for kainoid natural products
TIMS works by “trapping” ions through a balance between the drag force from a forward-flowing gas and an opposing electric field gradient (Figure 2A). Ions are sequentially released by “ramping down” the electric field, allowing separation based on ion mobility (K), often reported as the inverse reduced mobility (1/K0) for TIMS (Figure 2A).^53^ A key feature for our platform development was optimizing the resolution of the peaks (Figure 2B). Resolution in TIMS is primarily governed by the ramp rate—the rate at which the electric field is decreased during ion release—which is determined by both the ramp time (the duration over which the electric field is reduced) and the width of the 1/K0 range being scanned.^54,55^ Increasing the ramp time allows ions with similar mobilities more time to separate in the electric field. Likewise, narrowing the 1/K0 range while keeping the ramp time constant effectively reduces the ramp rate, enhancing resolution by generating a shallower electric field gradient over a narrow mobility window. For high-throughput screening applications, these parameters can be balanced to optimize both resolution and acquisition speed.
To optimize TIMS parameters in real time, we performed direct infusion electrospray ionization (DI-ESI)-TIMS-MS on a 1:1 mixture of pure 10 μM KA and KAL across m/z 50–700 in negativeion mode. Given their structural similarity, we hypothesized that KA and KAL would have nearly identical ion mobilities, supported by predicted collisional cross-section (CCS) values of 148.3 and 147.4 Å^2^, respectively.^56^ We initially monitored a broad 1/K0 range (0.3–1.5 Vs/cm^2^) to identify the mobility of m/z 212.09. Under these conditions, the mobilogram showed only a single peak, indicating insufficient TIMS resolution to separate the two isomers. Increasing the ramp time from 50 to 100 ms revealed a shoulder peak indicative of partial resolution. Extending the ramp time further to 200 ms enhanced separation, making the second peak distinct; however, substantial overlap remained, limiting reliable relative quantification.
Since our analysis was highly targeted for KA and KAL, we opted to reduce the 1/K0 range to further enhance resolution and suppress chemical noise. By narrowing the 1/K0 range in 0.20 Vs/cm^2^ increments centered between the peaks of KA and KAL, we observed a substantial improvement in TIMS resolution. Under these conditions, we achieved near-baseline separation of KA and KAL using a range that encompassed only the analytes of interest (Figure 2A). Comparing mobilograms of pure standards confirmed the peak at 0.693 Vs/cm^2^ (147.7 Å^2^) to be KA and the peak at 0.713 Vs/cm^2^ (152.0 Å^2^) to be KAL.
It is worth noting that the use of a narrow mobility range relies heavily on accurate calibration of the 1/K0 range prior to data collection. Because it is challenging to find TIMS calibrants that span such narrow 1/K0 windows, we performed calibration over a broader range (0.60–1.30 Vs/cm^2^) using Agilent Tune Mix and subsequently reduced the mobility range for data collection. This method proved consistent, with minimal variation in the measured 1/K0 values for both KA and KAL across runs. To maintain consistency, we continued using a 1:1 KA and KAL mixture to tune TIMS parameters prior to data acquisition.
After optimizing the TIMS parameters via ESI, we translated our settings to MALDI-TIMS-MS method for high-throughput screening. Following the evaluation of several matrices in the negative ion mode, 1,5-diaminonaphthalene (1,5-DAN) was selected for MALDI analysis, as it was the only matrix that generated a signal for m/z 212.09 (Figure S9). The TIMS parameters were transferred directly to the MALDI-TIMS-MS method, and tune parameters were subsequently adjusted to improve sensitivity for KA and KAL (Table S4). Notably, during DI-ESI of the 1:1 KA and KAL mixture, the signal intensity for KAL was significantly lower than that of KA (28% relative intensity), suggesting a difference in ionization efficiency between the two analytes under ESI conditions (Figure 2C). Interestingly, when the same 1:1 mixture was analyzed via MALDI-TIMS-MS, the intensities of KA and KAL were comparable (93% relative intensity for KAL), suggesting that MALDI ionization offers a more reliable platform for relative quantitation, particularly when KAL concentrations are low. It is important to note that the 10 μM 1:1 KA:KAL mixture was prepared in neutral LC-MS grade H_2_O without acidic modifiers. The addition of 1,5-DAN matrix in the MALDI-TIMS-MS experiment may further promote KAL deprotonation relative to ESI-TIMS-MS conditions. Additionally, because KA contains two carboxylic acid moieties, it is plausible that it ionizes more efficiently under ESI negative-mode conditions relative to KAL.
Despite narrowing the mobility range, considerable chemical noise persisted in our MALDI-TIMS-MS spectra, convoluting data analysis. We hypothesized that coupling quadrupole isolation with TIMS separation could selectively isolate the kainoid signals, simplifying downstream data analysis of microbial colony material.^57^ Parallel reaction monitoring-parallel accumulation serial fragmentation (PRM-PASEF) has recently been described as a method that synchronizes quadrupole ion isolation with TIMS separation, enabling the targeted acquisition of multiple MS/MS spectra within a single TIMS ramp. Although primarily used for MS/MS, we postulated that PRM-PASEF could function as a highly targeted pseudo-MS^1^ method when operated at low collision energy to prevent fragmentation, thereby filtering out all ions except KA and KAL. The default collision cell voltage on the Bruker timsTOF fleX is 10 eV, even during MS^1^ acquisition. By setting the PRM-PASEF collision energy table to 10 eV, the method effectively operates as an MS^1^ scan with both TIMS and quadrupole ion isolation. Under these conditions, MALDI-PRM-PASEF showed only the kainoid product signal (m/z 212.0918) (Figure 2D). We carried this forward into our subsequent analyses of biological samples and mutagenic libraries. Lastly, we recently applied PRM-PASEF to resolve and characterize coproporphyrin isomers directly from microbial co-cultures on agar, demonstrating that this analytical platform can be extended to other classes of isomeric natural products beyond the kainoids.^57^
MALDI-TIMS-MSI for optimization of on-agar KabC expression
After optimizing the MALDI-TIMS-MS method to monitor KA and KAL, we turned to biological sample preparation. Mutagenic libraries are typically expressed in microbial hosts and assayed via whole-cell lysates or biotransformations, with scalable methods for KA production already reported.^26^ Although faster than purifying enzymes for in vitro assays, these approaches require multiple handling steps that can take several days, and the resulting mixtures usually require cleanup (e.g., chromatography or extraction) prior to high-throughput screening. Moreover, MALDI-MS is highly sensitive to ion suppression from salts and buffers in lysates, often necessitating additional cleanup prior to analysis, which we sought to address.^58^
Several methods have been developed for direct colony-based analysis of biotransformation reactions and secondary metabolite production across various MS modalities.^59–61^ Our lab contributed to one such platform: “IDBac,” a MALDI-time of flight (TOF)-MS-based tool designed to reduce taxonomic and chemical redundancy in the construction of microbial drug discovery libraries.^60^ Drawing on our experience with direct colony-transfer MS analysis,^62^ we adapted the sample preparation workflow from IDBac to enable monitoring of kainoid isomer formation directly from E. coli colonies spread onto MALDI target plates. We directly transformed E. coli expressing WT DsKabC and its Y347F variant (as a positive control for increased KAL production) onto lysogeny broth (LB) agar plates containing 1 mM PKA substrate (Table S1), allowing protein expression and reaction conversion to proceed overnight.
Before adopting a colony-based approach, we used MALDI-mass spectrometry imaging (MSI) to optimize on-plate induction and conversion, focusing on isopropyl β-D-1-thiogalactopyranoside (IPTG) concentration and temperature. The T7 promoter in the pET28a(+) plasmid allows basal expression even without an inducer, and although the lac operator largely mitigates this, some uninduced expression persists.^63^ To assess its impact, DsKabC was expressed both with 100 μM IPTG—based on established agar plate protocols—and without IPTG. Cultures were grown at 37° C for 10 h to promote colony formation, then shifted to 30° C for 6 h to support protein expression and product conversion. No conversion was observed at 18° C, whereas incubation at 30° C supported DsKabC expression and subsequent product formation. Interestingly, product ion intensity (m/z 212.09) was higher when IPTG was omitted, regardless of the temperature shift. The combination of IPTG omission and temperature transfer produced the highest median intensity and greatest spread in values, suggesting enhanced product conversion and higher DsKabC expression, while the temperature shift alone increased variability irrespective of IPTG presence (Figures 3A, 3B, and S9). While the signal at m/z 212.09 appears in the surrounding agar in Figure 3A, the reaction substrate (PKA) and products (KA and KAL) have been previously reported to passively diffuse across the E. coli cell membrane when grown in minimal media, which explains the observed gradient of signal.^26^ We note that minor variations in temperature or incubation time may further influence the extent of diffusion.
MSI enabled a spatial, semi-quantitative analysis of kainoid isomers in E. coli expressing either wild-type DsKabC or the Y347F mutant. The mutant showed a modest but consistent increase in KAL relative to KA, consistent with LC-MS results (Figures 1E and 3C). In the TIMS range of 0.67–0.73 Vs/cm^2^, two distinct signals at 0.693 (KA) and 0.709 Vs/cm^2^ (KAL) matched prior standard analyses for m/z 212.09 (Figure 2). Ion images confirmed differences in KA and KAL signal intensities between WT and Y347F strains (Figure 3C). This trend was further supported by plotting the average relative abundance of KA and KAL across all spectra collected for both genotypes during the MALDI-TIMS-MSI experiment (Figure 3D). Ultimately, MALDI-TIMS-MSI proved effective for optimizing on-agar expression conditions and enabling robust relative quantitation of KA and KAL, establishing a foundation for a higher-throughput, colony-based MALDI-TIMS-MS screening platform.
With biological sample preparation optimized, the next challenge was adapting our workflow for high-throughput analysis. The key modification involved transferring biomass from agar plates onto MALDI target plates, rather than analyzing biomass directly on the agar surface. This approach reduces chemical background from the media, facilitates systematic cataloging of mutant colonies for organized library screening, and enables the rapid, high-throughput “dried droplet” format typical of MALDI-MS workflows—an advantage not available in single-sample MALDI imaging experiments.
Generation of KabC chimeric library and high-throughput screening via MALDI-TIMS-MS
To build on the limited success of our rational mutagenesis approach, we next generated a random mutagenic KabC library using error-prone PCR (epPCR). Our initial epPCR library encompassed 736 KabC variants, but only modest improvements in relative KAL production were observed in first-pass screening (Figures S12–S14). To drive a more substantial shift in kainoid isomer distribution, we turned to DNA shuffling, which has been successfully employed to improve activity and alter function in other Fe/αKG systems.^64,65^ The nucleotide sequence of gfKabC was optimized from 64.9% to 82.7% dsKabC identity to improve their compatibility at degenerate codons.^64^ The two sequences were subjected to DNA shuffling (Tables S5–S8) and used to generate a chimeric library of variants with shuffled dsKabC and gfKabC sequences (Figure 4A).
To screen our chimeric KabC library in high throughput, we acquired MALDI-TIMS-MS data using the MALDI PharmaPulse (MPP) software, which is specifically designed for rapid evaluation of reactant and product screening campaigns via MALDI-MS (Figure 4B). To ensure robust analysis from single colonies, the MALDI laser was rastered randomly across each spot position (5× bursts) on the MALDI target to account for sample heterogeneity. The spectra from each raster point were summed into a consensus spectrum for each MALDI spot. The consensus spectrum was used for subsequent data analysis. The results were exported as a *.csv file, and an in-house Python script was used to visualize chimeric KabC activity as heatmaps of the fold change (FC) in relative KAL intensity with respect to the average of four DsKabC template enzyme control spots (DsKabC normalized to FC = 1 (Figures 4C and 4D).
In total, we screened 318 variants (Figures S14 and S15) from the shuffle library in <30 min (~5 s per sample). DNA shuffling enriched for active variants, many of which had fold change values ≥ 4. For sequencing, we selected 6 variants with FC values > 4, 10 variants with FC values between 3 and 4—potentially representing mutations that may lead to moderate changes in isomeric selectivity—and 2 variants with FC values < 3 that, despite lower calculated fold changes, exhibited raw mobilograms suggestive of high relative KA production (Figure S17). When plotting the total intensity of all variants, 57 had total kainoid mobilogram peak areas greater than our selection threshold (Figure S16A). Subsequently, plotting these 57 variants in terms of their relative KAL production revealed a clear split in the distribution of active variants, suggesting that 21 active variants primarily produced KA, 36 primarily produced KAL, and interestingly, none produced KA and KAL in near-equal measure (Figure S16B). All 18 variants selected for sequencing were broadly distributed across the 36 variants, selective for KAL production. Of these 18 variants, 5 corresponded to wild-type gfKabC, while 7 were determined to have shuffled sequences. The remaining 6 hits yielded poor sequencing reads.
To confirm the activity of the shuffle library hits, each of the 7 chimeric enzymes was cloned into a pET28a(+) vector, heterologously expressed in E. coli, and purified via nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (Figure S18). Each purified variant was incubated in vitro with Fe^2+^, αKG, L-ascorbate, and PKA and allowed to react for 18 h in an aerobic environment.^24^ Following conventional LC-MS analysis (Figures S19 and S20), five of the shuffled variants demonstrated improved KAL production (~94 to >99% relative KAL abundance) compared to GfKabC (~93% relative KAL abundance). Moreover, two variants (SL44 and SL306) exhibited a higher substrate consumption (>99% and ~98%, respectively) compared to GfKabC (~86%) (Figure 5A). The calculated relative abundance of KAL from the MALDI-TIMS-MS data correlated strongly with that calculated for the in vitro LC-MS data of the 7 tested shuffle variants (R^2^ = 0.95; Figure 5B). Notably, SL44 and SL306 were well-expressed with an N-terminal His tag, over-coming the solubility and expression limitations of GfKabC, which requires a maltose-binding protein tag for adequate production in E. coli.^26^ SL306 retained activity at variable enzyme concentrations (0.5–5.0 mol %) compared to GfKabC and SL44 after storage at −70° C for 4 months and multiple freeze-thaw cycles, suggesting increased stability over both GfKabC and SL44 (Figure S21).
We generated AlphaFold 3^66^ models of all seven chimeric enzymes to investigate structural features that might explain differences in activity among the variants (Figures S21 and S22). Although no specific interactions were identified to account for the observed reaction outcomes, mapping regions of sequence conservation from DsKabC and GfKabC onto a predicted consensus AlphaFold 3 model allowed us to visualize conserved features across all seven shuffle variants (Figures 5C and 5D). The protein environment surrounding the active site was consistently derived from GfKabC in all seven shuffle variants that were high KAL producers. However, increased variability was observed at both the N and C termini, suggesting that the amino acid sequences in these regions are less responsible for product distribution, corroborating the rationally designed fusion constructs, vide supra. The enzymatic preference for KAL formation is likely influenced by complex secondary-sphere interactions that affect both the active-site geometry and the substrate binding pocket, as observed in other Fe/αKG enzymes.^64,67^ Efforts are underway to examine these interactions in greater detail to provide deeper mechanistic insight, identify structural determinants of KAL production, and leverage this for broader non-native kainoid biocatalysis.
In summary, we have created and employed a high-throughput MALDI-TIMS-MS method for screening the activity of kainoid synthase biocatalysts on their native PKA substrate. Using this rapid assay that does not require enzyme purification, we generated and screened a 318-variant DNA shuffle library in ~24 h. This is a dramatic improvement over liquid-culture enzyme expression followed by LC-MS analysis, saving both time and labor. We identified seven KabC variants with improved conversion to KAL relative to GfKabC. Notably, two shuffled variants (SL44 and SL306) exhibited an almost complete shift toward KAL production, accompanied by enhanced substrate consumption, with SL306 exhibiting improved in vitro stability relative to GfKabC. Engineering kainoid synthases to mimic the isomeric output of GfKabC while retaining the favorable expression profile of DsKabC provides both a practical advantage for biocatalyst development and a robust model for analytical screening. Importantly, the combination of MALDI-TIMS-MS with enzyme engineering leverages the convenience and speed of direct colony MS analysis without sacrificing detailed analytical information, decreases reliance on LC, and enables the rapid assessment of large libraries for small molecule products. Further, PRM-PASEF enables the simultaneous analysis of multiple analytes with distinct mobility values, offering improved sensitivity and multiplexing capabilities, which is particularly advantageous for monitoring isomeric products. While demonstrated here for constitutional isomers, additional optimization can extend this platform to diastereomers and, with chemical derivatization, potentially enantiomeric products.^68^ Importantly, this strategy enables direct product readouts in enzymatic systems that are less compatible with conventional high-throughput screening (e.g., lacking chromophores, using non-chromogenic substrates, or requiring coupled assays). Finally, ongoing efforts to further engineer kainoid synthase variants will deepen our understanding of structure-function relationships and leverage this enzyme family for the construction of bioactive non-native kainoid natural products.
METHODS
Reagents
PKA, KA, and KAL were all prepared using previously established protocols.^24,26^ Phosphorus red was purchased from Spectrum Chemical MFG Corporation (New Brunswick, NJ), and Agilent Tune Mix was purchased from Agilent Technologies Inc. (Santa Clara, CA).
Preparation of kainoid standard solutions for MS analysis
A small amount of KA, KAL, and PKA was suspended in 1:1 LC/MS-grade ACN and Milli-Q ultrapure H_2_O to form 1 mM stock solutions. The stocks were diluted 100-fold to form 0.01 mM (10 μM) solutions for analysis. For MALDI-TIMS-MS analysis, equal parts of the 10 μM KA and KAL solutions were mixed and further suspended in an equal volume of 50 mM 1,5-DAN (in 1:1 ACN/H_2_O). The matrix-analyte solution (1 μL) was spotted in duplicate onto a MALDI target plate and allowed to dry in a chemical fume hood before analysis on the timsTOF fleX mass spectrometer. The same 1:1 mixture of KA and KAL (without MALDI matrix) was directly infused into the timsTOF fleX and subjected to ESI-TIMS-MS analysis (Figure 2A).
MALDI-TIMS-MSI sample preparation
pET-28a(+) plasmid containing DsKabC was transformed into E. coli BL21(DE3) competent cells, and the recovery culture was incubated at 37° C until reaching an optical density 600 (OD600) of 0.1. A 5 μL aliquot of the recovery culture was then spotted in duplicate onto thin LB agar plates containing 50 μg/mL kanamycin, 1 mM PKA, and either with or without 0.1 mM IPTG. E. coli cultures were incubated inverted for 10 h at 37° C to promote colony formation. The plates were then transferred to a 30° C incubator for an additional 6 h.
Following incubation, the E. coli colonies along with the surrounding agar (~0.5 cm around the edge of each bacterial colony) were excised from the agar plates using a sterile razor blade and gently transferred to a ground steel MALDI target plate as outlined by Yang et al.^69^ An optical image of the target plate with the colonies was taken prior to matrix application. A standard solution of 10 μM 1:1 KA:KAL was spotted on an empty spot for tuning the TIMS before acquisition. A 3:1 mixture of 1,5-DAN and 2,5-dihydroxybenzoic acid (3:1 1,5-DAN:2,5-DHB) was used as MALDI matrix and applied with a 53 μm stainless steel sieve (Hogentogler & Co., USA). The target plates were allowed to dry at 35° C for 4 h using a homemade spinning apparatus.^70^ Following drying, excess matrix was removed using compressed air, and 1 μL of saturated red phosphorus in 1:1 ACN/H_2_O was spotted as a calibrant and allowed to dry. Finally, a second optical image of the target plate was taken following matrix application for subsequent MSI acquisition.
Translation of biological sample preparation to colony-based format
To test sample preparation, we performed transformations of DsKabC, vide supra. However, instead of spotting a small volume of recovery culture, we spread the culture evenly across the agar surface to promote colony formation across the entire plate, allowing for colony picking and direct transfer to MALDI targets. After incubation, single colonies were then transferred to spots on a MALDI target using sterile toothpicks, taking care to spread as uniformly as possible to minimize sample heterogeneity between spots. When screening mutant libraries, control spots should also be included for normalization. For library generation and storage, the residual material from each colony was inoculated into the corresponding well of a 96-deepwell plate containing LB broth with 50 μg/mL kanamycin. These 96-well culture plates were then incubated overnight at 37° C, and 50:50 glycerol-water was subsequently added to allow for storage at −70° C until ready for sequencing. Once colony transfer was complete, the colony material was allowed to dry on the surface of the MALDI target for 10 min prior to matrix application. A 5 mg/mL solution of 3:1 1,5-DAN:2,5-DHB in 1:1 MilliQ H_2_O:ACN was applied using a TM Sprayer (HTX Imaging). Matrix was sprayed rather than sieved or spotted to ensure the highest possible uniformity between spots, enabling reliable relative quantitation of KA and KAL. When assessing experimental reproducibility among 92 replicates of E. coli expressing DsKabC Y347F, the coefficient of variation was ~17%, indicating variation adequate for first-pass screening (Figure S11).
RESOURCE AVAILABILITY
Lead contact
Requests for further information and resources should be directed to and will be fulfilled by the lead contact, Laura M. Sanchez ([email protected]).
Materials availability
All primers, plasmids, and generated mutant DNA are available from the lead contact upon request.
Supplementary Material
1
2
Supplemental information can be found online at https://doi.org/10.1016/j.xcrp.2025.103092.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Xiao H, Bao Z, and Zhao H (2015). High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res 54, 4011–4020.26074668 10.1021/ie 503060 a PMC 4461044 · doi ↗ · pubmed ↗
- 2Sellés Vidal L, Isalan M, Heap JT, and Ledesma-Amaro R (2023). A Primer to Directed Evolution: Current Methodologies and Future Directions. RSC Chem. Biol 4, 271–291.37034405 10.1039/d 2cb 00231 k PMC 10074555 · doi ↗ · pubmed ↗
- 3Kan SBJ, Huang X, Gumulya Y, Chen K, and Arnold FH (2017). Genetically Programmed Chiral Organoborane Synthesis. Nature 552, 132–136.29186119 10.1038/nature 24996 PMC 5819735 · doi ↗ · pubmed ↗
- 4Mc Intosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, and Arnold FH (2013). Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P 450 Enzymes in Vitro and in Vivo. Angew. Chem. Int. Ed. Engl 52, 9309–9312.23893546 10.1002/anie.201304401 PMC 3988694 · doi ↗ · pubmed ↗
- 5Caen O, Schütz S, Jammalamadaka MSS, Vrignon J, Nizard P, Schneider TM, Baret J-C, and Taly V (2018). High-Throughput Multiplexed Fluorescence-Activated Droplet Sorting. Microsyst. Nanoeng 4, 33.31057921 10.1038/s 41378-018-0033-2PMC 6220162 · doi ↗ · pubmed ↗
- 6Vallejo D, Nikoomanzar A, Paegel BM, and Chaput JC (2019). Fluorescence-Activated Droplet Sorting for Single-Cell Directed Evolution. ACS Synth. Biol 8, 1430–1440.31120731 10.1021/acssynbio.9b 00103 PMC 6942126 · doi ↗ · pubmed ↗
- 7Delagrave S, Murphy DJ, Pruss JL, Maffia AM 3rd, Marrs BL, Bylina EJ, Coleman WJ, Grek CL, Dilworth MR, Yang MM, and Youvan DC (2001). Application of a Very High-Throughput Digital Imaging Screen to Evolve the Enzyme Galactose Oxidase. Protein Eng. 14, 261–267.11391018 10.1093/protein/14.4.261 · doi ↗ · pubmed ↗
- 8KonéFMT, Le Béchec M, Sine J-P, Dion M, and Tellier C (2009). Digital Screening Methodology for the Directed Evolution of Transglycosidases. Protein Eng. Des. Sel 22, 37–44.18996967 10.1093/protein/gzn 065 · doi ↗ · pubmed ↗